

Abstract
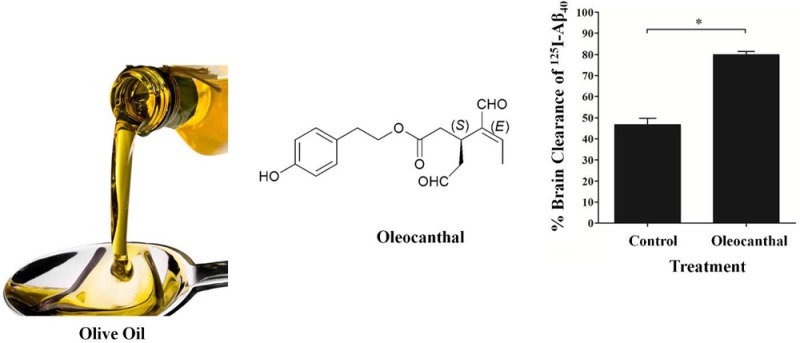
Oleocanthal, a phenolic component of extra-virgin olive oil, has been recently linked to reduced risk of Alzheimer’s disease (AD), a neurodegenerative disease that is characterized by accumulation of β-amyloid (Aβ) and tau proteins in the brain. However, the mechanism by which oleocanthal exerts its neuroprotective effect is still incompletely understood. Here, we provide in vitro and in vivo evidence for the potential of oleocanthal to enhance Aβ clearance from the brain via up-regulation of P-glycoprotein (P-gp) and LDL lipoprotein receptor related protein-1 (LRP1), major Aβ transport proteins, at the blood-brain barrier (BBB). Results from in vitro and in vivo studies demonstrated similar and consistent pattern of oleocanthal in controlling Aβ levels. In cultured mice brain endothelial cells, oleocanthal treatment increased P-gp and LRP1 expression and activity. Brain efflux index (BEI%) studies of 125I-Aβ40 showed that administration of oleocanthal extracted from extra-virgin olive oil to C57BL/6 wild-type mice enhanced 125I-Aβ40 clearance from the brain and increased the BEI% from 62.0 ± 3.0% for control mice to 79.9 ± 1.6% for oleocanthal treated mice. Increased P-gp and LRP1 expression in the brain microvessels and inhibition studies confirmed the role of up-regulation of these proteins in enhancing 125I-Aβ40 clearance after oleocanthal treatment. Furthermore, our results demonstrated significant increase in 125I-Aβ40 degradation as a result of the up-regulation of Aβ degrading enzymes following oleocanthal treatment. In conclusion, these findings provide experimental support that potential reduced risk of AD associated with extra-virgin olive oil could be mediated by enhancement of Aβ clearance from the brain.
Keywords: Oleocanthal, β-amyloid clearance, β-amyloid degradation, P-glycoprotein, LRP1, BBB
The Mediterranean diet is associated with beneficial health properties against Alzheimer’s disease (AD), a neurodegenerative disease that affects about 30 million people worldwide.1 Epidemiological studies indicate that the prevalence of AD and cognitive decline is low among the Mediterranean area populations compared to those of other geographical regions of the world.2−4 One integral component of the Mediterranean dietary pattern is the consumption of extra-virgin olive oil (EVOO).5 Typically, the intake of EVOO ranges from 25 to 50 mL per day in the Mediterranean diet.6 Therefore, the apparent health benefits have been partially attributed to the dietary consumption of EVOO by Mediterranean populations.
Historically, the health promoting properties of EVOO were attributed to the high concentration of monounsaturated fatty acids, in particular oleic acid, contained in EVOO. However, other seed oils (i.e., sunflower, soybean, and rapeseed), which also contain high concentrations of oleic acid, do not exhibit the same health benefits as EVOO.7−9 In addition to oleic acid, EVOO contains a minor, yet significant phenolic fraction that other seed oils lack and this fraction has generated much interest regarding its health promoting properties. Currently, 36 phenolic compounds have been identified in EVOO and in vitro and in vivo studies have demonstrated that olive oil phenolics have positive effects on certain physiological parameters such as plasma lipoproteins, oxidative damage, inflammatory markers, platelet and cellular function, antimicrobial activity, and bone health.10
Among the phenolic olive oil constituents, (−)-oleocanthal, a naturally occurring phenolic secoiridoid isolated from EVOO, has shown an anti-inflammatory and antioxidant properties similar to the nonsteroidal anti-inflammatory drug ibuprofen.11 (−)-Oleocanthal is the dialdehydic form of (−)-deacetoxyligstroside glycoside responsible for the bitter taste of EVOO, and its chemical structure is related to the secoiridoid glycosides ligstroside and oleuropein, which are also common in EVOO. Chemical structure of oleocanthal is shown in Figure 1.
Figure 1.
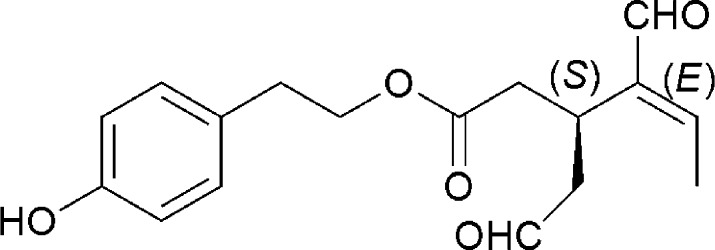
Chemical structure of (−)-oleocanthal.
Recently, oleocanthal has been demonstrated to have potential neuroprotective properties and contribute to preventing cognitive decline due to neurodegenerative diseases.12−14 This has been supported by population-based studies indicating that Mediterranean diet, rich in olive oil and monounsaturated fats, protects against age-related cognitive decline.15,16 One cohort study performed on 1880 elders in the United States showed a 40% decrease in the incidence of AD in populations consuming Mediterranean style diet.4
Different mechanisms have been proposed to describe the role that oleocanthal plays in the reduced incidence of AD; Li and co-workers showed that oleocanthal inhibits the formation of neurofibrillary tangles, a key hallmark in the pathogenesis of AD, by acting on microtubule associated proteins known as tau proteins, which are involved in the promotion of microtubule assembly and stability in the neurons.12 This study was followed by Monti et al., who investigated the mechanism by which oleocanthal inhibits tau fibrillization and aggregation in vitro via covalent chemical interaction with the fibrillogenic fragment of tau proteins.13 Additional potential mechanism suggests that oleocanthal reduce the formation of β-amyloid (Aβ) senile plaques, another pathological hallmark of AD, in the brain. Pitt et al. demonstrated that oleocanthal can interact with Aβ and alter the oligomerization state of Aβ oligomers and protect the neurons from the synaptopathological effects associated with Aβ aggregation and plaque formation.14
Several mechanisms have been proposed to account for the clearance of Aβ, including enzymatic degradation by a variety of proteases such as neprilysin (NEP) and insulin degrading enzyme (IDE), removal through the brain ISF bulk flow into the bloodstream, perivascular lymphatic drainage, and transport across the BBB.17,18 Increasing evidence from the literature show that AD mainly develop due to the excessive accumulation of Aβ in the brain as a result of its faulty clearance across the BBB.19 It has been reported that Aβ removal from the brain across the BBB is mediated by two major transport proteins; P-glycoprotein (P-gp) and LDL lipoprotein receptor related protein-1 (LRP1).20−23 Moreover, recent evidence indicated a progressive decline in the levels of P-gp and LRP1 at the BBB during normal aging, and this decline was positively correlated with accumulation of Aβ in AD.24,25 On the other hand, the receptor for advanced glycation end products (RAGE), a multiligand receptor in the immunoglobulin superfamily, mediates and regulates Aβ influx to the brain.26,27 Numerous attempts have been made to decrease Aβ accumulation, and the enhanced Aβ clearance across BBB was one potential therapeutic approach that has been thoroughly reviewed.28,29
In the present study, we aimed to demonstrate, using in vitro and in vivo studies, the role of oleocanthal in enhanced clearance of Aβ from the brain as an additional possible mechanism for its neuroprotective effect via its potential to up-regulate P-gp and LRP1 at the BBB, and its ability to enhance Aβ degradation. Based on the findings, we hope to provide a new insight on the protective effect of oleocanthal against AD by enhancing Aβ clearance from the brain.
Results and Discussion
Available experimental data strongly suggest impaired clearance of Aβ across the BBB might largely contribute to the formation of Aβ brain deposits and AD progression. Also, it has been demonstrated that P-gp and LRP1 play substantial role in the elimination of Aβ from the brain across the BBB.20−23,30 Thus, in the current study we aimed to investigate the effect of oleocanthal treatment on the levels of P-gp, LRP1 and Aβ degrading enzymes, and then the consequences of such treatment on the clearance of Aβ across the BBB.
The ability of oleocanthal at different concentrations to induce P-gp and LRP1 expressions at the BBB was assessed by Western blotting using bEnd3 cells as a representative model of mouse BBB. As shown in Figure 2, treatment of bEnd3 cells with oleocanthal resulted in significant increase in P-gp (1.2–1.35-fold increase, P < 0.05) and LRP1 levels (1.25–1.30-fold increase, P < 0.05). Similar pattern has been observed in immunofluorescence studies, where oleocanthal treatment resulted in 2.8- and 2.2-fold increase in P-gp and LRP1 expressions, respectively, when compared to control cells (Figure 3). While both methods provide semiquantitative results, the greater fold increase in P-gp and LRP1 expressions observed by the immunofluorescence method compared to Western blotting could be related to methods sensitivity.31
Figure 2.

Representative Western blots for P-gp (A) and LRP1 (B) in bEnd3 cells treated with oleocanthal. Cells were treated for 72 h with increasing concentrations of the indicated compounds in the range of 0.5–50 μM.
Figure 3.

Representative fluorescent micrographs of (A) P-gp (green) and (B) LRP1 (red) for control and bEnd3 cells treated with 25 μM of oleocanthal. Quantitative folds change in P-gp and LRP1 expression were measured using ImageJ version 1.44. The data are expressed as mean ± SD (n = 4). *P < 0.05 compared to control untreated cells. Scale bar = 50 μm.
To determine the effect of oleocanthal treatment on the accumulation of Aβ, cellular uptake studies were conducted. While both Aβ peptide species, Aβ40 and Aβ42, have been implicated in the pathogenesis of AD,32 Aβ40 was used in this work because it is practically feasible as it has much faster clearance rate than Aβ42.33 The results showed conclusive evidence for the specific roles of P-gp and LRP1 in 125I-Aβ40 cellular uptake, and their up-regulation on 125I-Aβ40 cellular level. Unlike in transport studies where cells are polarized and P-gp and LRP1 work in the same direction (i.e., from abluminal to luminal side), in uptake studies P-gp and LRP1 are localized at the cell membrane and function in opposite directions where P-gp limits 125I-Aβ40 cellular entry due to its efflux function, while LRP1 enhances cellular uptake of 125I-Aβ40. The cellular uptake of 125I-Aβ40 was evaluated following oleocanthal treatment in the presence and absence of 100 μM verapamil (P-gp inhibitor), 1 μM RAP (LRP1 inhibitor), or RAGE (N-16) antibody (as RAGE inhibitor) at a dilution ratio 1:100. The results showed significant alteration in the activities of P-gp and LRP1 but not RAGE caused by oleocanthal treatment (Figure 4). In oleocanthal treated cells, P-gp inhibition caused significant increase in 125I-Aβ40 cellular uptake by 69% (from 812 ± 12 to 1376 ± 132 cpm/mg protein, P < 0.01) compared to 28% in vehicle treated cells (from 837 ± 36 to 1068 ± 102 cpm/mg protein, P < 0.01), indicating enhanced P-gp function by oleocanthal. In contrast to the function of P-gp, LRP1 functions by cellular internalization, thus its inhibition by RAP caused a significant reduction (P < 0.02) in the cellular uptake of 125I-Aβ40 by 34% (from 837 ± 36 to 555 ± 8 cpm/mg protein) for vehicle treated cells, compared to only 17% (from 812 ± 12 to 671 ± 30 cpm/mg protein) for oleocanthal treated cells. The percent reduction in 125I-Aβ40 uptake in oleocanthal treated cells was significantly lower than that of vehicle treated cells (P < 0.05; Figure 4). In concept, assuming lack of LRP1 induction by oleocanthal, RAP inhibition should reduce 125I-Aβ40 uptake equally in both oleocanthal and control treated cells. However, the reduction in 125I-Aβ40 cellular level in oleocanthal treated cells was less than that observed with control suggesting that oleocanthal possibly induced an unknown uptake mechanism that is not inhibited by RAP. This finding is further supported by the in vivo data discussed below. With regard to RAGE, unlike P-gp and LRP1, oleocanthal has no significant effect on its expression or activity (Figure 5).
Figure 4.

Effect of treatment of bEnd3 cells with vehicle (CTRL) or oleocanthal (OLC) in presence or absence of inhibitors on the intracellular accumulation of radiolabeled 125I-Aβ40. The data are expressed as mean ± SEM (n = 3 independent experiments). *Significantly different from no inhibitor treated cells (P < 0.02). #Significantly different from control treated cells with inhibitors (P < 0.05).
Figure 5.

Quantitative analysis for RAGE in bEnd3 cells treated with oleocanthal. Cells were treated for 72 h with 0, 25, and 50 μM concentrations of oleocanthal.
Next, we aimed to investigate the effect of oleocanthal treatment on the clearance of 125I-Aβ40 from mouse brain. To our knowledge, this study is the first to in vivo investigate oleocanthal. The dosage regimen selected to conduct these studies was intraperitoneal administration at 10 mg/kg/day twice daily for 2 weeks to wild-type mice. At this dose, all animals were healthy and no weight loss was observed. Given the aldehyde structure of oleocanthal, metabolically it could be unstable suggesting formation of active metabolites that may exert the up-regulation effect.34 Further studies are currently in progress to investigate oleocanthal metabolism. In line with the in vitro results, Western blot analysis (Figure 6) revealed significant increase in the expression of P-gp and LRP1 in the brain microvessels of C57BL/6 mice by oleocanthal treatment. Densitometric analysis of the bands showed that oleocanthal increased P-gp and LRP1 expression by 1.30- and 1.25-fold compared to control, respectively. No significant change in the expression of RAGE has been observed in control or treated animals (data not shown).
Figure 6.

Western blot analysis of P-gp and LRP1 in mice brain microvessels. Significantly higher expression levels of P-gp and LRP1 were detected in oleocanthal (OLC) treated mice compared to control group (CTRL). (A) Representative Western blot lanes for P-gp, LRP1, and protein loading control (β-actin). (B) Quantitative fold increase in P-gp and LRP1 expressions. The data are expressed as mean ± SEM of n = 3 independent experiments (*P < 0.05).
Aβ is cleared from the brain by nonsaturable (passive) and saturable pathways (transport and metabolism).35 Nonsaturable pathway involves passive removal of soluble Aβ through bulk flow of cerebrospinal fluid (CSF) (i.e., CSF turnover). Saturable pathways involve degradation and BBB efflux components. The brain efflux index (BEI) method was used to study brain 125I-Aβ40 clearance with 14C-inulin as a reference compound.36 The BEI% method is commonly used to investigate molecules’ brain clearance via degradation/metabolism and/or transport across BBB.37 Thus, changes in BEI% of 125I-Aβ40 following oleocanthal treatment compared to control demonstrates alteration in 125I-Aβ40 removal from the brain by enhanced enzymatic degradation and/or clearance across BBB via the transport system. Consequently, both mechanisms were investigated.
Clearance BEI% experiments were performed at 24 h after the last injection of oleocanthal or vehicle (normal saline). As shown in Figure 7A, BEI% analysis demonstrated about 18% increase in the clearance of 125I-Aβ40 in oleocanthal treated mice (79.9 ± 1.6%) compared to control mice (62.0 ± 3.0%; P < 0.05). To study whether the enhanced brain clearance of 125I-Aβ40 is specifically due to increased expression of P-gp and LRP1 at the BBB, inhibitory studies of each protein were performed. The role of P-gp in 125I-Aβ40 clearance across the BBB of C57BL/6 mice was evaluated by preinjection of valspodar, a specific P-gp inhibitor,38 into the brain of C57BL/6 mice 5 min before the intracerebral microinjection of 125I-Aβ40/14C-inulin solution. Valspodar was administered first to allow its brain distribution and interaction with P-gp prior to 125I-Aβ40 microinjection which has rapid brain clearance.33 P-gp inhibition significantly lowered BEI% value of 125I-Aβ40 in control and oleocanthal-treated group (Figure 7B). Thirty minutes post microinjection of 125I-Aβ40, the pretreatment of control and oleocanthal-treated mice with valspodar resulted in about 13% (from 62.0 ± 3.0% to 49.3 ± 4.6%; P < 0.05) and 40% (79.9 ± 1.6% to 39.4 ± 2.8%; P < 0.001) reduction in BEI% values, respectively. The reduction in BEI% following P-gp inhibition by valspodar in oleocanthal treated group compared to control group was statistically insignificant (P > 0.05). Similarly, LRP1 inhibition by RAP significantly reduced 125I-Aβ40 BEI% (P < 0.05, Figure 7B) in oleocanthal-treated mice. Pretreatment of control and oleocanthal-treated mice with RAP resulted in about 33% (from 62.0 ± 3.0% to 29.1 ± 1.5%; P < 0.001) and 21% (79.9 ± 1.6% to 59.2 ± 3.5%; P < 0.05) reduction in BEI% values of 125I-Aβ40, respectively. These results are in line with similar studies demonstrating the role of P-gp and LRP1 in the clearance of Aβ across the BBB.20,22,23
Figure 7.

(A) Brain efflux index (BEI) of 125I-Aβ40 in control (CTRL), and oleocanthal (OLC) treated groups measured 24 h after the last injection of oleocanthal or normal saline. Significantly higher BEI% was observed in oleocanthal treated mice compared to control group. (B) Effect of P-gp and LRP1 inhibition by valspodar (24 ng/0.5 μL injection) and RAP (19.5 ng/0.5 μL injection), respectively, on BEI% of 125I-Aβ40 in CTRL and OLC treated groups. Both valspodar and RAP caused a significant reduction in the BEI% in oleocanthal treated group. The data are expressed as mean ± SEM of n = 4–6 (*P < 0.05, ***P < 0.001).
To characterize the effect of oleocanthal treatment on the degradation of 125I-Aβ40 in C57BL/6 mice brain, we performed TCA degradation assay and measured the expression of Aβ degrading enzymes NEP and IDE. The percent of degraded Aβ peptide (cpm in supernatant) were significantly higher in oleocanthal treated group (60.0 ± 2.3%) compared to control (40.1 ± 1.2%, P < 0.05, Figure 8A). Further, treatment of mice with oleocanthal resulted in a significant increase (1.6-fold) in the expression of IDE and small but insignificant increase in NEP (1.1-fold) in mice brain microvessels, respectively (Figure 8B, C). Consistent with in vivo findings, in vitro treatment of bEnd3 cells resulted in significant increase in the expression of IDE (1.6-fold, P < 0.05) and NEP (1.3-fold, P < 0.05) enzymes (data not shown). Thus, oleocanthal treatment enhanced the degradation of Aβ40 in mice brain, mostly via induction of IDE enzyme and possibly NEP.
Figure 8.

(A) Effect of oleocanthal (OLC) treatment on Aβ degradation in mice brain homogenate compared to control (CTRL) measured using TCA assay. Significantly higher Aβ degradation% was observed in OLC treated mice compared to control. The data are expressed as mean ± SEM of n = 4–6 (*P < 0.05). (B) Representative Western blots lanes for IDE, NEP, and protein loading control (β-actin) in mice brain microvessels. Significantly higher expression levels of IDE but not NEP were detected in OLC treated mice compared to control. (C) Quantitative fold increase in IDE and NEP expressions. The data are expressed as mean ± SEM of n = 3 independent experiments (*P < 0.05).
Collectively, the above results corroborate the assumption that oleocanthal improves 125I-Aβ40 clearance by enhancing P-gp and LRP1 expression/activity at the BBB. However, the inhibition of LRP1 by RAP was more apparent in control mice than oleocanthal-treated mice, which is consistent with the data obtained from the in vitro inhibition studies. Yet, Aβ clearance from the brain of oleocanthal-treated mice was greater than control mice even in the presence of RAP, suggesting the induction of another mechanism(s) contributing to the clearance of 125I-Aβ40, which could be an unknown transporter(s)/receptor(s),23 most likely at the abluminal side, and/or enhanced degradation of 125I-Aβ40 as a result of the treatment. Our results confirm both, induction of an unknown transport mechanism, supported by the in vitro data, and degradation. Beside its role in the up-regulation of P-gp and LRP1, oleocanthal enhanced the clearance of 125I-Aβ40 by up-regulating NEP (in vitro studies) and IDE, two peptidases that have been reported to cleave and degrade Aβ implicated in AD. Several studies reported overexpression of NEP to protect hippocampal neurons from Aβ-mediated toxicity in vitro,39 and the up-regulation of NEP or IDE has been proposed as a potential means of protecting the brain against Aβ accumulation, prevention of amyloid plaque formation, and consequent cognitive decline.40−42
In support of its putative health benefits, oleocanthal has more recently been demonstrated to possess therapeutic activities in the treatment of AD disease.12,14 These studies support research surveys showing a 40% decrease in AD in populations consuming a Mediterranean style diet comprising olive oil.4 Our findings described in this study provide further evidence on the role of oleocanthal as a neuroprotective agent against AD in vivo in wild-type mice brains by increasing 125I-Aβ40 clearance, which substantiate the in vitro findings reported by us and other research laboratories. Additional research is required to determine the therapeutic benefits of oleocanthal using AD animal model, and its bioavailability which has not been investigated.
In conclusion, our findings demonstrated that the beneficial effect of oleocanthal against AD could be extended to its ability to induce P-gp and LRP1, which are responsible for Aβ clearance across the BBB. Also, this study provides conclusive evidence for the role of oleocanthal on Aβ degradation as shown by the up-regulation of Aβ degrading enzymes IDE and possibly NEP. Furthermore, our results show that extra-virgin olive oil-derived oleocanthal associated with the consumption of Mediterranean diet has the potential to reduce the risk of AD or related neurodegenerative dementias.
Methods
Reagents and Antibodies
1,1,1,3,3,3-Hexafluoro-2-propanol (HFP) and Tween 20 were purchased from Sigma-Aldrich (St. Louis, MO). RIPA buffer was purchased from Thermo Scientific (Rockford, IL). Synthetic monoiodinated and nonoxidized 125I-Aβ40 (human, 2200 Ci/mmol) was purchased from PerkinElmer (Boston, MA). 14C-Inulin (2.2 mCi/g) was purchased from American Radiolabeled Chemicals (St. Louis, MO). The human receptor associated protein (RAP) was purchased from Oxford Biomedical Research (Oxford, MI). Valspodar was obtained from XenoTech (Lenexa, KS). The reagents and supplements required for Western blotting were purchased from Bio-Rad (Hercules, CA). For Western blot, the mouse monoclonal antibody C-219 against P-gp was obtained from Covance Research Products (Dedham, MA); mouse monoclonal antibody against light chain LRP1 (5A6) was obtained from Calbiochem (Gibbstown, NJ); rabbit monoclonal antibody against light chain LRP1 was obtained from Epitomics (Burlingame, CA); goat polyclonal antibodies against, RAGE (N-16), β-actin (C-11), and HRP-labeled secondary antibodies were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA); rabbit polyclonal antibody against insulin degrading enzyme (IDE) and mouse monoclonal antibody against neprilysin (NEP, anti-CD10) were obtained from Abcam Inc. (Cambridge, MA). For immunohistochemistry, the rabbit polyclonal antibody against P-gp was purchased from Rockland Immunochemicals Inc. (Gilbertsville, PA). Donkey anti-rabbit Alexa Flour 594 was purchased from Invitrogen (Carlsbad, CA), and goat anti-mouse IgG-FITC labeled secondary antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Formaldehyde 16% (Methanol Free, Ultrapure EM grade) was purchased from Polysciences, Inc. (Warrington, PA). Ketamine and xylazine for mice anesthetization were purchased from Henry Schein Inc. (Melville, NY). All other reagents and supplies were purchased from VWR (West Chester, PA).
Extraction of Oleocanthal from Extra-Virgin Olive Oil
Oleocanthal was extracted as reported by Elnagar et al.43 from EVOO (Member’s Mark, batch no. VF1_US102808, Italy) on lipophilic Sephadex LH20 (Sigma Aldrich, bead size 25–100 μm) using n-hexane-CH2Cl2 (1:9) and finally purified on C-18 reversed-phase Bakerbond octadecyl (40 μm; Mallinckrodt Baker, Inc.) using isocratic CH3CN–H2O (40:60). A purity of >90% was established for oleocanthal as assessed by TLC, 1H NMR spectroscopy, and/or HPLC analysis.
Cell Culture
The mouse brain endothelial cells (bEnd3; ATCC, Manassas, VA), passage 25–30, were cultured in DMEM growth medium supplemented with 10% fetal bovine serum (FBS), and the antibiotics penicillin G (100 units/mL) and streptomycin (100 μg/mL), 1% w/v nonessential amino acids, glutamine 2 mM. The cells were grown to confluence in 75 cm2 cell culture flasks for 1–2 days in a humidified atmosphere (5%CO2/95% air) at 37 °C.
In Vitro Induction of P-gp and LRP1 Expression by Oleocanthal in bEnd3 Cells
Cells were seeded in 10 mm cell culture dishes (Corning, NY) at a density of 1 × 106 cells per dish. The cells were allowed to grow up to 50% confluence and then treated with oleocanthal or control (vehicle) in a humidified atmosphere (5%CO2/95% air) at 37 °C. Methanolic stock solution of oleocanthal was diluted to a final concentration of 0.5–50 μM in growth medium before use. Media containing oleocanthal at different concentrations in addition to control medium were added to the respective treatment cells in duplicate at a maximum methanol concentration of 0.2%. The cells were then incubated for 72 h in a humidified atmosphere (5% CO2/95% air) at 37 °C. At the end of treatment period, cells were harvested and total protein was extracted for Western blot analysis.
Western Blot Analysis of P-gp, LRP1, RAGE, IDE, and NEP Proteins in bEnd3 Cells
At the end of treatment period with oleocanthal or control, total protein was extracted from bEnd3 cells with lysis buffer (RIPA buffer; 25 mM Tris.HCl, 150 mM NaCl, 1% NP-40, 1% sodium deoxylate, 0.1% SDS, pH 7.6). For Western blot analysis, 25 μg of cellular protein was resolved on 7.5% SDS-polyacrylamide gels and transferred onto nitrocellulose membrane. Blotting membranes were blocked with 2% BSA and incubated overnight with antibodies for LRP1 (light chain), P-gp (C-219), RAGE (N-16), β-actin (C-11), IDE, or NEP at dilutions 1:1500, 1:200, 1:200, 1:3000, 1:100, and 1:100, respectively. For protein detection, the membranes were washed and incubated with HRP-labeled secondary IgG antibody for LRP1, P-gp, NEP (antimouse), IDE (antirabbit) and RAGE, β-actin (antigoat) at 1:5000 dilution. The bands were visualized using SuperSignal West Femto Maximum Sensitivity substrate detection kit (Thermo Scientific; Rockford, IL). Quantitative analysis of the immunoreactive bands was performed using GeneSnap luminescent image analyzer (Scientific Resources Southwest Inc., Stafford, TX) and band intensity was measured by densitometric analysis. Three independent Western blotting experiments were carried out for each treatment group.
In Vitro Uptake Study of 125I-Aβ40 in bEnd3 Cells
The aim of this experiment was to investigate the effect of oleocanthal treatment on the accumulation of 125I-Aβ40 in bEnd3 cells. An uptake study using 125I-Aβ40 was conducted as described previously.31 Briefly, bEnd3 cells cultured onto 48 well plates at a density of 5 × 104 cells/well and were grown to 50–60% confluence. Following 72 h of treatment with 25 μM of oleocanthal or vehicle, the medium was aspirated and the cells were incubated in fresh growth medium for 4 h. After treatment, cells were washed three times with phosphate buffer-saline (PBS) solution. The cells were then preincubated with or without 100 μM verapamil (P-gp inhibitor), 1 μM RAP (LRP1 inhibitor), or RAGE (N-16) antibody (RAGE inhibitor) at a dilution ratio 1:100, in complete culture medium for 30 min. The activity experiments were started by the addition of 0.07 nM of 125I-Aβ40 in complete culture media with or without 100 μM verapamil, 1 μM RAP, or 1:100 dilution ratio of RAGE (N-16) antibody for 15 min in a humidified atmosphere (5%CO2/95% air) at 37 °C. The activity experiment was then terminated by washing the cells three times with ice-cold PBS. The cells were then disrupted with the lysis buffer containing complete mammalian protease inhibitor for 15 min at 4 °C. The radioactivity of 125I-Aβ40 accumulated inside the cells was measured using Wallac 1470 Wizard Gamma Counter (PerkinElmer Inc., Waltham, MA). The data were normalized for the protein content. The protein concentrations were determined using the Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Scientific, Rockford, IL) according to the manufacturer’s instruction with bovine serum albumin (BSA) as a standard.
Immunofluorescence Staining and Imaging of P-gp and LRP1 in bEnd3 Cells
To confirm effect of drugs treatment on the expression and up-regulation of P-gp and LRP1, both proteins were visualized using confocal microscopy. bEnd3 cells (5 × 104) were seeded on 35 mm poly-d-lysine coated glass bottom plates no. 1.5 (MatTek Corporation, Ashland, MA) and treated with 25 μM of oleocanthal or vehicle as described above. After incubation, the cells were washed three times with PBS and fixed with 4% formaldehyde for 10 min. The cells were then washed with PBS and blocked for 30 min with 10% of normal donkey and goat sera in 0.3% Triton X-100/PBS. The cells were then incubated overnight at 4 °C with a 1:200 dilution of primary antibody against P-gp or LRP1 in solution composed of 1% normal donkey and goat sera in PBS. After washing with PBS, the cells were incubated for 30 min with antirabbit Alexa flour 594 conjugated secondary IgG for the detection of LRP1 and antimouse FITC for the detection of LRP1, both at 1:250 dilution in PBS. Images for P-gp and LRP1 were captured using Zeiss LSM 5 Pascal confocal microscope equipped with 543 nm line of HeNe Laser and 63X oil immersion objective lens with numerical aperture = 1.4 (Carl Zeiss MicroImaging, LLC, Thornwood, NY). Negative controls for each treatment that were processed without primary antibody showed negligible background fluorescence. P-gp and LRP1 membrane immunofluorescence for each sample was quantified using ImageJ version 1.44 software (Research Services Branch, NIMH/NIH, Bethesda, MD).
Animals
C57BL/6 wild-type male mice were purchased from Harlan Laboratories (Houston, TX). The mice were 6–7 weeks old with an average body weight of 20 g. Mice were kept under standard environmental conditions (22 °C, 35% relative humidity, 12 h dark/light cycle) with free access to tap water and standard rodent food. After shipping, mice were allowed to adapt to the new environment for one week before initiating the experiments. All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Louisiana at Monroe and all surgical and treatment procedures were consistent with the IACUC policies and procedures.
In Vivo Induction of P-gp and LRP1 Expression by Oleocanthal
Animals were divided into two treatment groups: control and oleocanthal groups. Each group contained at least four animals. Drug administration was started at 7–8 weeks of age and continued for 2 weeks. Mice of control group received intraperitoneal vehicle (normal saline) twice daily. Mice of oleocanthal group received intraperitoneal oleocanthal at dose of 10 mg/kg twice daily.
Brain Efflux Index Study
At the end of treatment period, mice were prepared for brain efflux index study (BEI) to assess the clearance of Aβ 24 h after the last dose of oleocanthal. The in vivo Aβ clearance experiments (BEI%) were performed using the intracerebral microinjection technique reported previously.23,36,44,45 In brief, mice were anesthetized with intraperitoneal xylazine and ketamine (20 and 125 mg/kg, respectively) and placed in a stereotaxic apparatus (Stoelting Co., Wood Dale, IL) to determine the coordinates of the mice brain that coincide with the right caudate nucleus. A stainless steel guide cannula was implanted stereotaxically at 0.9 mm anterior, 1.9 mm lateral to bregma, and 2.9 mm below the surface of the brain as previously reported.20 The guide cannula and screw were fixed to the skull with binary dental cement, and a stylet was introduced into the guide cannula. Once the cement was firm, the animal was removed from the stereotaxic device, and the wound was closed anterior and posterior to the guide assembly using 1.75 mm Michel suture clips. Animals were then allowed for 12 h recovery from acute brain injury due to insertion of guide cannula in order to restore BBB integrity.45 Animals were reanesthetized with intraperitoneal xylazine/ketamine and an injector cannula connecting via Teflon tubing to a 1.0 μL gastight Hamilton microsyringe was inserted into the guide cannula. The applied tracer fluid (0.5 μL) containing 125I-Aβ40 (30 nM) and 14C-inulin (0.02 μCi) prepared in extracellular fluid buffer (ECF; 122 mM NaCl, 25 mM NaHCO3, 3 m M KCl, 1.4 mM CaCl2, 1.2 mM MgSO4, 0.4 mM K2HPO4, 10 mM d-glucose, and 10 mM HEPES, pH 7.4) was administered into the caudate nucleus over a period of 3 min. To prevent aggregation, 125I-Aβ40 was initially solubilized in HFP, dried, and resolubilized in ECF buffer prior to the experiment. The intactness and quality of 125I-Aβ40 was initially confirmed by trichloacetic acid (TCA) precipitation assay.20 After the microinjection, the microsyringe was left in place for 3 min to minimize any backflow. At the designated time, 30 min post 125I-Aβ40 injection,20,22,23 plasma and brain tissues were rapidly collected for Aβ measurements. To characterize the role of LRP1 and P-gp function, 0.5 μL of ECF containing RAP, an LRP1 inhibitor20,23,46 or valspodar, a well-established P-gp inhibitor23,47 were intracerebrally preadministered 5 min prior to 125I-Aβ40 injection at concentrations of 1 μM (19.5 ng/0.5 μL injection volume) and 40 μM (24 ng/0.5 μL injection volume), respectively.
Calculation of 125I-Aβ40 Clearance
125I-Aβ40 is characterized by its rapid clearance across the BBB,33 thus brain was collected 30 min postinjection to determine Aβ BEI%.20,22,23 Brain tissues were excised and homogenized by Dounce tissue grinder with seven strokes in two volumes of DPBS buffer (2.7 mM KCl, 1.46 mM KH2PO4, 136.9 mM NaCl, 8.1 mM Na2HPO4, 0.9 mM CaCl2, and 0.5 mM MgCl2 supplemented with 5 mM d-glucose and 1 mM sodium pyruvate, pH 7.4) containing mammalian protease inhibitor cocktail. About half of the brain homogenate was used for 125I-Aβ40 and 14C-inulin quantification and the second half for microvessels isolation and protein expression study. Trichloroacetic acid (TCA) precipitation method was used to calculate the amount of intact 125I-Aβ40 remained in the brain.20,23 To measure intact 125I-Aβ40, one volume of TCA (20%) was added to the sample, and then samples were vortexed, incubated in ice for 30 min, and centrifuged at 14 000 rpm (4 °C) for 30 min. Following centrifugation, gamma radioactivity of precipitated 125I-Aβ40 (intact peptide) and TCA supernatant (degraded peptide) were measured using Wallac 1470 Wizard Gamma Counter. The supernatant and precipitate were then mixed with 5 mL scintillation cocktail and the beta radioactivity of 14C-inulin was measured using Wallac 1414 WinSpectral Liquid Scintillation Counter (PerkinElmer Inc.). The BEI% was defined by eq 1 and the percentage of substrate remaining in the brain (100 – BEI%) was determined using eq 2.46
| 1 |
 |
2 |
The percent degradation for each sample was calculated by dividing the supernatant cpm by the total cpm (sum of the cpm in the precipitate and the supernatant cpm) and the resulting percent were subtracted from the percent of free 125I which determined from preinjected sample by spiking mice brain homogenate with 0.5 μL injectate solution and then processed as sample homogenate.
Isolation of Brain Microvessels
Brain microvessels were isolated as described previously by us.23 Briefly, after decapitation of mouse, brain was immediately put in ice-cold normal saline, homogenized and divided into halves as described above. One volume Ficoll (30%) was added to the half of brain homogenate to a final concentration of 15% and the mixture was mixed, and then centrifuged (5000 rpm for 10 min, 4 °C). The resulting pellets were suspended in ice-cold DPBS containing 1% bovine serum albumin (BSA) and passed over glass bead column. Microvessels adhering to the glass beads were collected by gentle agitation in 1% BSA in DPBS. Isolated microvessels were used for Western blotting and immunohistochemistry studies of LRP1 and P-gp.
Western Blot Analysis of P-gp, LRP1, and RAGE in Isolated Brain Microvessels
Protein expression levels in isolated brain microvessels were analyzed by Western blotting as described above. Total protein was extracted from isolated brain microvessels by homogenization with lysis buffer (RIPA buffer) containing protease inhibitor. Homogenized samples were centrifuged at 13 000 rpm for 10 min and supernatant was used for further Western blot analysis as described above.
Statistical Analysis
Wherever possible, the experimental results were analyzed for statistically significant difference using Two-tailed unpaired Student’s t-test to evaluate differences between controls and treated groups. A p-value less than 0.05 was considered to be statistically significant. All results were expressed as means and standard error of mean (SEM).
Author Contributions
All in vitro and in vivo experiments and data analysis were performed by A.H.A. and H.Q. Oleocanthal extraction from EVOO and characterization were performed by B.A.B. and supervised by K.A.E. All experiments were designed and supervised by A.K.
This research work was funded by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under Grant Number P20GM103424.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Hebert L. E.; Scherr P. A.; Bienias J. L.; Bennett D. A.; Evans D. A. (2004) State-specific projections through 2025 of Alzheimer disease prevalence. Neurology 62, 1645. [DOI] [PubMed] [Google Scholar]
- Panza F.; Solfrizzi V.; Colacicco A. M.; D’Introno A.; Capurso C.; Torres F.; Del Parigi A.; Capurso S.; Capurso A. (2004) Mediterranean diet and cognitive decline. Public Health Nutr. 7, 959–963. [DOI] [PubMed] [Google Scholar]
- Solfrizzi V.; Panza F.; Capurso A. (2003) The role of diet in cognitive decline. J. Neural. Transm. 110, 95–110. [DOI] [PubMed] [Google Scholar]
- Scarmeas N.; Luchsinger J. A.; Schupf N.; Brickman A. M.; Cosentino S.; Tang M. X.; Stern Y. (2009) Physical activity, diet, and risk of Alzheimer disease. JAMA, J. Am. Med. Assoc. 302, 627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicerale S., Lucas L. J., and Keast R. S. J. (2012) Oleocanthal: A Naturally Occurring Anti-Inflammatory Agent in Virgin Olive Oil. In Olive Oil - Constituents, Quality, Health Properties and Bioconversions (Dimitrios B., Ed.), pp 357–374, InTech, Rijeka, Croatia. [Google Scholar]
- Corona G.; Spencer J. P.; Dessi M. A. (2009) Extra virgin olive oil phenolics: absorption, metabolism, and biological activities in the GI tract. Toxicol. Ind. Health 25, 285–293. [DOI] [PubMed] [Google Scholar]
- Lopez-Miranda J.; Perez-Jimenez F.; Ros E.; De Caterina R.; Badimon L.; Covas M. I.; Escrich E.; Ordovas J. M.; Soriguer F.; Abia R.; de la Lastra C. A.; Battino M.; Corella D.; Chamorro-Quiros J.; Delgado-Lista J.; Giugliano D.; Esposito K.; Estruch R.; Fernandez-Real J. M.; Gaforio J. J.; La Vecchia C.; Lairon D.; Lopez-Segura F.; Mata P.; Menendez J. A.; Muriana F. J.; Osada J.; Panagiotakos D. B.; Paniagua J. A.; Perez-Martinez P.; Perona J.; Peinado M. A.; Pineda-Priego M.; Poulsen H. E.; Quiles J. L.; Ramirez-Tortosa M. C.; Ruano J.; Serra-Majem L.; Sola R.; Solanas M.; Solfrizzi V.; de la Torre-Fornell R.; Trichopoulou A.; Uceda M.; Villalba-Montoro J. M.; Villar-Ortiz J. R.; Visioli F.; Yiannakouris N. (2010) Olive oil and health: summary of the II international conference on olive oil and health consensus report, Jaen and Cordoba (Spain) 2008. Nutr., Metab. Cardiovasc. Dis. 20, 284–294. [DOI] [PubMed] [Google Scholar]
- Harper C. R.; Edwards M. C.; Jacobson T. A. (2006) Flaxseed oil supplementation does not affect plasma lipoprotein concentration or particle size in human subjects. J. Nutr. 136, 2844–2848. [DOI] [PubMed] [Google Scholar]
- Aguilera C. M.; Mesa M. D.; Ramirez-Tortosa M. C.; Nestares M. T.; Ros E.; Gil A. (2004) Sunflower oil does not protect against LDL oxidation as virgin olive oil does in patients with peripheral vascular disease. Clin. Nutr. 23, 673–681. [DOI] [PubMed] [Google Scholar]
- Cicerale S.; Lucas L.; Keast R. (2010) Biological activities of phenolic compounds present in virgin olive oil. Int. J. Mol. Sci. 11, 458–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchamp G. K.; Keast R. S.; Morel D.; Lin J.; Pika J.; Han Q.; Lee C. H.; Smith A. B.; Breslin P. A. (2005) Phytochemistry: ibuprofen-like activity in extra-virgin olive oil. Nature 437, 45–46. [DOI] [PubMed] [Google Scholar]
- Li W.; Sperry J. B.; Crowe A.; Trojanowski J. Q.; Smith A. B. 3rd; Lee V. M. (2009) Inhibition of tau fibrillization by oleocanthal via reaction with the amino groups of tau. J. Neurochem. 110, 1339–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti M. C.; Margarucci L.; Tosco A.; Riccio R.; Casapullo A. (2011) New insights on the interaction mechanism between tau protein and oleocanthal, an extra-virgin olive-oil bioactive component. Food Funct. 2, 423–428. [DOI] [PubMed] [Google Scholar]
- Pitt J.; Roth W.; Lacor P.; Smith A. B. 3rd; Blankenship M.; Velasco P.; De Felice F.; Breslin P.; Klein W. L. (2009) Alzheimer’s-associated Abeta oligomers show altered structure, immunoreactivity and synaptotoxicity with low doses of oleocanthal. Toxicol. Appl. Pharmacol. 240, 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhart M. J.; Geerlings M. I.; Ruitenberg A.; van Swieten J. C.; Hofman A.; Witteman J. C.; Breteler M. M. (2002) Dietary intake of antioxidants and risk of Alzheimer disease. JAMA, J. Am. Med. Assoc. 287, 3223–3229. [DOI] [PubMed] [Google Scholar]
- Solfrizzi V.; Colacicco A. M.; D’Introno A.; Capurso C.; Torres F.; Rizzo C.; Capurso A.; Panza F. (2006) Dietary intake of unsaturated fatty acids and age-related cognitive decline: a 8.5-year follow-up of the Italian Longitudinal Study on Aging. Neurobiol. Aging 27, 1694–1704. [DOI] [PubMed] [Google Scholar]
- van Assema D. M.; Lubberink M.; Bauer M.; van der Flier W. M.; Schuit R. C.; Windhorst A. D.; Comans E. F.; Hoetjes N. J.; Tolboom N.; Langer O.; Muller M.; Scheltens P.; Lammertsma A. A.; van Berckel B. N. (2012) Blood-brain barrier P-glycoprotein function in Alzheimer’s disease. Brain 135, 181–189. [DOI] [PubMed] [Google Scholar]
- Weller R. O.; Subash M.; Preston S. D.; Mazanti I.; Carare R. O. (2008) Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 18, 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic B. V., and Frangione B. (2003) Transport-clearance hypothesis for Alzheimer’s disease and potential therapeutic implications. In Aβ Metabolism in Alzheimer’s Disease (Saido T. C., Ed.), pp 114–122, Landes Bioscience, Georgetown, TX. [Google Scholar]
- Shibata M.; Yamada S.; Kumar S. R.; Calero M.; Bading J.; Frangione B.; Holtzman D. M.; Miller C. A.; Strickland D. K.; Ghiso J.; Zlokovic B. V. (2000) Clearance of Alzheimer’s amyloid-beta(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Invest. 106, 1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhnke D.; Jedlitschky G.; Grube M.; Krohn M.; Jucker M.; Mosyagin I.; Cascorbi I.; Walker L. C.; Kroemer H. K.; Warzok R. W.; Vogelgesang S. (2007) MDR1-P-Glycoprotein (ABCB1) Mediates Transport of Alzheimer’s amyloid-beta peptides--implications for the mechanisms of Abeta clearance at the blood-brain barrier. Brain Pathol. 17, 347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito J. R.; Deane R.; Fagan A. M.; Spinner M. L.; Parsadanian M.; Finn M. B.; Jiang H.; Prior J. L.; Sagare A.; Bales K. R.; Paul S. M.; Zlokovic B. V.; Piwnica-Worms D.; Holtzman D. M. (2005) P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J. Clin. Invest. 115, 3285–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qosa H.; Abuznait A. H.; Hill R. A.; Kaddoumi A. (2012) Enhanced brain amyloid-beta clearance by rifampicin and caffeine as a possible protective mechanism against Alzheimer’s disease. J. Alzheimer’s Dis. 31, 151–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverberg G. D.; Messier A. A.; Miller M. C.; Machan J. T.; Majmudar S. S.; Stopa E. G.; Donahue J. E.; Johanson C. E. (2010) Amyloid efflux transporter expression at the blood-brain barrier declines in normal aging. J. Neuropathol. Exp. Neurol. 69, 1034–1043. [DOI] [PubMed] [Google Scholar]
- Vogelgesang S.; Warzok R. W.; Cascorbi I.; Kunert-Keil C.; Schroeder E.; Kroemer H. K.; Siegmund W.; Walker L. C.; Pahnke J. (2004) The role of P-glycoprotein in cerebral amyloid angiopathy; implications for the early pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 1, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S. D.; Chen X.; Fu J.; Chen M.; Zhu H.; Roher A.; Slattery T.; Zhao L.; Nagashima M.; Morser J.; Migheli A.; Nawroth P.; Stern D.; Schmidt A. M. (1996) RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 382, 685–691. [DOI] [PubMed] [Google Scholar]
- Zlokovic B. V.; Martel C. L.; Matsubara E.; McComb J. G.; Zheng G.; McCluskey R. T.; Frangione B.; Ghiso J. (1996) Glycoprotein 330/megalin: probable role in receptor-mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid beta at the blood-brain and blood-cerebrospinal fluid barriers. Proc. Natl. Acad. Sci. U.S.A. 93, 4229–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron M. (2010) Alzheimer’s disease: strategies for disease modification. Nat. Rev. Drug Discovery 9, 387–398. [DOI] [PubMed] [Google Scholar]
- Abuznait A. H.; Kaddoumi A. (2012) Role of ABC Transporters in the Pathogenesis of Alzheimer’s Disease. ACS Chem. Neurosci. 3, 820–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam F. C.; Liu R.; Lu P.; Shapiro A. B.; Renoir J. M.; Sharom F. J.; Reiner P. B. (2001) beta-Amyloid efflux mediated by p-glycoprotein. J. Neurochem. 76, 1121–1128. [DOI] [PubMed] [Google Scholar]
- Abuznait A. H.; Cain C.; Ingram D.; Burk D.; Kaddoumi A. (2011) Up-regulation of P-glycoprotein reduces intracellular accumulation of beta amyloid: investigation of P-glycoprotein as a novel therapeutic target for Alzheimer’s disease. J. Pharm. Pharmacol. 63, 1111–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J. (2006) A hundred years of Alzheimer’s disease research. Neuron 52, 3–13. [DOI] [PubMed] [Google Scholar]
- Zlokovic B. V.; Yamada S.; Holtzman D.; Ghiso J.; Frangione B. (2000) Clearance of amyloid beta-peptide from brain: transport or metabolism?. Nat. Med. 6, 718. [DOI] [PubMed] [Google Scholar]
- Pinto J.; Paiva-Martins F.; Corona G.; Debnam E. S.; Jose Oruna-Concha M.; Vauzour D.; Gordon M. H.; Spencer J. P. (2011) Absorption and metabolism of olive oil secoiridoids in the small intestine. Br. J. Nutr. 105, 1607–1618. [DOI] [PubMed] [Google Scholar]
- Banks W. A.; Robinson S. M.; Verma S.; Morley J. E. (2003) Efflux of human and mouse amyloid beta proteins 1–40 and 1–42 from brain: impairment in a mouse model of Alzheimer’s disease. Neuroscience 121, 487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakee A.; Terasaki T.; Sugiyama Y. (1996) Brain efflux index as a novel method of analyzing efflux transport at the blood-brain barrier. J. Pharmacol. Exp. Ther. 277, 1550–1559. [PubMed] [Google Scholar]
- Castellano J. M.; Deane R.; Gottesdiener A. J.; Verghese P. B.; Stewart F. R.; West T.; Paoletti A. C.; Kasper T. R.; DeMattos R. B.; Zlokovic B. V.; Holtzman D. M. (2012) Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood Abeta clearance in a mouse model of beta-amyloidosis. Proc. Natl. Acad. Sci. U.S.A. 109, 15502–15507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidefelt U.; Liliemark J.; Gruber A.; Liliemark E.; Sundman-Engberg B.; Juliusson G.; Stenke L.; Elmhorn-Rosenborg A.; Mollgard L.; Lehman S.; Xu D.; Covelli A.; Gustavsson B.; Paul C. (2000) P-Glycoprotein inhibitor valspodar (PSC 833) increases the intracellular concentrations of daunorubicin in vivo in patients with P-glycoprotein-positive acute myeloid leukemia. J. Clin. Oncol. 18, 1837–1844. [DOI] [PubMed] [Google Scholar]
- El-Amouri S. S.; Zhu H.; Yu J.; Gage F. H.; Verma I. M.; Kindy M. S. (2007) Neprilysin protects neurons against Abeta peptide toxicity. Brain Res. 1152, 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemming M. L.; Patterson M.; Reske-Nielsen C.; Lin L.; Isacson O.; Selkoe D. J. (2007) Reducing amyloid plaque burden via ex vivo gene delivery of an Abeta-degrading protease: a novel therapeutic approach to Alzheimer disease. PLoS Med. 4, e262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier R.; Wolfer D. P.; Welzl H.; Tracy J.; Galsworthy M. J.; Nitsch R. M.; Mohajeri M. H. (2006) Neuronal neprilysin overexpression is associated with attenuation of Abeta-related spatial memory deficit. Neurobiol. Dis. 24, 475–483. [DOI] [PubMed] [Google Scholar]
- Leissring M. A.; Farris W.; Chang A. Y.; Walsh D. M.; Wu X.; Sun X.; Frosch M. P.; Selkoe D. J. (2003) Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron 40, 1087–1093. [DOI] [PubMed] [Google Scholar]
- Elnagar A. Y.; Sylvester P. W.; El Sayed K. A. (2011) (−)-Oleocanthal as a c-Met inhibitor for the control of metastatic breast and prostate cancers. Planta Med. 77, 1013–1019. [DOI] [PubMed] [Google Scholar]
- Shinohara M.; Sato N.; Kurinami H.; Takeuchi D.; Takeda S.; Shimamura M.; Yamashita T.; Uchiyama Y.; Rakugi H.; Morishita R. (2010) Reduction of brain beta-amyloid (Abeta) by fluvastatin, a hydroxymethylglutaryl-CoA reductase inhibitor, through increase in degradation of amyloid precursor protein C-terminal fragments (APP-CTFs) and Abeta clearance. J. Biol. Chem. 285, 22091–22102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito J. R.; May P. C.; O’Dell M. A.; Taylor J. W.; Parsadanian M.; Cramer J. W.; Audia J. E.; Nissen J. S.; Bales K. R.; Paul S. M.; DeMattos R. B.; Holtzman D. M. (2003) In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J. Neurosci. 23, 8844–8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S.; Ohtsuki S.; Terasaki T. (2006) Functional characterization of the brain-to-blood efflux clearance of human amyloid-beta peptide (1–40) across the rat blood-brain barrier. Neurosci. Res. 56, 246–252. [DOI] [PubMed] [Google Scholar]
- Twentyman P. R.; Bleehen N. M. (1991) Resistance modification by PSC-833, a novel non-immunosuppressive cyclosporin [corrected]. Eur. J. Cancer 27, 1639–1642. [DOI] [PubMed] [Google Scholar]