

Abstract
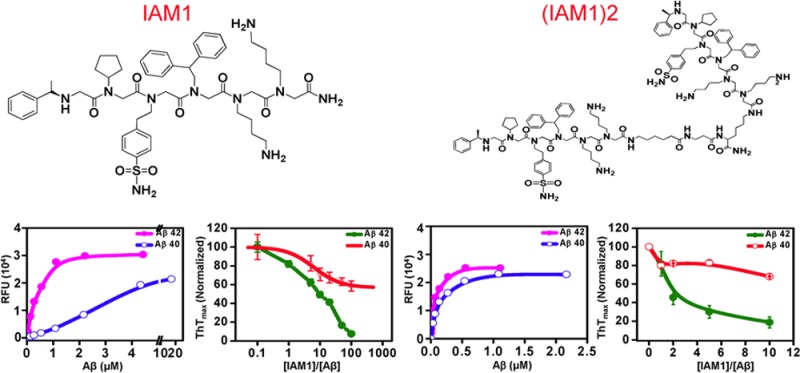
Alzheimer’s disease (AD) is the most common form of dementia and currently affects 5.4 million Americans. A number of anti-Aβ (beta amyloid) therapeutic agents have been developed for AD, but so far all of them failed in clinic. Here we used peptoid chemistry to develop ligands selective for Aβ42. Peptoids are N-substituted glycine oligomers, a class of peptidomimics. We synthesized an on-bead peptoid library consisting of 38 416 unique peptoids. The generated peptoid library was screened and arrays of Aβ42-selective peptoid ligands were identified. One of those peptoid ligands, IAM1 (inhibitor of amyloid), and the dimeric form (IAM1)2 were synthesized and evaluated in a variety of biochemical assays. We discovered that IAM1 selectively binds to Aβ42, while the dimeric derivative (IAM1)2 has a higher affinity for Aβ42. Furthermore, we demonstrated that IAM1 and (IAM1)2 were able to inhibit the aggregation of Aβ42 in a concentration-dependent manner, and that (IAM1)2 protected primary hippocampal neurons from the Aβ-induced toxicity in vitro. These results suggest that IAM1 and (IAM1)2 are specific Aβ42 ligands with antiaggregation and neuroprotective properties. IAM1, (IAM1)2, and their derivatives hold promise as Aβ42 detection agents and as lead compounds for the development of AD therapeutic agents.
Keywords: Alzheimer’s disease, peptoids, beta-amyloid peptide (42&40), scyllo-inositol, hippocampal neurons
Alzheimer’s disease (AD) is a neurodegenerative disease and a form of dementia in the elderly, which is characterized by a progressive damage to the brain cells that leads to cognitive dysfunction. Most cases of AD are sporadic and occur in the aging population, but in approximately 1–2% of cases AD segregates as an autosomal dominant trait in families. Although the cause of AD is not clear, studies have uncovered that the beta amyloid (Aβ) peptides of various lengths (typically 40 and 42 amino acids) which are generated from the amyloid precursor protein (APP) tend to aggregate and deposit as plaques in the brain of AD patients.1 The level of Aβ40 is higher in the brain, but Aβ42 is more pathogenic as it is more prone to aggregate and is more abundant in the plaques.1 These finding led to formulation of the “amyloid cascade hypothesis” that states that accumulation of synaptotoxic and neurotoxic Aβ42 oligomers cause AD.1 The “amyloid cascade hypothesis” is the major driving force behind developing anti-Aβ therapeutics for AD. A main strategy for such therapeutics is to target Aβ42, by reducing its formation, preventing its aggregation or facilitating its removal from the brain.2,3 Most efforts in those areas are focused on anti-Aβ antibodies which have demonstrated positive effects in animal models of AD.4,5 However, severe adverse effects such as encephalitis and brain inflammation accounted for the clinical failure of active immunization with Aβ.6 Poor penetration across the blood-brain-barrier (BBB) also creates significant problems in applying immunotherapy to AD.7 Experimental evidence has suggested that inhibitors which selectively prevent Aβ42 from forming aggregates or oligomers while keeping Aβ40 unperturbed may be effective therapeutic agents for AD.1,8−11 Although various small molecules were reported as Aβ ligands that inhibit Aβ aggregation or oligomerization3,12−14 and some even demonstrated in vitro or in vivo neuroprotective effects,15−19 majority of them bind to Aβ42 with low affinity and without selectivity.
To address this problem, we utilized novel chemical modality (peptoids) to develop selective and high affinity Aβ42 ligands. Peptoids are N-substituted glycine oligomers, a class of peptidomimics. They are similar to peptides in the chemical structure except that in peptoids, side chains are attached to the nitrogen instead of the α-carbon as in peptides. Our laboratory recently developed HQP09 peptoid ligand that specifically binds to expanded polyglutamine proteins20 and we applied a similar approach in the current study. To identify specific Aβ42 peptoid ligands, we synthesized an on-bead peptoid library consisting of a total of ∼38 416 unique compounds. The generated peptoid library was screened and arrays of Aβ42-selective peptoid ligands were identified. One of those peptoid ligands IAM1 (Inhibitor of Amyloid) was synthesized. A dimeric form of IAM1, (IAM1)2, was also derived. These peptoids were evaluated for their binding ability to Aβ peptides, for their inhibitory ability toward the aggregation of Aβ peptides, and for neuroprotection activity toward primary hippocampal neuronal cultures. The obtained results suggested that IAM1, (IAM1)2, and their derivatives hold promise for the development of Aβ42 detection agents and as lead compounds for the development of AD therapeutic agents.
Results and Discussion
AD is a fatal and common neurodegenerative disease. Neuronal accumulation and aggregation of Aβ42 peptides is considered to be one of the major pathological events in AD.1 A number of agents that bind and sequester Aβ42 have been developed as potential AD therapeutics.2,3 These agents include anti-Aβ antibodies and small molecules. So far not one of these agents was successful in clinic. Potential reasons for clinical setbacks include adverse side effects, poor brain permeability, and low selectivity of these agents. In the present study, we used a novel chemical modality (peptoids) to develop selective and high affinity Aβ42 ligands. These peptoids are more resistant to proteolytic degradation21 and demonstrate higher membrane permeability.22−25 When compared to peptides, the synthesis of peptoids is facile and cost-effective. As a result, peptoids can readily reach higher chemical diversity, and their bioavailability and BBB permeability can be readily adjusted by chemical modification.24,26 Both academia and the biotech industry have been developing peptoids as potential therapeutics for more than two decades.24 The peptoids have been used as scaffolds for the generation of chemically diverse libraries of novel molecules24,26 and proven as a rich source of ligands possessing biological functions for various proteins.24,26−28 The peptoids are in particularly advantage for development of novel protein–protein interaction inhibitors, something that is difficult to achieve by using small molecules.29,30 The peptoids were used to develop novel antibacterial agents31 and as a potential therapy for neonatal respiratory distress syndrome.32 A number of peptoids were studied for pharmacokinetic properties,33,34 and some even advanced into preclinical and clinical trials. To utilize peptoids more efficiently, on-bead peptoid library screening has recently been developed and applied successfully in identifying inhibitors for a variety of protein targets.35,36
Despite all these potential advantages, peptoids have not been widely used in neurodegenerative disease field. Our laboratory recently developed HQP09 peptoid ligand that specifically binds to expanded polyglutamine proteins.20 The HQP09 peptoid can potentially be useful as a therapeutic agent for treatment of Huntington’s disease (HD) or spinocerebellar ataxias resulting from polyglutamine expansion mutations.20 In another application, peptoids were developed as antibody-detection agents for AD biomarker studies.37 In the present paper, we report development of selective Aβ42-binding peptoid agent.
The peptoid library was synthesized according to a solid-phase submonomer method,28,38 which consists of an acylation step followed by a nucleophilic displacement with a primary amine. The same synthesis procedure was used in our previous studies of polyglutamine-binding peptoids.20 An essential feature of the library is that a single TentaGel macrobead is attached to multiple copies of a unique peptoid, which is achieved via combinatorial library synthesis.39 The general structure of our library (Figure 1A) included a spacer composed of two Nlys residues at the C-terminus. The other four residues (R1–R4) were variable and randomized using different amino acids (Figure 1A). The fixed spacer was used to facilitate the sequencing of R1–R4 in peptoids and to increase their water solubility, whereas the four variable residues were aimed at interacting with Aβ42. The NMR structures of Aβ42 fibrils have shown that residues 18–42 of Aβ42 form a β-turn-β-fold that is stabilized by hydrophobic interactions among three hydrophobic regions (residues 18–21, 31–36, and 39–42) and a salt bridge between Asp23 and Lys28.40 Accordingly, we reasoned that four residues would allow a peptoid to interact effectively with any hydrophobic region in Aβ42 in order to disrupt the formation of hydrophobic interactions among Aβ42 monomers, thus preventing Aβ42 from aggregating. The selection of primary amines for the construction of peptoids (Figure 1B) was done with the following considerations: (i) to maximize hydrophobic interactions between peptoids and Aβ42, four aromatic amines were chosen, as it has been shown that aromatic rings commonly exist in CNS active drug molecules and the addition of aromatic groups facilitates BBB permeability;40,41 (ii) three aliphatic amines were also selected to facilitate hydrophobic interactions; (iii) three hydrophilic amines were chosen because their hydrophilic side groups can form electrostatic interactions or hydrogen bonds with Aβ42; (iv) four additional amines were included to increase chemical diversity of the library. These 14 various amines (Figure 1B) were randomly incorporated in 4 positions to form side chains R1–R4, resulting in a combinatorial diversity of 144 = 38 416 peptoids in the library. The synthesized library was of high quality and diversity, as confirmed by sequencing of peptoid beads randomly picked out from this library.
Figure 1.
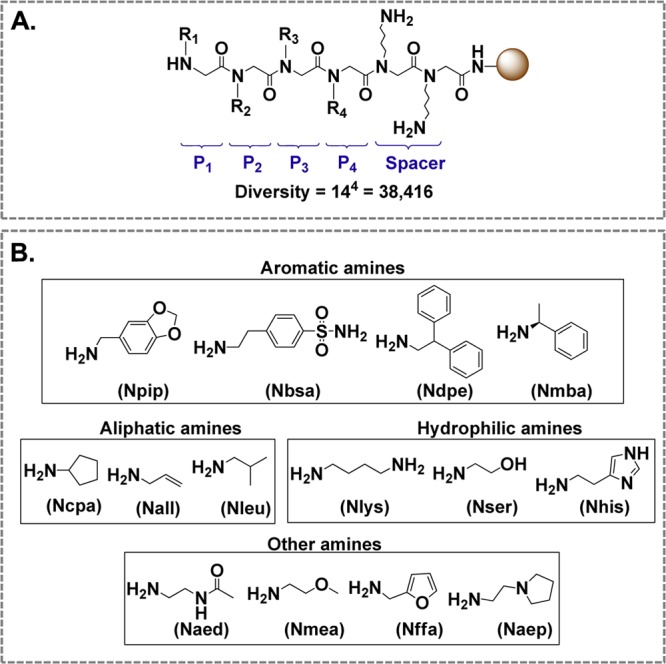
Structure and composition of peptoid library. (A) General structure of the peptoid library containing two constant monomers and four variable monomers (R1–R4). (B) Primary amines used for the synthesis of monomers. The abbreviation in the parentheses under each amine refers to the corresponding monomer formed from that amine.
To identify Aβ42 selective ligands from the library, aliquots of the library TentaGel macrobeads were screened using previously reported bead-based screening method.30 A similar screening procedure was used in our previous screen for mutant Huntingtin-binding peptoids.20 The beads were incubated with 1 μM biotin-labled Aβ42 (biotin-Aβ42) in the presence of unlabeled Aβ40 at increasing concentrations. Following incubation with biotin-Aβ42:Aβ40 mixture, the beads were incubated with streptavidin-Qdot 655 and the beads that associated with biotin-Aβ42 were visualized under a fluorescence microscope. Once excited, Qdot 655 emitted red fluorescence that appeared as a red halo around the attached bead under the lens of the microscope. The red halo appeared due to biotin-Aβ42 interaction with streptavidin. Beads that displayed such red-halos were considered as “hits”.
In our experiments, the generated peptoid library was divided into 4 equal parts of approximately 9600 beads each. Each portion of the library was incubated with 1 μM biotin-Aβ42 in the blocking buffer overnight (see Methods for details). Following overnight incubation, the beads were washed and incubated with streptavidin-Qdot 655 to visualize the Aβ42-binding peptoids. In the initial experiments, we discovered that approximately 25% of the beads were binding to Aβ42 (Table 1). To identify peptoids which are selective for Aβ42 versus Aβ40, we repeated the screens in the presence of increasing amounts of unlabeled Aβ40 (1, 10, and 20 μM) in the blocking buffer. As the molar ratio of Aβ40:Aβ42 was increased, the number of potential hits was decreased (Table 1). To identify most selective Aβ42 peptoid ligands, we focused on the screen preformed in the presence of the highest concentration of Aβ40 (20 μM) and isolated 37 potential hits obtained in this screen (Table 1). These 37 hits were designated as IAMs. Fourteen beads out of selected 37 were selected at random and sequenced by Edman degradation. Resulting peptoids (IAM1–IAM14) showed high similarity in their chemical structures (Supporting Information Figure 1). IAM1 possessed the four most frequently recurring residues critical in binding to Aβ42; therefore, we selected IAM1 (Figure 2A) for further evaluation. The random peptoid (RP) from the library was chosen to be used in control experiments (Figure 2B).
Table 1. Hit Frequency in the Peptoid Library Screens with Biotin-Aβ42 as Bait.
| screen 1 | screen 2 | screen 3 | screen 4 | |
|---|---|---|---|---|
| biotin-Aβ42:Aβ40 (mol:mol) | 1:0 | 1:1 | 1:10 | 1:20 |
| hit rate | >25% | ∼20% | ∼1.3% | ∼0.4% |
| no. of hits | >2400 | ∼1920 | ∼125 | ∼37 |
Figure 2.
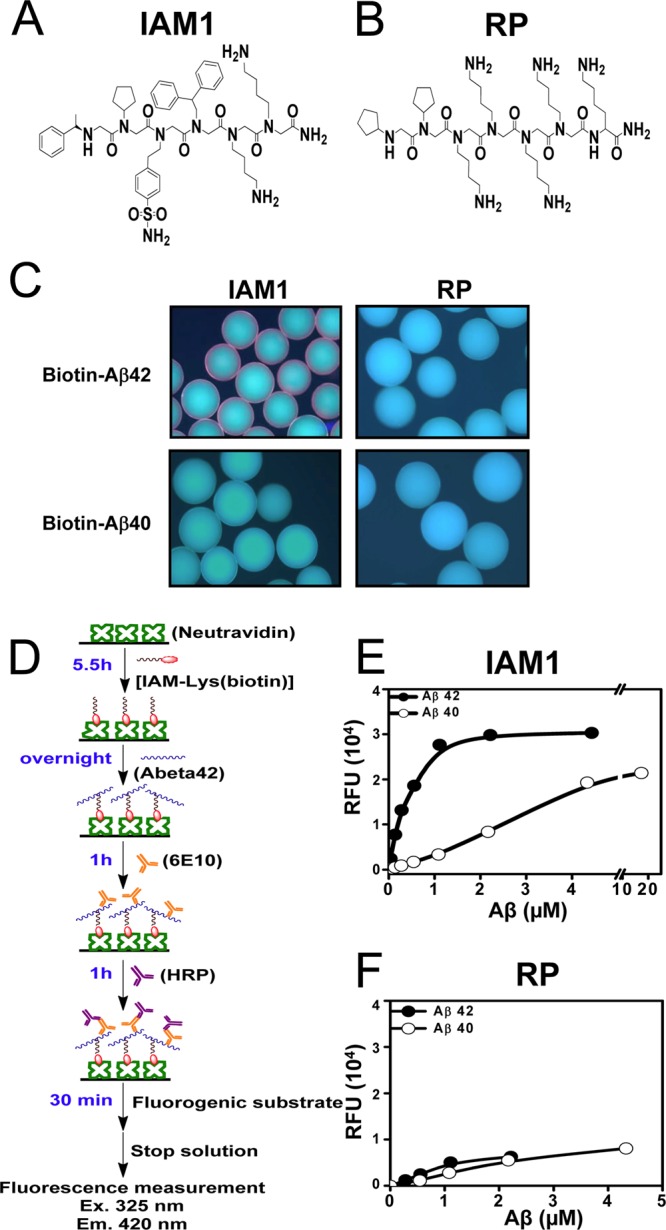
Quantitative binding affinity of peptoid IAM1 to Aβ42 and Aβ40. (A) Chemical structure of the peptoid IAM1 identified from the screen of the library. (B) Chemical structure of a random peptoid (RP) used as a negative control. (C) Biotin-Aβ42 and biotin-Aβ40 binding assay with IAM1 and RP peptoid beads. (D) Principle of solid phase binding assay with fluorescent readout. (E) Solid-state binding curve for IAM1 peptoid using synthetic Aβ42 and Aβ40. (F) Solid-state binding curve for RP peptoid using synthetic Aβ42 and Aβ40. In panels (E) and (F), the average fluorescence reading at each Aβ concentration is shown as mean ± SE (n = 3). The average fluorescence data were fitted with a nonlinear regression curve using one site binding equation.
The IAM1 and RP peptoids were resynthesized on TentaGel beads. Using the same conditions as used in the screening, the IAM1 and RP beads were incubated with biotin, biotin-Aβ42, or biotin-Aβ40 overnight. Unbound ligands were washed, and the beads were incubated with streptavidin-Qdot 655. When observed under the fluorescence microscope, only IAM1-displaying beads incubated with biotin-Aβ42 showed red fluorescent halo (Figure 2C). No red fluorescent signal was observed when IAM1 beads were incubated with biotin-Aβ40 (Figure 2C). The negative control peptoid RP was also tested in parallel. No fluorescent signal was observed when RP beads were incubated with biotin-Aβ42 or biotin-Aβ40 (Figure 2C). Incubation of IAM1 or RP with biotin did not generate any fluorescent signal (data not shown). The results obtained in on-beads binding assay confirmed that IAM1 is a specific ligand for Aβ42 and can be considered a genuine hit from the screening procedure used in our study.
In order to quantitatively determine the binding affinity of IAM1 to Aβ42, we developed a solid phase binding assay that had its origin in ELISA (Figure 2D). Similar binding assay was used in previous studies of peptoids.42,43 For these experiments, biotin-IAM1 was synthesized on Rink Amide AM resin, cleaved from the resin and purified by HPLC (Supporting Information Methods). The biotin-IAM1 was used to coat commercially available 96-well NeutrAvidin plates (Figure 2D). The solutions of increasing concentrations of Aβ42 or Aβ40 were incubated in IAM1-coated wells overnight with shaking (Figure 2D). The unbound Aβ was decanted and washed away. To measure the amount of Aβ bound to IAM1, each well was incubated sequentially with an anti-Aβ antibody 6E10 and a secondary antibody conjugated to horseradish peroxidase (HRP) (Figure 2D). The QuantaBlu was used as a substrate for HRP; and the resulting fluorescence at 325 nm excitation and 420 nm emission was recorded for each well and taken as an indication of the amount of bound Aβ (Figure 2D).
When these experiments were performed with IAM1-covered plates, a single site binding curve was observed for both Aβ42 and Aβ40 (Figure 2E). The fit to the binding curves (Figure 2E, curves) resulted in IAM1 Kd values equal to 0.43 ± 0.05 μM (n = 4) for Aβ42 and 4.12 ± 1.45 μM (n = 4) for Aβ40 (Table 2). Thus, consistent with conditions used for library screening, IAM1 is approximately 10-fold more selective for Aβ42 than for Aβ40 (Table 2). This implied that the last two residues (IA) at the C-terminus of Aβ42 contribute significantly to its binding to IAM1. Although it is unknown how those residues influence the interaction between IAM1 and Aβ42, the reported NMR structures of Aβ42 and Aβ4044−46 suggested that residues (IA) result in higher rigidity of the C-terminus of Aβ42 in comparison to the C-terminus of Aβ40. The increased rigidity may facilitate the binding of IAM1 to Aβ42. The NMR studies of IAM1 and Aβ42 complex may shed light on their interactions and are currently underway. In control experiments with biotin-RP-coated plates we failed to observe specific binding of Aβ42 or Aβ40 (Figure 2F), confirming specificity of Aβ42 association with IAM1 in a solid phase binding assay.
Table 2. Binding Affinities of Peptoids for Aβ42 and Aβ40 As Determined by Solid Phase Binding Assay.
| IAM1 | (IAM1)2 | ASR1 | |
|---|---|---|---|
| Kd (Aβ42) (μM) | 0.43 ± 0.05 | 0.06 ± 0.04 | 1.50 ± 0.34 |
| Kd (Aβ40) (μM) | 4.12 ± 1.45 | 0.12 ± 0.08 | 3.61 ± 1.16 |
| Kd (Aβ40)/Kd (Aβ42) | 9.6 | 2.1 | 2.4 |
Aggregation of Aβ42 and formation of amyloid plaques is considered one of the important pathological events in AD.1 A number of potential therapeutic compounds have been developed as inhibitors of Aβ42 aggregation.2,3 To determine if IAM1 also acts as an inhibitor of Aβ42 aggregation, we applied an in situ kinetic thioflavin T (ThT) assay.47−49 This assay is based on increase in ThT fluorescence resulting from its binding to amyloid aggregates.47 The assay is performed in a multiwell plate, and progression of Aβ42 aggregation in each well is measured by monitoring ThT fluorescence (emission 440 nm and excitation 485 nm) in each well every 10 min. A typical time course of Aβ42 aggregation in control conditions (in the presence of DMSO) is shown in Figure 3A. A similar ThT fluorescence assay was also used to monitor Aβ40 aggregation, with the typical time course of Aβ40 aggregation in control conditions shown in Figure 3B.
Figure 3.

Inhibitory ability of IAM1 toward the aggregation of Aβ42 and Aβ40 using the in situ kinetic thioflavin T (ThT) assay. (A, B) Time course of the fluorescence of aggregate-bound ThT in the presence of Aβ42 (A) or Aβ40 (B) and different compounds. The RP (100:1) was used as a negative control and anti-Aβ antibody 6E10 (20-fold dilution) as a positive control. The scyllo-Inositol (500:1) was used as a reference compound. (C, D) Time courses of the fluorescence of aggregate-bound ThT in the aggregation processes of Aβ42 (C) or Aβ40 (D) in the presence of IAM1 at different concentrations. Molar ratio of IAM1:Aβ in the range from 1:1 to 100:1 as indicated. (E) The normalized ThTmax values for the Aβ42 and Aβ40 aggregation processes are plotted as a function of IAM1 concentration. The data in each aggregation experiment were normalized to the ThTmax value obtained in the presence of DMSO, averaged and shown as mean ± SEM (n = 3).
As a positive control in these experiments, we used anti-Aβ antibody 6E10 that binds Aβ with extremely high affinity (Kd ∼ 300 pM).50 Indeed, addition of 6E10 antibodies to the aggregation mixture (at 1:20 dilution of antisera) resulted in strong suppression of Aβ42 aggregation (Figure 3A) and complete inhibition of Aβ40 aggregation (Figure 3B). Addition of 100-fold molar excess of RP had minor effect on Aβ42 aggregation (Figure 3A) and no effect on Aβ40 aggregation (Figure 3B). Scyllo-inositol (SCI) was developed recently as potential amyloid ligand51,52 and was utilized in AD clinical trials under the name AZD-103 or ELN005.53 Thus, we used SCI as a reference compound in our studies. Surprisingly, SCI exerted no effect on Aβ42 or Aβ40 aggregation in our experiments even when added at 500-fold molar excess (Figure 3A and B). In recently published findings, scyllo-inositol in comparison to other inhibitors came out as a weaker inhibitor.54
The fluorescence properties of IAM1 incubated with ThT (in the absence of Aβ42 or Aβ40) were similar to ThT in PBS (data not shown). To evaluate antiaggregation activity of IAM1, increasing concentrations of IAM1 were added to Aβ42 and Aβ40 aggregation reactions. We discovered that IAM1 inhibited Aβ42 aggregation in a concentration-dependent manner (Figure 3C). To quantify these effects, we plotted the maximum ThT fluorescence reached at the end of aggregation experiment as a function of IAM1 concentration (Figure 3E). The fit to the obtained data suggested that IAM1 can completely prevent Aβ42 aggregation with half-maximal effects observed at 10-fold molar excess of IAM1 and complete inhibition of aggregation at 100-fold molar excess of IAM1 (Figure 3C and E). To confirm these conclusions, samples of Aβ42 aggregation reaction were collected at the end of the experiment and images were taken by transmission electron microcopy (TEM). TEM reported formation of Aβ42 fibrils in the presence of DMSO (Supporting Information Figure 2). Consistent with ThT data, formation of the Aβ42 fibers was disrupted by 6E10 antibodies or in the presence of 10-fold molar excess of IAM1 (Supporting Information Figure 2). In contrast to Aβ42, IAM1 was a much less potent inhibitor of Aβ40 aggregation (Figure 3D). Even at highest concentration of IAM1 used (100-fold molar excess), ThTmax was reduced by only 40% when compared to solvent control in experiments with Aβ40 (Figure 3D, E). These results indicated that IAM1 is able to inhibit Aβ42 aggregation much more effectively that Aβ40 aggregation, in agreement with the solid-phase binding data (Figure 2E, Table 2).
IAM1 binds Aβ42 selectively, but with the modest affinity (Figure 2E, Table 2). To develop higher affinity ligand for Aβ42, we took advantage of dimerization and generated (IAM1)2, a dimeric derivative of IAM1 (Figure 4A). The dimerization was achieved by using linker between two monomeric units of parent compound. Similar dimerization strategy was used in the previous studies to develop high affinity peptoid antagonist to VEGFR2.35 (IAM1)2 was also evaluated as parent compound IAM1. To measure the binding affinity of (IAM1)2 toward Aβ42, we utilized solid state binding assay using the same procedure as for IAM1 (Figure 2D). We discovered that (IAM1)2-covered plates bound Aβ42 with high affinity (Figure 4B). The binding curve (Figure 4B, line) yielded Kd value of 0.06 ± 0.04 μM for Aβ42 (Table 2), that is 7.4-fold increase in affinity for Aβ42 when compared to parent peptoid IAM1. The higher affinity of (IAM1)2 was possibly due to the avidity effect resulted from dimerization. In the case of (IAM1)2, multiple binding sites simultaneously interacted with a target. Many binding interactions present at the same time significantly enhance avidity and selectivity is observed in many systems.55 The proposed mechanism is similar to an increase in the affinity for Aβ oligomers resulting from bridging of the two Aβ-binding peptides.56 The precedence in which nanomolar Kd values resulted from dimerization was also found in vascular endothelial growth factor receptor 2 (VEGFR2)-binding peptoids as reported by Udugamasooriya and co-workers.35 (IAM1)2 covered plates bound Aβ40 with Kd of 0.12 ± 0.08 μM (Figure 4B, Table 2), that is a 34-fold increase in affinity for Aβ40 when compared to IAM1. Thus, the increase in affinity for Aβ42 resulted in some loss of specificity of the peptoid.
Figure 4.

Evaluation of the dimeric derivative (IAM1)2. (A) Chemical structure of the dimeric derivative (IAM1)2. (B) The binding curves of (IAM1)2 with Aβ42 and Aβ40 using fluorescence solid phase binding assay. The average fluorescence reading at each Aβ concentration is shown as mean ± SE (n = 3). The average fluorescence data were fitted with a nonlinear regression curve using one site binding equation. (C, D) Time courses of the fluorescence of aggregate-bound ThT in the aggregation processes of Aβ42 (C) or Aβ40 (D) in the presence of (IAM1)2 at different concentrations. Molar ratio of (IAM1)2:Aβ in the range from 1:1 to 10:1 as indicated. (E) The normalized ThTmax values for the aggregation processes of Aβ42 and Aβ40 is plotted as a function of (IAM1)2 concentration. The data in each aggregation experiment were normalized to ThTmax value obtained in the presence of DMSO, averaged and shown as mean ± SEM (n = 3).
To further characterize activity of (IAM1)2, we evaluated the inhibitory effects of (IAM1)2 in Aβ42 and Aβ40 aggregation assays as measured in situ by ThT fluorescence. We found that (IAM1)2 efficiently inhibited Aβ42 aggregation (Figure 4C), with half-maximal inhibitory effect achieved at 2:1 molar ratio of (IAM1)2:Aβ42. That is, (IAM1)2 is approximately 5-fold more effective inhibitor of Aβ42 aggregation than IAM1, in agreement with the solid state binding results. Interestingly, (IAM1)2 was not effective in inhibiting Aβ40 aggregation (Figure 4D), with only 20% inhibition achieved even at 10-fold molar excess of the peptoid (Figure 4E). Thus, Aβ42 vs Aβ40 selectivity appears to be retained by (IAM1)2 in aggregation assay but partially lost in solid-state binding assay.
While our paper was in preparation, another group reported a development of amyloid-binding peptoid ASR1 (Figure 5A).57 It was suggested that ASR1 peptoid can be used to capture Aβ. To compare ASR1 with IAM1, we synthesized ASR1 based on published structure57 and evaluated activities of ASR1 in solid-state binding and aggregation assays with Aβ42 and Aβ40. Using solid-state binding assay we discovered that Aβ42 and Aβ40 indeed associated with ASR1-covered plates yielded Kd values of 1.50 ± 0.34 μM for Aβ42 and 3.61 ± 1.16 μM for Aβ40 (Figure 5B, Table 2). These results reflected the lower affinity of ASR1 toward Aβ42 in comparison to IAM1 and much lower than (IAM1)2 in solid-state binding assay. We further discovered that ASR1 was not able to inhibit aggregation of Aβ42 or Aβ40, even when tested in 50:1 molar excess (Figure 5C, D). Thus, we concluded that IAM1 and (IAM1)2 are much more effective Aβ42 ligands than ASR1.
Figure 5.

Evaluation of ASR1. (A) Chemical structure of ASR1. (B) The binding curves of ASR1 with Aβ42 and Aβ40 using fluorescence solid phase binding assay. The average fluorescence reading at each Aβ concentration is shown as mean ± SE (n = 3). The average fluorescence data were fitted with a nonlinear regression curve using one site binding equation (C, D) Time courses of the fluorescence of aggregate-bound ThT in the aggregation processes of Aβ42 (C) or Aβ40 (D) in the presence of ASR1 (50:1).
As hippocampus is affected most severely in AD patients,58 we evaluated the neuroprotective effects of Aβ42-binding peptoids in amyloid toxicity assay with cultured primary hippocampal neurons. In this assay, Aβ-containing conditioned media was generated by infecting cultured mouse cortical neurons with lentivirus encoding human amyloid-precusor protein (hAPP) with Swedish mutation (hAPPsw). Four or five days after infection with Lenti-APPsw, the neuronal culture media was collected and used as a source of amyloid. We reasoned that conditioned media prepared this way represents most biologically relevant source of human amyloid. A similar strategy was used in the previous studies of amyloid toxicity59 but with the conditional media from the hAPP transgenic mice cortical cultures used in the studies. By using commercial ELISA assay, we demonstrated that the concentration of Aβ42 in the conditioned media is in the range 700–800 pg/mL and concentration of Aβ40 is in the range 2000–2500 pg/mL.
The amyloid-containing conditioned media was added to DIV10 wild type mouse hippocampal neurons cultures at 1:1 dilution. In control experiments, conditional media from noninfected cortical neurons was added at 1:1 dilution. Three days after addition of conditioned media, the neurons were fixed, permeabilzed, and stained for neuronal marker MAP2. The intensity of MAP2 staining in each well was quantified by laser-based scanner. For neurons incubated with Aβ conditional medium, the loss of MAP2 staining was clearly visible when compared to control cultures (Figure 6A). On average, neuronal viability was only about 73% relative to the control (Figure 6B). Addition of Aβ-specific 6E10 antibody (at 100-fold dilution) alleviated toxic effects of amyloid conditional media and restored the neuronal viability to about 95% (Figure 6A, B). This result is in agreement with the known neuroprotective effects of this antibody. Addition of IAM1 yielded variable results and did not result in consistent neuroprotective effects (data not shown), most likely because of relatively low affinity of IAM1 for Aβ42 (Table 2). However, addition of (IAM1)2 resulted in significant and dose-dependent neuroprotective effects. (IAM1)2 had no effect on neuronal survival at concentration of 10 nM (Figure 6B), but restored neuronal viabilty to 87% at 100 nM (Figure 6A, B) and 92% at 1 μM (Figure 6B). For the neurons incubated with control media addition of 6E10 antibody or (IAM1)2 had no effect on MAP2 signals (Figure 6A), indicating that these agents are not neurotoxic. The effect of (IAM1)2 was specific, as RP peptoid had no protective effect in Aβ-toxicity assay and actually resulted in some additional toxicity for hippocampal cultures (Figure 6A, B).
Figure 6.

Neuroprotective effects of (IAM1)2 in amyloid toxicity assay. (A) MAP2 staining of neurons incubated in the control medium (upper panel) and in Aβ-containing conditioned medium (lower panel). The images are shown for DMSO, 6E10 antibodies, 100 nM RP, and 100 nM (IAM1)2 as indicated. (B) The normalized MAP2 signals for hippocampal neurons treated with Aβ-containing conditioned medium in the presence of 6E10 antibodies, RP or (IAMI)2 at increasing concentrations. The MAP2 signals were normalized to the signals in the wells that received only control medium in sister cultures. The normalized and averaged data presented as mean ± SE (n = 3). *p < 0.05; **p < 0.01 when compared to DMSO control.
Conclusion
In this study, we used a novel chemical modality (peptoids) to develop selective and potent ligand for Aβ42. By screening on-bead peptoid library with biotinylated Aβ42 in the presence of 20-fold molar excess of Aβ40, we identified an array of Aβ42-selective ligands and selected one of these peptoids (IAM1) for detailed evaluation. We confirmed that IAM1 selectively binds Aβ42 in solid state binding assay and inhibits Aβ42 aggregation in vitro. The potency of IAM1 in these assays was superior to the known Aβ-binder scyllo-inositol (SCI, AZD-103, ELND005)53 and to the recently developed Aβ-binding peptoid ASR1.57 We further evaluated dimeric derivative (IAM1)2 and demonstrated that this molecule binds Aβ42 with affinity of 60 nM, efficiently inhibits Aβ42 aggregation and exerts neuroprotective effects in amyloid-toxicity assay with cultured hippocampal neurons. Our results indicate that IAM1, (IAM1)2, and derivatives hold promise for the development of selective detection agents for Aβ42 and as lead compounds for the development of anti-Aβ therapeutic agents. Further in vivo studies to characterize the compounds BBB permeability will be necessary to validate these claims.
Methods
Materials, equipment and peptoid synthesis methods are described in the Supporting Information.
Synthesis of the Peptoid Library
Synthesis of the Two Constant Nlys Monomers
TentaGel macrobeads (1 g; capacity: 0.48 mmol/g) were incubated with 20 mL of N,N-dimethylformamide (DMF, anhydrous) at room temperature for 1 h. The beads were deprotected using 4 mL of20% piperidine in DMF for 20 min at room temperature. This step was repeated, and the beads were washed with DMF. The acylation step was carried out in a standard 25 mL glass peptide synthesis reaction vessel in an incubator shaker at 37 °C for 1 h. All the beads were incubated in a mixture of 10 mL of 1.0 M chloroacetic acid (CAA) and 10 mL of 1.0 M diisopropylcarbodiimide (DIC) in anhydrous DMF. The beads were washed with DMF. The N-(tert-butoxycarbonyl)-1,4-diaminobutane (3.8 g, 10 mmol) in 20 mL of 1-methyl-2-pyrrolidinone (NMP, anhydrous) was added to the beads and the reaction mixture was shaken at 37 °C for 2 h before beads were washed with DMF. The above reaction steps were repeated once in order to synthesize the second Nlys monomer. After the synthesis of the two constant monomers was completed, the beads were carried into the following reactions.
Synthesis of the Four Variable Monomers
CAA and DIC were added to the beads and the reaction mixture was shaken at 37 °C for 1 h. In the substitution step, the beads were split into 14 equal aliquots. Each aliquot was incubated with one of the 14 primary amines (1 M in 2 mL of NMP) chosen for this library (Figure. 1B) and the reaction was allowed to react at 37 °C for 2 h. All the aliquots of the beads were then combined and the two preceding steps were repeated three times in order to synthesize the rest three monomers. The beads were washed sequentially with DMF and dichloromethane. Protective groups were removed by incubating with a mixture of 95% trifluoroacetic acid (TFA), 2.5% triisopropylsilane (TIPS), and 2.5% water at room temperature for 1 h. The beads were then again washed sequentially with DMF and dichloromethane and stored at 4 °C until further use.
Screening of the Peptoid Library
TentaGel macrobeads of the synthesized peptoid library were allowed to swell in Tris-buffered saline (50 mM Tris, pH 7.4, 150 mM NaCl) with 0.1% Tween 20 (TBST) overnight at room temperature. Then the beads were incubated with a blocking buffer (10 mg/mL E. coli lysate and 0.5% BSA in TBST) for 1 h. The beads were then incubated with biotin-Aβ42 (1 μM in the blocking buffer, including 0, 1, 10, or 20 μM Aβ40) overnight. The unbound peptides were washed off with TBST. Finally, beads were incubated with streptavidin-Qdot 655 (1:200 dilution) in the blocking buffer for 3 h. The above reaction steps were done in a cold room. The unbound streptavidin-Qdot 655 was removed by washing with TBST. The beads were visualized under a fluorescence microscope. Thirty seven red beads were picked out as “hits” in the screen performed in the presence of 20 μM Aβ40. The 14 beads out of these 37 hits were chosen at random for sequencing. The streptavidin-Qdot 655 was stripped off the selected beads by incubating with 1% SDS at 90 °C for 20 min. The beads were sequenced by automated Edman degradation. Prior to screening, the library beads were prescreened in order to remove those that nonspecifically bound to streptavidin-Qdot 655.
Synthesis of Individual Peptoids
For on-bead binding assay, each individual peptoid was synthesized on the TentaGel beads following the same synthetic steps as in the synthesis of the peptoid library. For other assays in which peptoids were used free from the solid support, peptoids were synthesized on Rink Amide AM resin and then cleaved from the resin. Detailed synthesis procedures are presented in Supporting Information Methods. The molecular weight of each peptoid was confirmed by MALDI-TOF Mass spectrometry and presented in Supporting Information Table 1.
Preparation of Aβ Peptide Solution
Peptides solutions are prepared in this manner for the detection of beta amyloid in biological samples which requires disaggregated peptide.60 Aβ42 or Aβ40 (0.05 mg; rPeptide) was dissolved in 5 μL freshly opened 100% dimethyl sulphoxide (DMSO) (Sigma), vortexed for 1 min and sonicated for 3 min. To the DMSO solution of Aβ42 or Aβ40, ice-cold PBS buffer (filtered twice through 0.02 μm Anotop 10 syringe filters, Whatman) was added and resulted in 110 μM Aβ42 or Aβ40. This PBS solution of Aβ42 or Aβ40 was placed on ice for further use.
Solid Phase Binding Assay
A NeutrAvidin coated 96-well plate (black; Thermo Scientific) was washed three times with 200 μL/well wash buffer (Tris-buffered saline (25 mM Tris, 150 mM NaCl, pH 7.2; Thermo Scientific), 0.1% BSA, 0.05% Tween-20, 0.05% NaN3) and then incubated with IAM1-biotin, or (IAM1)2-biotin, or RP-biotin, or ASR1-biotin (62.5 nM in wash buffer, 100 μL/well) at room temperature for 5.5 h. Unbound compounds were decanted; the resulting IAM1/(IAM1)2/RP/ASR1-coated 96-well plate was washed three times with wash buffer, rinsed three times with 200 μL/well blocking Buffer (Super Block T2, Thermo Scientific) and again washed three times with wash buffer. An Aβ42 or Aβ40 peptide of increasing concentrations was added to corresponding wells (100 μL/well) and incubated at 4 °C overnight with shaking. The plate was again washed three times with wash buffer and incubated with anti-Aβ antibody 6E10 (Covance, 1:1000 dilution in blocking buffer) at room temperature for 1 h. After incubation, plate was again washed three times with wash buffer. The plate was then incubated with IgG-horseradish peroxidase (HRP, 1:1000 dilution in blocking buffer) at room temperature for 1 h. The plate was washed three times with wash buffer and incubated with (100 μL/well) of QuantaBlu fluorogenic peroxidase substrate (Thermo Scientific) at room temperature for 1 h. Stop solution (100 μL/well) was added to stop the enzymatic reaction. The fluorescence (excitation 325 nm and emission at 420 nm) of each well was measured by a Spectramax M5 microplate reader (Molecular Device).
In Situ Kinetic Thioflavin T (ThT) Aggregation Assay
The Aβ42 or Aβ40 solutions were prepared as described above. A volume of 18 μL of Aβ42 or Aβ40 (110 μM stock in PBS, 10 μM final concentration) was mixed with 2 μL stock solution of IAM1 (1, 5, 10, 20, 50, 100 mM), RP (100 mM), ASR1 (50 mM), 10 μL scyllo-inositol (500 mM), or 10 μL 6E10 (20-fold dilution). In assays with (IAMI)2, 18 μL of Aβ42 or Aβ40 (110 μM stock in PBS, 10 μM final concentration) was mixed with 2 μL stock solution of (IAM1)2 (1 mM, 2 mM, 5 mM, 10 mM). The mixtures were immediately added to corresponding wells each containing 180 μL ThT (22.2 μM stock in PBS, 20 μM final concentration) in a 96-well plate (black/clear bottom, BD Biosciences). Each sample was prepared in duplicate or triplicate. The plate was placed on a Spectramax M5 microplate reader at room temperature and the fluorescence (excitation at 440 nm and emission at 485 nm) in each well was measured every 10 min up to 24 h. The plate was automatically shaken for 5 s before each reading.
Amyloid Toxicity Assay
The hippocampal and cortical neurons of wild type (WT) mice were established respectively from postnatal day 0–1 pups and maintained in culture as described previously.61,62 The lentivirus encoding human amyloid-precusor protein with the Swedish mutation (K595N/M596L) was generated using standard molecular methods. Lenti-hAPPsw viruses were added to cortical cultures at day 4 in vitro (DIV 4). The neurons were washed and fresh medium was added on DIV 5. The Aβ conditioned media was collected at DIV 10–11. Half of the original medium for hippocampal neurons was removed and replaced with Aβ conditioned medium harvested as described above. Different concentrations of IAM1, (IAM1)2 or RP were added at DIV 7 and DIV 10. After incubation at 37 °C (5% CO2) for another 3 days, the hippocampal neurons were fixed with 4% sucrose and formaldehyde in PBS. The immunostaining of those neurons was performed as described previously,63,64 in which neurons were stained with monoclonal antimicrotubulin-associated protein-2 antibody, anti-MAP2 (Sigma, 1:1000 dilution), followed by staining with Alexa Fluor-488 anti-mouse IgG (1:1000 dilution). The quantitative analysis of MAP2 was performed blindly using the Isocyte laser scanner system (Molecular Devices). The average area was calculated automatically for each well (24-well plates) by using proprietary image analysis software (Molecular Devices). Triple wells of each group were quantified for the analysis.
Acknowledgments
Our thanks go to Dr. Kevin Luebke, Dr. Sara Chirayil, and Dr. Kathlynn Brown’s lab for technical help and the access to experimental instruments. Our thanks to Dr. Christopher Gilpin for assistance with electron microscopy analysis. Y.L. appreciates Dr. Thomas Kodadek for valuable training, Dr. Kerensa Broersen for technical advice, and Drs. Philip Thomas and Jiyong Lee for constructive discussions. I.B. is a holder of the Carl J. and Hortense M. Thomsen Chair in Alzheimer’s disease Research.
Supporting Information Available
Additional figure and methods as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
Y.L. and S.V. contributed equally to this work. I.B. designed research. Y.L., S.V., X.C., T.D., and E.P. performed research. S.S. and X.L. carried out amyloid toxicity assay. Y.L., S.V., E.P., and I.B. wrote the paper. All authors read and approved the final manuscript.
This work was supported by the Welch Foundation Grant I-1754 (I.B.), by NIH Grant R01AG030746 (I.B.), and by the contract with the Russian Ministry of Science 14.740.11.0924 (I.B.).
The international patent application for use of IAM1 and related compounds for AD treatment has been filed by UT Southwestern Medical Center.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hardy J.; Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Karran E.; Mercken M.; De Strooper B. (2011) The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discovery 10, 698–712. [DOI] [PubMed] [Google Scholar]
- Amijee H.; Scopes D. I. (2009) The quest for small molecules as amyloid inhibiting therapies for Alzheimer’s disease. J. Alzheimer’s Dis. 17, 33–47. [DOI] [PubMed] [Google Scholar]
- DeMattos R. B.; Bales K. R.; Cummins D. J.; Dodart J. C.; Paul S. M.; Holtzman D. M. (2001) Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 98, 8850–8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk D.; Barbour R.; Dunn W.; Gordon G.; Grajeda H.; Guido T.; Hu K.; Huang J.; Johnson-Wood K.; Khan K.; Kholodenko D.; Lee M.; Liao Z.; Lieberburg I.; Motter R.; Mutter L.; Soriano F.; Shopp G.; Vasquez N.; Vandevert C.; Walker S.; Wogulis M.; Yednock T.; Games D.; Seubert P. (1999) Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173–177. [DOI] [PubMed] [Google Scholar]
- Gilman S.; Koller M.; Black R. S.; Jenkins L.; Griffith S. G.; Fox N. C.; Eisner L.; Kirby L.; Rovira M. B.; Forette F.; Orgogozo J. M. (2005) Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 64, 1553–1562. [DOI] [PubMed] [Google Scholar]
- Banks W. A. (2008) Developing drugs that can cross the blood-brain barrier: applications to Alzheimer’s disease. BMC Neurosci. 9(Suppl 3), S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzi R. E.; Bertram L. (2005) Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell 120, 545–555. [DOI] [PubMed] [Google Scholar]
- McLaurin J.; Kierstead M. E.; Brown M. E.; Hawkes C. A.; Lambermon M. H.; Phinney A. L.; Darabie A. A.; Cousins J. E.; French J. E.; Lan M. F.; Chen F.; Wong S. S.; Mount H. T.; Fraser P. E.; Westaway D.; St George-Hyslop P. (2006) Cyclohexanehexol inhibitors of Abeta aggregation prevent and reverse Alzheimer phenotype in a mouse model. Nat. Med. 12, 801–808. [DOI] [PubMed] [Google Scholar]
- Shankar G. M.; Li S.; Mehta T. H.; Garcia-Munoz A.; Shepardson N. E.; Smith I.; Brett F. M.; Farrell M. A.; Rowan M. J.; Lemere C. A.; Regan C. M.; Walsh D. M.; Sabatini B. L.; Selkoe D. J. (2008) Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D. M.; Townsend M.; Podlisny M. B.; Shankar G. M.; Fadeeva J. V.; El Agnaf O.; Hartley D. M.; Selkoe D. J. (2005) Certain inhibitors of synthetic amyloid beta-peptide (Abeta) fibrillogenesis block oligomerization of natural Abeta and thereby rescue long-term potentiation. J. Neurosci. 25, 2455–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Ho L.; Zhao W.; Ono K.; Rosensweig C.; Chen L.; Humala N.; Teplow D. B.; Pasinetti G. M. (2008) Grape-derived polyphenolics prevent Abeta oligomerization and attenuate cognitive deterioration in a mouse model of Alzheimer’s disease. J. Neurosci. 28, 6388–6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Wu Z.; Butko P.; Christen Y.; Lambert M. P.; Klein W. L.; Link C. D.; Luo Y. (2006) Amyloid-beta-induced pathological behaviors are suppressed by Ginkgo biloba extract EGb 761 and ginkgolides in transgenic Caenorhabditis elegans. J. Neurosci. 26, 13102–13113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F.; Lim G. P.; Begum A. N.; Ubeda O. J.; Simmons M. R.; Ambegaokar S. S.; Chen P. P.; Kayed R.; Glabe C. G.; Frautschy S. A.; Cole G. M. (2005) Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 280, 5892–5901. [DOI] [PubMed] [Google Scholar]
- Austen B. M.; Paleologou K. E.; Ali S. A.; Qureshi M. M.; Allsop D.; El-Agnaf O. M. (2008) Designing peptide inhibitors for oligomerization and toxicity of Alzheimer’s beta-amyloid peptide. Biochemistry 47, 1984–1992. [DOI] [PubMed] [Google Scholar]
- Nerelius C.; Sandegren A.; Sargsyan H.; Raunak R.; Leijonmarck H.; Chatterjee U.; Fisahn A.; Imarisio S.; Lomas D. A.; Crowther D. C.; Stromberg R.; Johansson J. (2009) Alpha-helix targeting reduces amyloid-beta peptide toxicity. Proc. Natl. Acad. Sci. U.S.A. 106, 9191–9196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. Y.; Kim D. S. (2002) Discovery of natural products from Curcuma longa that protect cells from beta-amyloid insult: a drug discovery effort against Alzheimer’s disease. J. Nat. Prod. 65, 1227–1231. [DOI] [PubMed] [Google Scholar]
- Takahashi T.; Mihara H. (2008) Peptide and protein mimetics inhibiting amyloid beta-peptide aggregation. Acc. Chem. Res. 41, 1309–1318. [DOI] [PubMed] [Google Scholar]
- Gervais F.; Paquette J.; Morissette C.; Krzywkowski P.; Yu M.; Azzi M.; Lacombe D.; Kong X.; Aman A.; Laurin J.; Szarek W. A.; Tremblay P. (2007) Targeting soluble Abeta peptide with Tramiprosate for the treatment of brain amyloidosis. Neurobiol. Aging. 28, 537–547. [DOI] [PubMed] [Google Scholar]
- Chen X.; Wu J.; Luo Y.; Liang X.; Supnet C.; Kim M. W.; Lotz G. P.; Yang G.; Muchowski P. J.; Kodadek T.; Bezprozvanny I. (2011) Expanded polyglutamine-binding peptoid as a novel therapeutic agent for treatment of huntington’s disease. Chem. Biol. 18, 1113–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S. M.; Simon R. J.; Ng S.; Zuckermann R. N.; Kerr J. M.; Moos W. H. (1994) Proteolytic studies of homologous peptide and N-substituted glycine peptoid oligomers. Bioorg. Med. Chem. Lett. 4, 2657–2662. [Google Scholar]
- Cho S.; Choi J.; Kim A.; Lee Y.; Kwon Y. U. (2010) Efficient solid-phase synthesis of a series of cyclic and linear peptoid-dexamethasone conjugates for the cell permeability studies. J. Comb. Chem. 12, 321–326. [DOI] [PubMed] [Google Scholar]
- Miller S. M.; Simon R. J.; Ng S.; Zuckermann R. N.; Kerr J. M.; Moos W. H. (1995) Comparison of the proteolytic susceptibilities of homologous l-amino-acid, d-amino-acid, and N-substituted glycine peptide and peptoid oligomers. Drug Dev. Res. 35, 20–32. [Google Scholar]
- Zuckermann R. N.; Kodadek T. (2009) Peptoids as potential therapeutics. Curr. Opin. Mol. Ther. 11, 299–307. [PubMed] [Google Scholar]
- Lim H. S.; Reddy M. M.; Xiao X.; Wilson J.; Wilson R.; Connell S.; Kodadek T. (2009) Rapid identification of improved protein ligands using peptoid microarrays. Bioorg. Med. Chem. Lett. 19, 3866–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon R. J.; Kania R. S.; Zuckermann R. N.; Huebner V. D.; Jewell D. A.; Banville S.; Ng S.; Wang L.; Rosenberg S.; Marlowe C. K.; Spellmeyer D. C.; Tans R.; Frankelo A. D.; Santi D. V.; Cohen F. E.; Bartletts P. A. (1992) Peptoids: a modular approach to drug discovery. Proc. Natl. Acad. Sci. U.S.A. 89, 9367–9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodadek T.; Bachhawat-Sikder K. (2006) Optimized protocols for the isolation of specific protein-binding peptides or peptoids from combinatorial libraries displayed on beads. Mol. BioSyst. 2, 25–35. [DOI] [PubMed] [Google Scholar]
- Zuckermann R. N.; Martin E. J.; Spellmeyer D. C.; Stauber G. B.; Shoemaker K. R.; Kerr J. M.; Figliozzi G. M.; Goff D. A.; Siani M. A.; Simon R. J.; et al. (1994) Discovery of nanomolar ligands for 7-transmembrane G-protein-coupled receptors from a diverse N-(substituted)glycine peptoid library. J. Med. Chem. 37, 2678–2685. [DOI] [PubMed] [Google Scholar]
- Hara T.; Durell S. R.; Myers M. C.; Appella D. H. (2006) Probing the structural requirements of peptoids that inhibit HDM2-p53 interactions. J. Am. Chem. Soc. 128, 1995–2004. [DOI] [PubMed] [Google Scholar]
- Alluri P. G.; Reddy M. M.; Bachhawat-Sikder K.; Olivos H. J.; Kodadek T. (2003) Isolation of protein ligands from large peptoid libraries. J. Am. Chem. Soc. 125, 13995–14004. [DOI] [PubMed] [Google Scholar]
- Bremner J. B.; Keller P. A.; Pyne S. G.; Boyle T. P.; Brkic Z.; David D. M.; Garas A.; Morgan J.; Robertson M.; Somphol K.; Miller M. H.; Howe A. S.; Ambrose P.; Bhavnani S.; Fritsche T. R.; Biedenbach D. J.; Jones R. N.; Buckheit R. W.; Watson K. M.; Baylis D.; Coates J. A.; Deadman J.; Jeevarajah D.; McCracken A.; Rhodes D. I. (2010) Binaphthyl-based dicationic peptoids with therapeutic potential. Angew. Chem., Int. Ed. 49, 537–540. [DOI] [PubMed] [Google Scholar]
- Brown N. J.; Dohm M. T.; de la Serna J. B.; Barron A. E. (2011) Biomimetic N-terminal alkylation of peptoid analogues of surfactant protein C. Biophys. J. 101, 1076–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland C. L.; Lynn K. D.; Toombs J. E.; Dineen S. P.; Udugamasooriya D. G.; Brekken R. A. (2009) Cytokine levels correlate with immune cell infiltration after anti-VEGF therapy in preclinical mouse models of breast cancer. PloS One 4, e7669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn K. D.; Udugamasooriya D. G.; Roland C. L.; Castrillon D. H.; Kodadek T. J.; Brekken R. A. (2010) GU81, a VEGFR2 antagonist peptoid, enhances the anti-tumor activity of doxorubicin in the murine MMTV-PyMT transgenic model of breast cancer. BMC Cancer 10, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udugamasooriya D. G.; Dineen S. P.; Brekken R. A.; Kodadek T. (2008) A peptoid ″antibody surrogate″ that antagonizes VEGF receptor 2 activity. J. Am. Chem. Soc. 130, 5744–5752. [DOI] [PubMed] [Google Scholar]
- Lim H. S.; Archer C. T.; Kodadek T. (2007) Identification of a peptoid inhibitor of the proteasome 19S regulatory particle. J. Am. Chem. Soc. 129, 7750–7751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy M. M.; Wilson R.; Wilson J.; Connell S.; Gocke A.; Hynan L.; German D.; Kodadek T. (2011) Identification of candidate IgG biomarkers for Alzheimer’s disease via combinatorial library screening. Cell 144, 132–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckermann R. N.; Kerr J. M.; Kent S. B. H.; Moos W. H. (1992) Efficient Method for the Preparation of peptoids [Oligo(N-substituted glycines)] by submonomer solid-phase synthesis. J. Am. Chem. Soc. 114, 10646–10647. [Google Scholar]
- Figliozzi G. M.; Goldsmith R.; Ng S. C.; Banville S. C.; Zuckermann R. N. (1996) Synthesis of N-substituted glycine peptoid libraries. Methods Enzymol. 267, 437–447. [DOI] [PubMed] [Google Scholar]
- Andrews P. R.; Lloyd E. J. (1982) Molecular conformation and biological activity of central nervous system active drugs. Med. Res. Rev. 2, 355–393. [DOI] [PubMed] [Google Scholar]
- Andrews P. R.; Lloyd E. J. (1983) A common structural basis for c.n.s. drug action. J. Pharm. Pharm. 35, 516–518. [DOI] [PubMed] [Google Scholar]
- Udugamasooriya D. G.; Ritchie C.; Brekken R. A.; Kodadek T. (2008) A peptoid antagonist of VEGF receptor 2 recognizes a ’hotspot’ in the extracellular domain distinct from the hormone-binding site. Bioorg. Med. Chem. 16, 6338–6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeVine H. III. (2006) Biotin-avidin interaction-based screening assay for Alzheimer’s beta-peptide oligomer inhibitors. Anal. Biochem. 356, 265–272. [DOI] [PubMed] [Google Scholar]
- Shen L.; Ji H. F.; Zhang H. Y. (2008) Why Is the C-terminus of Abeta(1–42) more unfolded than that of Abeta(1–40)? Clues from hydrophobic interaction. J. Phy. Chem. B 112, 3164–3167. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Liu J.; McCallum S. A.; Yang D.; Wang C. (2007) Methyl dynamics of the amyloid-beta peptides Abeta40 and Abeta42. Biochem. Biophys. Res. Commun. 362, 410–414. [DOI] [PubMed] [Google Scholar]
- Yam A. Y.; Wang X.; Gao C. M.; Connolly M. D.; Zuckermann R. N.; Bleu T.; Hall J.; Fedynyshyn J. P.; Allauzen S.; Peretz D.; Salisbury C. M. (2011) A universal method for detection of amyloidogenic misfolded proteins. Biochemistry 50, 4322–4329. [DOI] [PubMed] [Google Scholar]
- Baine M.; Georgie D. S.; Shiferraw E. Z.; Nguyen T. P.; Nogaj L. A.; Moffet D. A. (2009) Inhibition of Abeta42 aggregation using peptides selected from combinatorial libraries. J. Pept. Sci. 15, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuperstein I.; Broersen K.; Benilova I.; Rozenski J.; Jonckheere W.; Debulpaep M.; Vandersteen A.; Segers-Nolten I.; Van Der Werf K.; Subramaniam V.; Braeken D.; Callewaert G.; Bartic C.; D’Hooge R.; Martins I. C.; Rousseau F.; Schymkowitz J.; De Strooper B. (2010) Neurotoxicity of Alzheimer’s disease Abeta peptides is induced by small changes in the Abeta42 to Abeta40 ratio. EMBO J. 29, 3408–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolder S. G.; Sagis L. M.; Venema P.; van der Linden E. (2007) Thioflavin T and birefringence assays to determine the conversion of proteins into fibrils. Langmuir 23, 4144–4147. [DOI] [PubMed] [Google Scholar]
- Canovi M.; Markoutsa E.; Lazar A. N.; Pampalakis G.; Clemente C.; Re F.; Sesana S.; Masserini M.; Salmona M.; Duyckaerts C.; Flores O.; Gobbi M.; Antimisiaris S. G. (2011) The binding affinity of anti-Abeta1–42 MAb-decorated nanoliposomes to Abeta1–42 peptides in vitro and to amyloid deposits in post-mortem tissue. Biomaterials 32, 5489–5497. [DOI] [PubMed] [Google Scholar]
- McLaurin J.; Golomb R.; Jurewicz A.; Antel J. P.; Fraser P. E. (2000) Inositol stereoisomers stabilize an oligomeric aggregate of Alzheimer amyloid beta peptide and inhibit abeta -induced toxicity. J. Biol. Chem. 275, 18495–18502. [DOI] [PubMed] [Google Scholar]
- Townsend M.; Cleary J. P.; Mehta T.; Hofmeister J.; Lesne S.; O’Hare E.; Walsh D. M.; Selkoe D. J. (2006) Orally available compound prevents deficits in memory caused by the Alzheimer amyloid-beta oligomers. Ann. Neurol. 60, 668–676. [DOI] [PubMed] [Google Scholar]
- Ma K.; Thomason L. A.; McLaurin J. (2012) scyllo-Inositol, preclinical, and clinical data for Alzheimer’s disease. Adv. Pharmacol. 64, 177–212. [DOI] [PubMed] [Google Scholar]
- Sinha S.; Du Z.; Maiti P.; Klarner F. G.; Schrader T.; Wang C.; Bitan G. (2012) Comparison of three amyloid assembly inhibitors: The sugar scyllo-inositol, the polyphenol epigallocatechin gallate, and the molecular tweezer CLR01. ACS Chem. Neurosci. 3, 451–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiessling L. L.; Gestwicki J. E.; Strong L. E. (2006) Synthetic multivalent ligands as probes of signal transduction. Angew. Chem., Int. Ed. 45, 2348–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke A. A.; Ung P. M.; Quintero J. J.; Carlson H. A.; Gestwicki J. E. (2010) Chemical probes that selectively recognize the earliest Aβ oligomers in complex mixtures. J. Am. Chem. Soc. 132, 17655–17657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C. M.; Yam A. Y.; Wang X.; Magdangal E.; Salisbury C.; Peretz D.; Zuckermann R. N.; Connolly M. D.; Hansson O.; Minthon L.; Zetterberg H.; Blennow K.; Fedynyshyn J. P.; Allauzen S. (2010) Abeta40 oligomers identified as a potential biomarker for the diagnosis of Alzheimer’s disease. PloS One 5, e15725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H.; Braak E. (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. [DOI] [PubMed] [Google Scholar]
- Wu H. Y.; Hudry E.; Hashimoto T.; Kuchibhotla K.; Rozkalne A.; Fan Z.; Spires-Jones T.; Xie H.; Arbel-Ornath M.; Grosskreutz C. L.; Bacskai B. J.; Hyman B. T. (2010) Amyloid {beta} induces the morphological neurodegenerativetriad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J. Neurosci. 30, 2636–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broersen K.; Jonckheere W.; Rozenski J.; Vandersteen A.; Pauwels K.; Pastore A.; Rousseau F.; Schymkowitz J. (2011) A standardized and biocompatible preparation of aggregate-free amyloid beta peptide for biophysical and biological studies of Alzheimer’s disease. Protein Eng., Des. Sel. 24, 743–750. [DOI] [PubMed] [Google Scholar]
- Tang T. S.; Slow E.; Lupu V.; Stavrovskaya I. G.; Sugimori M.; Llinas R.; Kristal B. S.; Hayden M. R.; Bezprozvanny I. (2005) Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 102, 2602–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Fu Y.; Altier C.; Platzer J.; Surmeier D. J.; Bezprozvanny I. (2006) CaV1.2 and CaV1.3 neuronal L-type calcium channels: differential targeting and signaling to pCREB. Eur. J. Neurosci. 23, 2297–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang T. S.; Guo C.; Wang H.; Chen X.; Bezprozvanny I. (2009) Neuroprotective effects of inositol 1,4,5-trisphosphate receptor C-terminal fragment in a Huntington’s disease mouse model. J. Neurosci. 29, 1257–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Tang T. S.; Tu H.; Nelson O.; Herndon E.; Huynh D. P.; Pulst S. M.; Bezprozvanny I. (2009) Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J. Neurosci. 29, 9148–9162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.