

Abstract
The inflammatory cytokine interleukin-1 (IL1) potentially plays a role in cognitive deterioration through pathology due to a dementing disorder or due to an aging process. Study of genetic variants in the IL1 genes has been mostly limited to diseases such as Alzheimer’s, however, there may be benefit to studying a continuous measure of cognition. Using data from the Cardiovascular Health Study, we evaluate genetic variation in the genes encoding inflammatory agonists IL1A and IL1B, and the antagonist IL1RN, with repeated measures of global cognition (3MS) and processing speed (DSST), using mixed effects models. We found statistically significant minor allele SNP associations with baseline performance on the 3MS in the IL1RN gene for Caucasians (rs17042917: beta=0.47, 95%CI=0.09, 0.85, p=0.016; rs4251961: beta=−0.36, 95%CI=−0.13, −0.60, p=0.0027; rs931471: beta=0.39, 95%CI=0.13, 0.65, p=0.0032), and the IL1B gene for African Americans (rs1143627: beta=1.6, 95%CI=0.48, 2.8; p=0.006 and rs1143634: beta=2.09, 95%CI=0.39, 3.8; p=0.016). Associations appear to be weaker in a subgroup with higher education level. Upon removing those diagnosed with dementia, effect sizes and statistical significance attenuated. These results provide supporting evidence that genetic variants in IL1 genes may be involved in inflammatory-related lowered cognition, that higher education may modify genetic predisposition, and that these associations may be driven by a dementia process.
Keywords: Interleukin-1, Inflammation, Cognition, Longitudinal study, Genetic epidemiology
1. Introduction
Cognitive decline predicts morbidity, dementia, and mortality, and is one of the major problems of an aging population (Morley, 2004). The idea that brain inflammation precedes and is intimately involved in degenerative neuronal processes has gained traction over the last few decades (McGeer and McGeer, 2004). The motivation for inflammatory hypotheses has been largely due to observational epidemiological studies suggesting that NSAIDs are protective for cognitive decline (Rozzini et al., 1996), and cognitive disorders such as Alzheimer’s disease (Szekely et al., 2004; Zandi and Breitner, 2001). Other lines of evidence to support the role of inflammation in bringing about cognitive deficits have been reviewed and include investigations of post-mortem brains, in vitro studies and animal models (Akiyama et al., 2000).
Inflammatory cytokines serve important neuroregulatory functions, and can affect cognition adversely through both degenerative and nondegenerative pathways (Wilson et al., 2002). Epidemiological evidence shows that peripheral cytokine levels are often elevated prior to cognitive decline (Weaver et al., 2002; Yaffe et al., 2003) and dementia (Engelhart et al., 2004). The pro-inflammatory cytokine interleukin-1 (IL1) is considered a key orchestrating protein involved in a number of biological events that influence cognitive deterioration through diverse mechanisms. First, IL1 plays a mediating role in neuronal processes including synaptic plasticity and neural transmission under normal, healthy conditions (Vitkovic et al., 2000), but may inhibit memory and learning under pathological conditions (Rachal Pugh et al., 2001). Second, IL1 has been involved in the dysregulation of temperature and sleep, and ultimately, impaired cognitive performance (Kronfol, 2003; Rachal Pugh et al., 2001; Ross et al., 2003). IL1 is also implicated in a number of inflammatory diseases of the periphery such as diabetes and atherosclerosis (Licinio and Wong, 1997) that have been shown to underlie cognitive disturbances (Cechetto et al., 2008; Knopman et al., 2009; Romero et al., 2009). It has been shown to mediate brain pathology in response to cerebral damage such a head injury or ischemic event (Rothwell, 1999). Finally, IL1 has been implicated in a number of neurodegenerative processes associated with Alzheimer’s disease (AD), including amyloid deposition, cholinergic deficit, dystrophic neurites, and hyperphosphoralation of tau protein; these events may create a self-perpetuating cycle, driven by IL1, that can spread from a local site to affect brain regions more broadly (Griffin et al., 1998).
Establishing whether IL1 protein dysfunction precedes or is a product of pathology leading to impaired cognition is difficult. Direct measurements of IL1 protein in brain can only be taken post mortem. Studies of serum IL1 in demented patients who have increased post mortem brain IL1 levels compared to controls have been mixed (Licastro and Chiappelli, 2003), and suffer from a failure to establish the timing of inflammation. Because inherited genetic variation precedes brain pathology, genetic evidence provides an important means of establishing the role of IL1 in cognitive decline. Specific IL1-related gene variants have been associated with increased serum levels (Hulkkonen et al., 2000; Pociot et al., 1992; Licastro et al., 2000) and inflammatory diseases such as rheumatoid arthritis (McDowell, Symons et al., 1995). Study of these variants with AD provide support for an association (Grimaldi et al., 2000; Nicoll et al., 2000; Wehr et al., 2006) but also yield inconclusive or negative findings (Déniz-Naranjo et al., 2008; Klimkowicz-Mrowiec et al., 2009; Giedraitis et al., 2009). Recent GWA also have failed to discover the IL1 region (Naj et al., 2011; Hollingworth et al., 2011).
Few studies have evaluated the genetic impact of IL1-specific gene variants on age-related cognitive changes independent of frank AD (Baune et al., 2008; Trompet et al., 2008). Further, the use of continuously measured cognitive test scores as an endpoint offers a potentially more informative way of evaluating the genetic role of this cytokine. Continuous, repeated measures of cognition can reflect cognitive impairment that arises from different pathways to pathology, and allows for assessment of baseline performance as well as change over time. Given this, we hypothesize that genetic variation in three IL1 genes will influence cognitive trajectories in older adults, and set out to test this hypothesis in the Cardiovascular Health Study population.
2. Materials and methods
2.1. Study population
The Cardiovascular Health Study (CHS) is a prospective, longitudinal study designed to investigate factors involved in cardiovascular disease and stroke. Details of the design have been published elsewhere (Fried et al., 1991). 5201 members of the original cohort were recruited using random sampling within defined age groups from HCFA Medicare files in each of four locations: Forsyth County, North Carolina; Washington County, Maryland; Pittsburgh, Pennsylvania; and Sacramento County, California. Selected participants were 65 years or older, were not in a nursing home and did not need a proxy informant at baseline. A second, African-American (AA), cohort began collection in 1992. Both cohorts were followed until 1999.
In 1992–1993, an ancillary study, The CHS Cognition Cohort Study (Lopez et al., 2002; Lyketsos et al., 2002), was launched. All participants were invited to undergo a brain MRI as well as additional cognitive testing, resulting in a sample of 3608 participants who were tested for this project. It has been previously reported that participants in the CHS Cognition Study compared to the overall CHS study are younger, better educated, more likely to have clinical cardiovascular disease, and performed better at baseline on the Modified Mini-Mental Exam 3MS (Kuller et al., 1998).
2.2. Measurements
2.2.1. Cognition
The MMSE (score ranges 0 to 30) was administered at baseline for the original cohort, after which the 3MS (score range 0 to 100) was administered annually; the baseline MMSE scores were adjusted to reflect the range of the 3MS. The 3MS (score range 0 to 100) (Teng and Chui, 1987) evaluates global cognition, and tests a variety of cognitive abilities including short-term and delayed recall, verbal fluency, as well as temporal and spatial orientation. A score of <80 is considered to be cognitively impaired, and a drop in score of 5 points over a one to two year time frame is considered to represent significant cognitive change (Kuller et al., 1998). The digit symbol substitution test (DSST) is a measure of psychomotor attention and speed (Salthouse, 1978), and a low score is considered to be <30 (Kuller et al., 1998). The DSST was administered at baseline and annually over the course of follow up.
2.2.2. Genetic measurements
Whole blood was collected at the baseline measurement for each cohort. DNA extraction and ApoE genotyping was carried out at the Core Molecular Genetics facility (Kuller et al., 1998; Yaffe et al., 2000). Genotyping for the three IL1 genes (IL1-beta, IL1- alpha, IL1-ra) was performed at the Johns Hopkins OAIC Genetics Core laboratory by a single technician using the Biotrove OpenArray™ SNP Genotyping System (Brennan et al., 2009).
IL1 SNP selection was carried out using HapMap Phase II, build 36. We placed emphasis on HapMap SNPs that were also widely reported in the literature for AD and other neurodegenerative disorders. We sought coverage of each gene, as well as the area of linkage disequilibrium (LD) surrounding the 5′ and 3′ ends of each gene, so that HapMap SNPs not selected to be genotyped were correlated with at least one of the genotyped SNPs at an r2 value ≥0.80 using the Tagger program (de Bakker et al., 2005) implemented in HaploView v3.1 software (Barrett et al., 2005). Where more than one SNP could be selected, we prioritized based on allele frequency and physical location across the gene. 32 SNPs were selected for genotyping.
2.2.3. Covariates
Age, gender and education were considered important confounders for their known strong correlations with cognitive test scores. Age was categorized into 5-year groups (<70, 70–74, 75–79, 80–84, 85+). Education was coded as in previous publications (Elkins et al., 2006) (<high school, high school, some college, college+). We hypothesized that IL1 genes may exert their effects on cognition through a number of vascular and health variables, likely resulting in an attenuation of the IL1 genetic associations. To explore this, we included in our models variables derived from information that was collected from annual interviews and clinical examinations. Baseline self-report of hypertension, diabetes, heart disease, stroke, and arthritis were considered, along with CRP values and white blood cell counts collected from the baseline clinical exam. We also hypothesized that NSAID drug use may mask any genetic effects by countering an upregulation of cytokine production, and that depression may enhance the genetic effects by providing a trigger for cytokine production. Therefore, we explored the modifying effects of NSAID drug use and depression on the IL1 genetic associations with cognition. Current NSAID drug use and physician- diagnosed current depression were self-reported at baseline.
2.3. Statistical analysis
2.3.1. Cognition and covariate diagnostics
The Caucasian and AA cohorts were analyzed separately out of concern for potential population stratification, or confounding by ethnicity. To compare basic demographics and covariate information by groups, we performed t-tests for continuous variables and Chi-squared tests for categorical and ordinal variables. After several diagnostic evaluations of the individual cognitive trajectories and after using the minimum BIC to guide considerations of spline and quadratic terms for the mean model, a general pattern for a linear decline over time was deemed appropriate for both the 3MS and the DSST.
2.3.2. Genotype diagnostics
Plots of the raw intensity data by array and by SNP were examined for gross deviations from expected clustering. Cluster-based genotype calls were generated via an in-house algorithm (contact authors for details). SNP allele and genotype frequencies were calculated and tests for Hardy Weinberg Equilibrium (HWE) performed. Allele frequencies were compared to those available on HapMap. Pairwise LD measures (D′ and r2) were calculated for each SNP pair within a gene in HaploView.
2.3.3. Mixed effects models
Initial mixed effects models of cognitive phenotypes were developed without consideration of SNP information. We considered a minimally adjusted model that incorporated a random term for the intercept and slope, and incorporated fixed terms for age, gender, education and their significant first order interactions. Differences at baseline (intercept) and annual rate of change (slope) in 3MS and DSST by SNP genotype were interpreted. We then developed a fully adjusted model by including vascular and health related variables. Each SNP was then included in both minimally and fully adjusted models. SNP genotypes were coded to reflect an additive model, with homozygotes of the common allele as the referent genotype. Mixed effects model parameter estimates were based on Wald tests. To test hypotheses about modification of genetic association by NSAID use and depression, we interpreted the parameter estimates for the product of these variables with SNP genotype.
To understand the role of IL1 genes on cognition per se, versus underlying disease pathology related to cognitive decline, we first considered all participants, regardless of dementia status. To understand whether any observed genetic associations with cognitive performance were a result of an underlying dementing process, we would ideally limit the analysis to those with diagnosed dementia and reassess the genetic association. Full cognitive evaluations and dementia adjudications, however, were only performed in a subset of the CHS that participated in the Cognition Cohort Study (Cognition Sub-sample). Therefore, we compared results for participants in the full CHS cohort versus results for this Sub-sample. Additionally, we sought to maintain power by reassessing the genetic association after removing those with baseline or incident dementia, rather than analyzing the smaller subset with dementia, which yielded non-significant p-values despite larger effect sizes. Thus, we have three levels of analysis: full CHS cohort, Cognition Sub-sample, and non-demented members of the Cognition Sub-sample. All analyses were carried out using SAS v9.1.
Haplotype analyses were also carried out for the mixed effect models, using haplotype blocks to create diplotype variables for analysis, as defined using the Solid Spine algorithm in HaploView v 3.1 (D′<0.80). All haplotypes with frequency<0.05 were grouped together as one “rare haplotype”. Diplotypes were coded according to an additive inheritance model with the most common diplotype homozygote as the referent group. Because software for direct inclusion of phase estimation and mixed effects model parameter estimation in a single analysis was not available, we modified HaploStats (haplo.glm) software to call upon SAS v9.1 to carry out mixed effects modeling during phase estimation iterations, allowing the predicted values from a mixed model to inform the subsequent iteration of haplotype phase estimation.
3. Results
3.1. Study sample
There were 5201 members of the original cohort, 4925 who reported as Caucasian with European descent. An additional 687 members were recruited into the African American (AA) cohort, 678 who reported African American ethnicity. Among Caucasians, 4297 gave consent to be genotyped, of whom 3878 were successfully genotyped for more than 50% of the SNPs. Among the AAs, 621 gave consent to be genotyped, of whom 590 were successfully genotyped. We excluded those who provided less than 3 measurements on the 3MS over the course of follow-up, leaving 3575 in the Caucasian cohort and 481 in the AA cohort included in analyses.
The CHS Cognition Study, reflecting the subset of CHS participants for whom dementia adjudication was available, collected 3608 participants, of whom 2761 who had genetic and phenotype data for inclusion in the analysis. Of these, there were 2426 Caucasians in our analyses, of whom 423 were demented. Of the 481 AAs in our analyses, 335 were in the CHS Cognition Study, of whom 71 were demented. The progression to a final analytic sample as well as the analytic group from the Cognition Sub-sample and their cognitive status are provided in Fig. 1.
Fig. 1.

Inclusion and exclusion of CHS participants to the analytic sample.
3.2. Measurement descriptions
3.2.1. Basic demographics and covariate status
Characteristics of the participants included in our report by cohort are provided in Table 1. Statistically significant differences between the Caucasian and AA cohorts were observed for most variables. Those included in our analyses were very similar to those not included due to lack of genotyping consent, across a wide range of characteristics, for both cohorts. Additional participants were not included, despite consent, due to lack of genotype or phenotype data. In Caucasians, those available for genotyping but excluded from analysis due to missing genotype or cognitive information were older (p<0.001), less educated (p-value from test for trend <0.001), more likely to be hypertensive (p=0.0001), more likely to report having heart disease at baseline (p=0.0001), more often reported poor or fair health (p-value for trend <.001), had higher peripheral white blood cell levels (p=0.0005) and had higher CRP levels (p=0.0022). Although prevalence was low, the excluded subjects also were more likely to report diabetes (p=0.0004) and stroke (p=0.0074) at baseline. In the AA cohort, of the participants who consented to be genotyped, those excluded from the analysis due to missing genotype or phenotype data were older (p<.001), more likely to report baseline heart disease (p=0.003) and more often reported poor or fair health (p-value for trend =0.017).
Table 1.
Baseline characteristics for participants of the Cardiovascular Health Study.
| Variable | Caucasian |
African American |
pval |
|---|---|---|---|
| (n=3575) | (n=481) | ||
| Age, years* | 72.67 (5.42) | 72.47 (5.59) | 0.45 |
| Female, n/N (%) | 2041/3575 (0.57) | 304/481 (0.63) | 0.011 |
| Education, n/N (%) | |||
| <HS | 888/3567 (0.25) | 201/478 (0.42) | |
| HS | 1345/3567 (0.38) | 133/478 (0.28) | |
| Some college | 544/3567 (0.15) | 65/478 (0.14) | |
| College or more | 790/3567 (0.22) | 79/478 (0.17) | <.0001 |
| Hypertension, n/N (%) | |||
| Normal | 1623/3571 (0.45) | 108/481 (0.22) | |
| Borderline | 518/3571 (0.15) | 60/481 (0.12) | |
| Hypertension | 1430/3571 (0.4) | 313/481 (0.65) | <.0001 |
| Diabetes, n/N (%) | 267/3562 (0.07) | 87/478 (0.18) | <.0001 |
| Stroke, n/N (%) | 66/3554 (0.02) | 25/478 (0.05) | <.0001 |
| Heart disease, n/N (%) | 754/3495 (0.22) | 113/466 (0.24) | 0.19 |
| Depression score | 4.29 (4.32) | 8.44 (2.86) | <.0001 |
| Social support score | 8.19 (2.58) | 5.9 (5.28) | 0.069 |
| Depressed, n/N (%) | 171/3398 (0.05) | 29/468 (0.06) | 0.29 |
| Healthy, n/N (%) | |||
| Excellent | 550/3568 (0.15) | 36/478 (0.08) | |
| Very good | 927/3568 (0.26) | 90/478 (0.19) | |
| Good | 1384/3568 (0.39) | 173/478 (0.36) | |
| Fair | 627/3568 (0.18) | 143/478 (0.3) | |
| Poor | 80/3568 (0.02) | 36/478 (0.08) | <.0001 |
| White blood cell count (x1000/cubic mm) | 6.3 (1.86) | 5.78 (1.87) | <.0001 |
| CRP (mg/L) | 4.35 (7.88) | 2.67 (2.41) | <.0001 |
| Adjusted cholesterol (mg/dl) | 212.6 (39.34) | 210.35 (37.01) | 0.24 |
| NSAID use, n/N (%) | 435/3572 (0.12) | 74/479 (0.15) | 0.043 |
| Arthritis, n/N (%) | 1785/3531 (0.51) | 268/476 (0.56) | 0.019 |
| ApoE ε4 status, n/N (%) | |||
| ε2/X | 541/3427 (0.16) | 104/453 (0.23) | |
| ε3/3 | 2150/3427 (0.63) | 219/453 (0.48) | |
| ε3/4, ε 4/4 | 736/3427 (0.21) | 130/453 (0.29) | <.0001 |
Unless otherwise specified, variable is given as a mean (sd).
3.2.2. Cognitive phenotype
All subjects in the final analysis contributed three or more measurements for the 3MS. Among Caucasians, about 5% had only three 3MS measurements, while 48% contributed all 10 measurements. Average follow up time was 7.31 years (sd=2.26), with a minimum of 1.9 years and a maximum of 9.9 years of follow up. For the AA cohort, 9% had only three 3MS measurements over the course of follow up, while 55% contributed all seven possible measurements. Average follow up time for the AA cohort was 5.26 years (sd=1.21) with a minimum of 1.8 years and a maximum of 6.3 years of follow up. The Caucasian cohort had an average baseline score for the 3MS of 91.0 (sd=5.41) and a DSST score of 38.6 (sd=12.7) (see Table 2). The AA cohort had an average baseline score for the 3MS of 84.15 (sd=11.5) and DSST score of 29.95 (sd=13.2)(see Table 2).
Table 2.
Description of available data on cognitive test scores for 3MS and DSST by visit in CHS participants.
| Milestone years | Visit | Caucasian cohort (N=3575) |
AA cohort (N=481) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| 3MS |
DSST |
3MS |
DSST |
||||||
| Median (interquartile range) | N missing | Median (interquartile range) | N missing | Median (interquartile range) | N missing | Median (interquartile range) | N missing | ||
| 1988/89 | 1/2 | 92.05 (88.6,94.7) | 46 | 39 (30,47) | 39 | – | – | – | – |
| 3 | 93 (88,97) | 62 | 41 (32,50) | 129 | – | – | – | – | |
| 4 | 94 (89,97) | 130 | 41 (32,49) | 221 | – | – | – | – | |
| 1992/93 | 5 | 94 (89,97) | 325 | 40 (32,49) | 421 | 87 (79,92) | 1 | 31 (20,39) | 27 |
| 6 | 95 (90,98) | 591 | 41 (32,50) | 725 | 89 (81.5,94) | 34 | 31 (22,40) | 73 | |
| 7 | 94 (88,97) | 721 | 41 (32,50) | 890 | 89 (80,93) | 40 | 31 (22,41) | 83 | |
| 8 | 94 (88,97) | 986 | 41 (32,50) | 1145 | 88 (80,94) | 59 | 31 (21.5,40.5) | 102 | |
| 9 | 95 (90,98) | 1198 | 40 (30,48) | 1349 | 89 (82,95) | 84 | 30 (22.25,40) | 127 | |
| 10 | 95 (88,98) | 1394 | 40 (31,49) | 1551 | 89 (80,95) | 110 | 31 (23,41) | 153 | |
| 1998/99 | 11 | 95 (89,98) | 1548 | 40 (30,49) | 1683 | 89 (79,94) | 152 | 31 (22,40) | 184 |
This table displays medians for the population by visit, and thus does not preserve the pattern of change within individuals. Due to drop out of sicker participants, medians do not appear to decline over time. The mixed effects models in our analyses address this by preserving the correlation within individuals over time. On average, a participant from the Caucasian Cohort declines −1.22 point per year on the 3MS and −0.67 per year on the DSST. The average decline per year in the AA Cohort for the 3MS is −0.80 and −0.36 for the DSST.
3.2.3. IL1 SNPs
Of the 32 genotyped SNPs, one SNP was dropped from analysis (rs315920) due to poor clustering, leaving 31 SNPs to be evaluated. The percentage of missing genotypes per SNP ranged from 1% to 10% in Caucasians and 0% to 23% in AAs. Allele and genotype frequencies, and tests of Hardy Weinberg proportions, for the 31 SNPs in our analyses are provided for both cohorts in Table 3 for whites and Table 4 for AAs. In both cohorts, significant Hardy Weinberg p-values resulted despite small differences in observed versus expected frequencies due to the large sample size. Of the six SNPs with statistically significant HWE departures at p<0.05, only two had meaningful departures from expected frequencies: rs1800587, excess homozygosity, and rs4849122, excess heterozygosity. Among the six SNPs flagged for HWE departure among AAs, rs2856838 showed moderate excess heterozygosity and both rs3213448 and rs17042998 showed excess homozygotes. The allele frequencies for the Caucasian and AA cohorts closely match those reported on HapMap for the CEU and YRI founders, respectively. All SNPs shown were included in subsequent association analyses and flagged for further consideration if associated. None of the SNPs with meaningful departures from HWE were associated with cognition in the association analyses.
Table 3.
Allele and genotype frequencies among white participants of the CHS (N=3575).
| Gene | SNP | Position | Alleles | MAF | −/− % | −/+ % | +/+ % | HWE Pval | % missing |
|---|---|---|---|---|---|---|---|---|---|
| IL1A | rs2048874 | 113240198 | C>T | 0.12 | 0.01 | 0.21 | 0.78 | 0.51 | 0.03 |
| IL1A | rs3783546 | 113251061 | G>C | 0.3 | 0.09 | 0.42 | 0.49 | 0.94 | 0.03 |
| IL1A | rs17561 | 113253454 | G>T | 0.3 | 0.08 | 0.42 | 0.49 | 0.39 | 0.05 |
| IL1A | rs3783538 | 113254426 | T>C | 0.03 | 0 | 0.06 | 0.94 | 0.11 | 0.08 |
| IL1A | rs2856838 | 113256203 | C>T | 0.4 | 0.15 | 0.49 | 0.36 | 0.23 | 0.03 |
| IL1A | rs1800587 | 113259191 | C>T | 0.32 | 0.15 | 0.33 | 0.52 | <0.00001 | 0.09 |
| IL1A | rs6722023 | 113262098 | C>T | 0.02 | 0 | 0.03 | 0.97 | 0.04 | 0.07 |
| IL1A | rs17042407 | 113275145 | T>C | 0.26 | 0.07 | 0.38 | 0.55 | 0.75 | 0.06 |
| IL1A | rs4849122 | 113277152 | A>G | 0.06 | 0.01 | 0.11 | 0.88 | 0.02 | 0.07 |
| IL1A | rs7585707 | 113285576 | T>C | 0.3 | 0.08 | 0.45 | 0.47 | 0.0001 | 0.05 |
| IL1B | rs4849125 | 113293146 | A>G | 0.29 | 0.08 | 0.42 | 0.49 | 0.29 | 0.09 |
| IL1B | rs7596684 | 113294434 | T>C | 0.23 | 0.05 | 0.36 | 0.59 | 0.39 | 0.02 |
| IL1B | rs3917386 | 113296097 | A>G | 0.03 | 0 | 0.06 | 0.94 | 0.37 | 0.02 |
| IL1B | rs3917368 | 113299013 | G>A | 0.36 | 0.12 | 0.47 | 0.41 | 0.44 | 0.02 |
| IL1B | rs3917365 | 113302940 | C>T | 0.09 | 0.01 | 0.16 | 0.84 | 0.58 | 0.03 |
| IL1B | rs1143643 | 113304533 | G>A | 0.35 | 0.12 | 0.45 | 0.42 | 0.91 | 0.04 |
| IL1B | rs1143634 | 113306621 | C>T | 0.24 | 0.05 | 0.37 | 0.58 | 0.28 | 0.04 |
| IL1B | rs1143627 | 113310618 | T>C | 0.33 | 0.11 | 0.44 | 0.45 | 0.62 | 0.02 |
| IL1RN | rs17042917 | 113586894 | G>A | 0.11 | 0.01 | 0.19 | 0.8 | 0.79 | 0.02 |
| IL1RN | rs4251961 | 113590698 | T>C | 0.38 | 0.16 | 0.44 | 0.4 | 0.00001 | 0.06 |
| IL1RN | rs2637988 | 113593010 | A>G | 0.41 | 0.16 | 0.49 | 0.35 | 0.45 | 0.07 |
| IL1RN | rs4251985 | 113593644 | G>T | 0.26 | 0.07 | 0.4 | 0.54 | 0.22 | 0.03 |
| IL1RN | rs3213448 | 113595528 | G>A | 0.13 | 0.02 | 0.23 | 0.76 | 1 | 0.04 |
| IL1RN | rs3087263 | 113601999 | G>A | 0.09 | 0.01 | 0.16 | 0.83 | 1 | 0.03 |
| IL1RN | rs4252022 | 113606392 | G>A | 0.01 | 0 | 0.99 | 0.01 | 1 | 0.01 |
| IL1RN | rs315951 | 113606817 | G>C | 0.29 | 0.08 | 0.42 | 0.5 | 0.2 | 0.03 |
| IL1RN | rs4252041 | 113606841 | C>T | 0.05 | 0 | 0.09 | 0.9 | 0.02 | 0.1 |
| IL1RN | rs315938 | 113612276 | T>A | 0.01 | 0 | 0.01 | 0.99 | 1 | 0.05 |
| IL1RN | rs17042998 | 113615756 | A>G | 0.02 | 0 | 0.04 | 0.96 | 0.11 | 0.02 |
| IL1RN | rs931471 | 113628924 | T>C | 0.33 | 0.1 | 0.46 | 0.44 | 0.05 | 0.08 |
| IL1RN | rs6739883 | 113630543 | G>A | 0.19 | 0.03 | 0.31 | 0.66 | 0.64 | 0.09 |
Table 4.
Allele and genotype frequencies among AA participants of the CHS (N=481).
| Gene | SNP | Position | Alleles | MAF | −/− % | −/+ % | +/+ % | HWE pval | % missing |
|---|---|---|---|---|---|---|---|---|---|
| IL1A | rs2048874 | 113240198 | C>T | 0.09 | 0.84 | 0.14 | 0.02 | 0.02 | 0.02 |
| IL1A | rs3783546 | 113251061 | G>C | 0.24 | 0.58 | 0.36 | 0.06 | 0.7 | 0.03 |
| IL1A | rs17561 | 113253454 | G>T | 0.18 | 0.04 | 0.28 | 0.68 | 0.34 | 0.04 |
| IL1A | rs3783538 | 113254426 | T>C | 0 | 0.99 | 0.01 | 0 | 1 | 0.02 |
| IL1A | rs2856838 | 113256203 | C>T | 0.39 | 0.17 | 0.44 | 0.39 | 0.1 | 0.03 |
| IL1A | rs1800587 | 113259191 | C>T | 0.4 | 0.25 | 0.3 | 0.45 | <.00001 | 0.15 |
| IL1A | rs6722023 | 113262098 | C>T | 0 | 0 | 0 | 0.99 | 0.01 | 0.04 |
| IL1A | rs17042407 | 113275145 | T>C | 0.2 | 0.02 | 0.34 | 0.63 | 0.05 | 0.05 |
| IL1A | rs4849122 | 113277152 | A>G | 0.01 | 0.97 | 0.03 | 0 | 1 | 0.03 |
| IL1A | rs7585707 | 113285576 | T>C | 0.36 | 0.13 | 0.46 | 0.41 | 0.92 | 0.02 |
| IL1B | rs4849125 | 113293146 | G>A* | 0.36 | 0.34 | 0.61 | 0.06 | <.00001 | 0.23 |
| IL1B | rs7596684 | 113294434 | T>C | 0.19 | 0.66 | 0.3 | 0.04 | 0.46 | 0.01 |
| IL1B | rs3917386 | 113296097 | A>G | 0.01 | 0.98 | 0.02 | 0 | 0.05 | 0.01 |
| IL1B | rs3917368 | 113299013 | G>A | 0.19 | 0.66 | 0.31 | 0.03 | 0.88 | 0.01 |
| IL1B | rs3917365 | 113302940 | C>T | 0.16 | 0.05 | 0.23 | 0.73 | 0.0009 | 0.04 |
| IL1B | rs1143643 | 113304533 | G>A | 0.19 | 0.66 | 0.31 | 0.04 | 1 | 0.02 |
| IL1B | rs1143634 | 113306621 | C>T | 0.12 | 0.02 | 0.2 | 0.78 | 0.08 | 0.03 |
| IL1B | rs1143627 | 113310618 | C>T* | 0.43 | 0.2 | 0.46 | 0.33 | 0.22 | 0.03 |
| IL1RN | rs17042917 | 113586894 | G>A | 0.18 | 0.03 | 0.3 | 0.67 | 0.88 | 0.01 |
| IL1RN | rs4251961 | 113590698 | T>C | 0.19 | 0.04 | 0.29 | 0.66 | 0.29 | 0.06 |
| IL1RN | rs2637988 | 113593010 | G>A* | 0.45 | 0.21 | 0.48 | 0.31 | 0.57 | 0.07 |
| IL1RN | rs4251985 | 113593644 | G>T | 0.09 | 0.83 | 0.16 | 0.01 | 1 | 0.01 |
| IL1RN | rs3213448 | 113595528 | G>A | 0.23 | 0.05 | 0.36 | 0.59 | 0.6 | 0.04 |
| IL1RN | rs3087263 | 113601999 | G>A | 0.02 | 0.96 | 0.04 | 0 | 0.22 | 0.03 |
| IL1RN | rs4252022 | 113606392 | G>A | 0.06 | 0 | 0.11 | 0.89 | 1 | 0 |
| IL1RN | rs315951 | 113606817 | C>G* | 0.39 | 0.39 | 0.45 | 0.16 | 0.24 | 0.03 |
| IL1RN | rs4252041 | 113606841 | C>T | 0.01 | 0.98 | 0.01 | 0 | 0.03 | 0.12 |
| IL1RN | rs315938 | 113612276 | T>A | 0.12 | 0.78 | 0.21 | 0.01 | 0.82 | 0.08 |
| IL1RN | rs17042998 | 113615756 | A>G | 0.01 | 0.98 | 0.02 | 0 | 1 | 0.02 |
| IL1RN | rs931471 | 113628924 | C>T* | 0.4 | 0.37 | 0.46 | 0.17 | 0.38 | 0.06 |
| IL1RN | rs6739883 | 113630543 | G>A | 0.14 | 0.02 | 0.25 | 0.73 | 0.56 | 0.08 |
Denotes when the minor allele in AAs is different than in Whites.
There are over 100 SNPs cataloged in dbSNP for IL1A and IL1B, and over 400 for IL1RN. Using our set of genotyped tag SNPs, we attained 100% coverage compared to HapMap for all three IL1 genes at a correlation of 0.80 for both Caucasians and AAs.
3.3. Association analyses
3.3.1. IL1 single SNP associations
P-values for tests of the effects of all SNPs on both baseline cognition and annual rate of cognitive change using the minimally adjusted mixed effects models are provided in Fig. 2 (Caucasians) and Fig. 3 (AAs). The effect sizes for statistically significant single SNP results (p<0.016 after correcting for 3 genes) are given in Table 5. The addition of covariates in the fully adjusted models did not alter our findings, thus, we show results only for the minimally adjusted models. For Causasians, almost all significant SNPs were located in the IL1RN gene and were associated with intercept performance values on the 3MS (see Fig. 4 for gene-specific results of 3MS intercept analyses). The strongest signal, for rs17042917 in the 5′ untranslated region, was associated with a 0.47 increase in baseline 3MS (95%CI =0.09, 0.85, pval=0.015) per copy of the minor allele. The intronic SNP rs4251961 is nearby and significant for lowered performance on the 3MS intercept (β (95%CI)= −0.36 (−0.60, −0.13), pval=0.0027). SNP rs17042917 is not strongly correlated with rs4251961 (r2=0.07). Two additional intronic SNPs, rs2637988 and rs3213448, were marginally associated, but did not meet our criteria for significance. Also for the IL1RN gene, SNP rs931471 is significant with the baseline 3MS score (β=0.39, 95%CI=0.13,0.65, pval=0.0032); this SNP lies outside the gene, and is modestly correlated with the other two SNPs (r2=0.22 for rs17042917and r2=0.22 for rs4251961). In addition to these IL1RN SNP associations with 3MS score, we detected an association with a missense SNP in the IL1A gene, rs17561, with increased annual rate of change on DSST score (β=0.08, 95%CI=0.01, 0.14, pval=0.016).
Fig. 2.
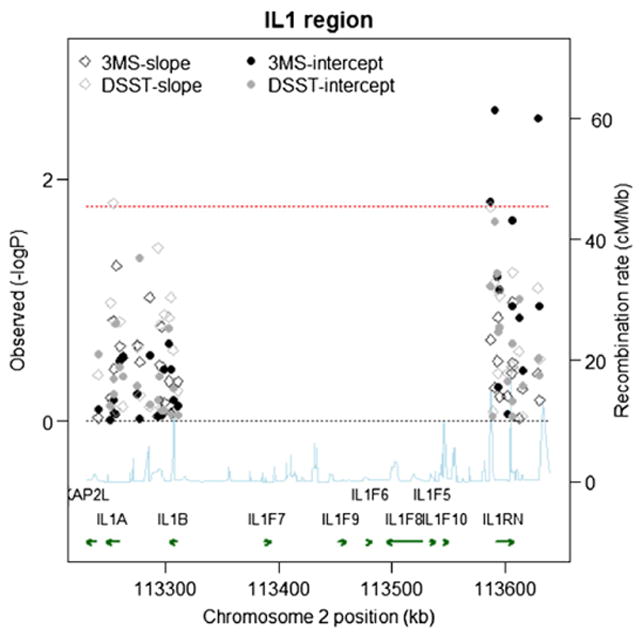
Single SNP Associations (−log10(pvalue)) by Position for 3MS and DSST scores in the White Cohort (n=3575). Chromosome position is shown in kilobases (kb). Genes annotations are shown on the bottom of the figure, where the direction of the arrow shows the 5′ to 3′ direction of transcription. Estimated recombination rates in centiMorgan per Megabase (cM/Mb) are obtained from HapMap data and are plotted directly above the genes to reflect the linkage disequilibrium structure. Position, annotations and estimated recombination rates use Build 36 coordinates. P-values are plotted as –log 10 values (−logP). Dashed lines are shown at values that correspond to a zero p-value and a p-value of 0.016. Solid symbols represent p-values from the fixed effect for the baseline term and open symbols represent p-values from the fixed effect for the slope term; symbols relating to the 3MS are in black, and symbols relating to the DSST are in gray. Software to create the figure is adapted from the R script provided for the Regional Association Plot on the Broad Institute website (http://www.broadinstitute.org/science/projects/diabetes-genetics-initiative/plotting-genome-wide-association-results).
Fig. 3.
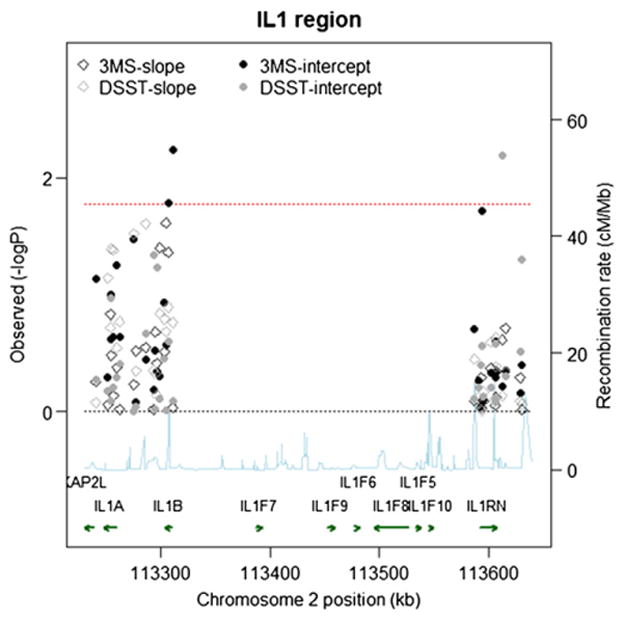
Single SNP Associations (−log10(pvalue)) by Position for 3MS and DSST scores in the AA Cohort (N=481). Chromosome position is shown in kilobases (kb). Genes annotations are shown on the bottom of the figure, where the direction of the arrow shows the 5′ to 3′ direction of transcription. Estimated recombination rates in centi-Morgan per Megabase (cM/Mb) are obtained from HapMap data and are plotted directly above the genes to reflect the linkage disequilibrium structure. Position, annotations and estimated recombination rates use Build 36 coordinates. P-values are plotted as −log 10 values (−logP). Dashed lines are shown at values that correspond to a zero p-value and a p-value of 0.016. Solid symbols represent p-values from the fixed effect for the baseline term and open symbols represent p-values from the fixed effect for the slope term; symbols relating to the 3MS are in black, and symbols relating to the DSST are in gray. Software to create the figure is adapted from the R script provided for the Regional Association Plot on the Broad Institute website (http://www.broadinstitute.org/science/projects/diabetes-genetics-initiative/plotting-genome-wide-association-results).
Table 5.
Significant single SNP associations with baseline cognition or annual rate of cognitive change among CHS and CHS cognition study participants.
| N | Test | Effect* | SNP | Type | CHS sample, minimally adjusted |
CHS cog sub-sample |
CHS cog sub-sample, non-demented only |
|||
|---|---|---|---|---|---|---|---|---|---|---|
| β (95%CI) | pval | β (95%CI) | pval | β (95%CI) | pval | |||||
| White cohort, IL1RN gene | ||||||||||
| 3515 | 3MS | Baseline | rs17042917 | 5′ UTR | 0.47 (0.1,0.85) | 0.016 | 0.27 (−0.14,0.68) | 0.19 | 0.08 (−0.27,0.43) | 0.64 |
| 3363 | 3MS | Baseline | rs4251961 | Intronic | −0.36 (−0.60, −0.13) | 0.0027 | −0.34 (−0.59, −0.09) | 0.008 | −0.08 (−0.29,0.14) | 0.48 |
| 3305 | 3MS | Baseline | rs931471 | Intergenic | 0.39 (0.13,0.65) | 0.0032 | 0.28 (0.00,0.55) | 0.050 | 0.08 (−0.15,0.32) | 0.49 |
| White cohort, IL1A gene | ||||||||||
| 3386 | DSST | Change | rs17561 | Non-synonymous | 0.08 (0.01, 0.14) | 0.016 | 0.06 (−0.01,0.13) | 0.12 | 0.09 (0.02, 0.16) | 0.02 |
| AA cohort, IL1B gene | ||||||||||
| 466 | 3MS | Baseline | rs1143627 | Upstream | 1.6 (0.48,2.8) | 0.006 | 1.12 (−0.10,2.33) | 0.073 | 0.60 (−0.55, 1.76) | 0.31 |
| 466 | 3MS | Baseline | rs1143634 | Synonymous | 2.1 (0.39,3.8) | 0.016 | 0.92 (−0.93,2.8) | 0.33 | −0.003 (−1.96,1.95) | 0.997 |
The SNP parameters presented in the table are interpreted from a mixed effects model that includes fixed effects for baseline cognition (intercept; β0), SNP genotype differences in cognition (β2), change in cognition over time (β1), and an interaction between SNP and change (differences in slope per SNP genotype; β3) and random effects for the baseline cognition (b1i) and the change (b2itij): Yij =β0 +β1tij +b1i +b2itij +β2IL1i +β3IL1i*tij +eij. Not shown are the fixed effects β terms for age, gender, education and all first order interactions terms that were significant.
Fig. 4.
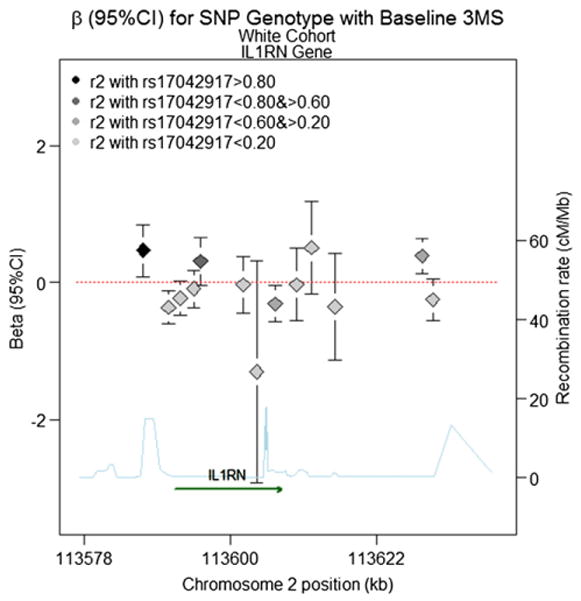
Beta (95%CI) for Baseline 3MS Score (fitted intercept) in the IL1RN gene for the White Cohort based on a model with additive inheritance. Chromosome position is shown in kilobases (kb). Genes annotations are shown on the bottom of the figure, where the direction of the arrow shows the 5′ to 3′ direction of transcription. Estimated recombination rates in centiMorgan per Megabase (cM/Mb) are obtained from HapMap data and are plotted directly above the gene to reflect the local linkage disequilibrium structure. Position, annotations and estimated recombination rates use Build 36 coordinates. The dashed line represents an effect size of zero. The point estimate for SNP rs17042917 is located on the far left, denoted by a black diamond, with error bars corresponding to the 95% confidence interval. For all other SNPs, the color of the diamond denoting the effect size reflects the pairwise correlation between a given marker and SNP rs17042917 (r2), so that darker shades of gray are highly correlated, and lighter shades of gray or not strongly correlated (see legend). Software to create the figure is adapted from the R script provided for the Regional Association Plot on the Broad Institute website (http://www.broadinstitute.org/science/projects/diabetes-genetics-initiative/plotting-genome-wide-association-results).
For the AA cohort, we observed two significant SNPs in the IL1B gene. Fig. 5 provides a schematic of the IL1B gene and regression estimates in AAs. Rs1143634 is a synonymous coding SNP that has been reported in the literature for AD (Hedley and others 2002; Wehr and others 2006). We observed a significant association with this SNP for the 3MS intercept (β (95%CI)=2.1 (0.39, 3.8), pval=0.016). The upstream SNP, rs1143627, is also significant (β (95%CI)=1.6 (0.48, 2.8), pval=0.006), and is not highly correlated with rs1143634 (r2=0.03).
Fig. 5.
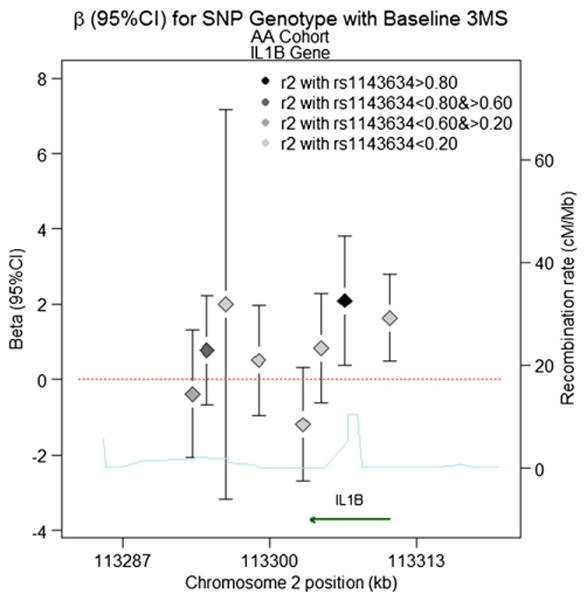
Beta (95%CI) for Baseline 3MS Score in the IL1B gene for the AA Cohort Models base on additive inheritance. Chromosome position is shown in kilobases (kb). Genes annotations are shown on the bottom of the figure, where the direction of the arrow shows the 5′ to 3′ direction of transcription. Estimated recombination rates in centi-Morgan per Megabase (cM/Mb) are obtained from HapMap data and are plotted directly above the gene to reflect the local linkage disequilibrium structure. Position, annotations and estimated recombination rates use Build 36 coordinates. The dashed line represents an effect size of zero. The point estimate for SNP rs1143634 is located second from the right, denoted by a black diamond, with error bars corresponding to the 95%confidence interval. For all other SNPs, the color of the diamond denoting the effect size reflects the pairwise correlation between a given marker and SNP rs1143634 (r2), so that darker shades of gray are highly correlated, and lighter shades of gray or not strongly correlated (see legend). Software to create the figure is adapted from the R script provided for the Regional Association Plot on the Broad Institute website (http://www.broadinstitute.org/science/projects/diabetes-genetics-initiative/plotting-genome-wide-association-results).
3.3.2. IL1 haplotype associations
Average haplotype block size for whites was 10.7 kb for whites and 10.9 kb in AAs. Haplotype block SNP membership was similar between the two cohorts, although the AA cohort had less linkage disequilibrium (LD) resulting in the designation of an extra haplotype block. None of the global haplotype tests yielded significant findings in the Caucasian or AA cohort.
3.3.3. Effect modification
We hypothesized that depression or NSAID use could modify the association between IL1 SNPs and cognition. We did not observe any statistically significant interaction for the AA cohort, or between SNPs and baseline use of NSAIDs in the Caucasian cohort. There was, however, a significant quantitative interaction with report of physician-diagnosed depression at baseline in the Caucasian cohort (interaction p value<0.05 for all three IL1RN SNPs with significant main effects). Supplementary Table 1 provides sample sizes, effect sizes and p-values for each SNP, stratified by the modifying variable for the Caucasian cohort. Associations for rs17042917, rs4251961, and rs931471 in the IL1RN gene, while significant among the non-depressed participants with effect size similar to the overall sample, were stronger among the approximate 171 individuals diagnosed with depression at baseline. Among these individuals, for rs17042917, those with a single copy of the minor allele performed several points better on the 3MS compared to those with a minor allele (β (95%CI)=3.99 (1.50, 6.5), pval=0.0017). Those depressed and possessing a single copy of rs931471 protective allele also performed better at baseline on the 3MS compared to those who were depressed with no protective alleles (β (95%CI)=1.97 (0.30, 3.7), pval=0.021). Finally, for rs4251961, interaction with depression at baseline was marginally significant; among those depressed at baseline, possession of the minor allele resulted in lowered cognitive performance on the 3MS compared to those with minor allele (β (95% CI)= −1.3 (−2.7, 0.11), pval=0.071). It should be noted that while the interaction with depression for these three SNPs are significant, genotype frequencies within the depressed group are small for the rare homozygotes. We did not observe any significant interactions with the IL1A SNP rs17561 in Caucasians or the IL1 SNPs in the AA cohort.
3.3.4. Specificity to dementia
It is important to further characterize whether the IL1 SNP associations identified were specific for those who were demented, rather than reflecting cognitive aging more generally. Because dementia was not adjudicated in the full sample, a comparison of “overall” results to only those without dementia was only possible among participants of the CHS Cognition Study. We first repeated the association analyses in this Cognition Sub-sample of participants, without regard to dementia status, to compare results for this subset versus the full CHS cohort presented in our primary results. Some of the associated SNP effects were attenuated among the CHS Cognition Study participants. For Caucasians, although effects sizes were consistent, SNPs rs17042917 and rs931471 were no longer statistically significant for their main effects (Table 5). We then compared results for the full Cognition sub-sample to results among only those without dementia. In general, when removing demented subjects from analysis, effect sizes attenuated and statistical significance decreased (Table 5).
4. Discussion
We evaluated several SNPs in three genes that are part of the interleukin-1 cluster on chromosome 2 for their association with cognitive test performance. The 3MS was used as a test of global cognition and the DSST as a measure of attention and processing speed. We found significant SNPs associated with baseline 3MS score in the IL1RN gene for Caucasians and in the IL1B gene for African Americans (AAs). Most of the variants in these genes lie in or near the promoter regions, and may affect cognition via alterations in the amount produced, rather than a conformational change in the protein. Supporting this, variants have been found for IL1RN (Zheng et al., 2000) and IL1B (Licastro et al., 2000; Pociot et al., 1992) that associate with increase serum levels of IL1, although SNPs in the IL1RN gene have not been identified to significantly increase gene expression (eQTLs). Only one significant association with annual rate of change on the DSST was found for a missense SNP in the IL1A gene. The lack of findings with change over time on cognitive test performance may be that these SNP associations affect only the intrinsic level of cognitive ability, but could also be due to the fact that this is an older cohort, and influences on cognitive change have already exerted their effects. It may also be that we were underpowered to detect very small effect sizes in the annual rate of change in test performance. Taken together, our results provide some supporting evidence that IL1 may precede brain-related changes that influence cognition.
For Caucasians, the pattern of SNP associations in IL1RN is consistent with LD attenuation of a true association signal. The strongest effect was for a SNP in the 5′ untranslated region (rs17042917) for baseline 3MS score. The next closest SNP (rs4251961) was also significant for baseline 3MS and baseline DSST; two other SNPs moderately correlated with these significant SNPs were also marginally associated. This pattern suggests that all associations may reflect a common signal from the 5′ untranslated SNP, rs17042917, or from an unmeasured variant in high LD with this SNP. In addition to these SNPs, an intergenic SNP 3′ of the IL1RN gene (rs931471) was also statistically significantly associated with baseline 3MS score. Given the modest correlation between this SNP and the 5′ untranslated SNP in our sample, it is possible that this SNP also reflects the 5′ untranslated SNP association. The association with increased performance over time in the DSST and the IL1A SNP rs17561, although modest, is interesting in that the DSST is perhaps more closely related to cognitive aging than to dementia (Joy et al., 2000). The persistence of the association after removing those with dementia supports this interpretation.
The SNP associations in IL1RN were stronger in those who reported being currently diagnosed by a physician with depression. Because IL1 levels have been shown to be elevated in older depressed adults (Thomas et al., 2005), and because animal models suggest that increased peripheral levels can increase brain levels of IL1 (Licinio and Wong, 1997), IL1 inflammation in depression could be a trigger that initiates the IL1-driven cytokine cycle in brain, where IL1 genetic predispositions can have greater influence. We chose a report of physician-diagnosed current depression rather than score on a 10-item shortened version of the Center for Epidemiological Studies Depression Scale (CES-D), which measures the number of depressive symptoms experienced in the previous week (Arbelaez et al., 2007). When using the CES-D >8 as a threshold for depression, overall agreement is 86%, but the effect modification is no longer significant. Thus, our observed quantitative effect modification depends on how depression is measured. We believe the ideal measurement would be long-standing clinical depression that results in chronically elevated cytokine levels, and acknowledge that measures of only current depression has limitation.
For the IL1B gene, we observed two significant SNPs in the AA sample. One lies in the promoter region (rs1143627) and the other is a synonymous coding SNP (rs1143634). Because IL1 genes are likely to affect cognition by increased cytokine production, a synonymous SNP could alter protein levels by affecting the translational stability, splicing, or transcriptional control of the codon. The synonymous IL1B coding SNP rs1143634 has been associated with AD in two previous reports (Hedley et al., 2002; Wehr et al., 2006). The promoter SNP rs1143627 (also referred to as the −31 IL1B promoter SNP) was shown to have a modest posterior probability for being an eQTL of 0.48 (Veyrieras et al., 2008). The rs1143627 SNP has not been investigated for AD, but was highly correlated with promoter SNPs that have been previously associated with AD: rs16944 (r2=0.96, also known as the IL1B −511 promoter SNP), rs3087258 (four base pairs away from rs16944), and rs1143623 (r2=0.86). This last SNP is interesting in that the rarer G allele was associated with weaker promoter activity and decreased gene expression which could be explained by lower binding at the GATA motif (Harrison et al., 2008; Lee et al., 2004). This SNP was also significantly associated with rheumatoid arthritis in a case–control study (Harrison et al., 2008). Finally, the rs16944 SNP correlated with memory performance in a population of older Caucasians (Baune et al., 2008).
Compared to the full CHS cohort, we saw an attenuation of the IL1 SNP associations when limiting to the Cognition Sub-sample. Allele and genotype frequencies do not explain this difference (data not shown), nor does a drop in precision due to decreased sample size. We posit that this attenuation is most likely due to the modifying effect of unmeasured cognitive reserve, which is likely higher in the participants in the Cognition Sub-sample compared to the full CHS. Evidence that supports the cognitive reserve hypothesis (Stern, 2003; Whalley, Deary et al., 2004) has been reported for CHS (Elkins et al., 2006), such that associations with cognitive tests and MRI-defined infarct were weaker among those with higher education levels, presumably because these participants have increased brain reserve and better compensatory strategies. Thus, we would expect IL1 SNP associations with cognitive tests to weaken among the CHS Sub-sample, which is better educated, assuming education is a good proxy for cognitive reserve. We would also expect SNP associations to be stronger in the low education group in the full CHS cohort. In fact, SNP association estimates among those with less than a high school education in the whole CHS cohort were stronger and more significant than those of higher education levels, although a test for statistical interaction was not significant (data not shown). This suggests education only imperfectly marks cognitive reserve. Even among similarly educated participants, 3MS scores in the Cognition Sub-sample were higher, and decline was slower, compared to the full CHS sample, supporting this idea. Thus, the attenuation of estimates when limiting to the Cognition Sub-sample may be explained by increased cognitive reserve in this sample, which cannot be fully accounted for by adjusting for education, or by including it as a modifier.
We could only address whether the IL1 gene effects were specific to dementia among the Cognition Sub-sample, since only these participants provided enough information for dementia adjudication. Within this group, IL1 effect sizes and statistical significance do attenuate when those diagnosed with dementia are removed from analysis. These findings provide some evidence to suggest that the genetic IL1 signals may be specific to a progressive or impending dementia process, as opposed to a cognitive aging process that is associated with more limited degenerative pathology (Albert, 2002). It should be noted, however, that the 3MS score was used in part to determine dementia adjudication so that those with low 3MS scores were most likely to be demented. Case-control studies have found IL1 gene associations with AD (Hedley et al., 2002; Wehr et al., 2006), further supporting our findings that IL1 genetic signals are specific to dementia.
Finally, we only observed one SNP to be associated with change in performance on the DSST, a test shown to be a robust predictor of cognitive aging (Joy et al., 2000; Salthouse, 1978), and this SNP association persisted in the non-demented group. The fact that most of our results are confined to the 3MS, and attenuated exclusively among the associations with 3MS, suggests that these IL1 SNPs could be specific to a dementing process.
Because the effects of IL1 on neurodegeneration are suspected to be diverse, global cognition may be a relevant general phenotype for these genes, as we hypothesize. However, the 3MS, while capturing global cognitive function, was intended primarily for detection of dementia (Teng and Chui, 1987) and may not fully represent global cognitive function associated with aging per se. In contrast, focus within separate cognitive domains may be useful to further characterize the role of IL1 variation. In fact, a study of older Caucasians found an association between an IL1B SNP and memory performance, but not speed and motor function (Baune et al., 2008). If a SNP is suspected to affect cognition through its purported role in an AD dementing process, we may benefit by measuring episodic memory or executive functioning, as these measures are thought to be early indicators of disease (Albert, 1996).
In our analyses of IL1 genes, we chose a modified Bonferroni correction where we assumed three independent tests for each of the three candidate genes (resulting in a threshold p-value of 0.016) to establish significance. No SNPs would have been significant at the more conservative study-wide Bonferroni value, which assumes 31 independent tests. Had we imposed a study-wide false discovery rate, three SNPs would be significant: rs4251961 and rs931471 in the IL1RN gene, and rs1143634 in the IL1B gene. We selected a less conservative criterion because of previous reported associations in these genes, and because we wanted to highlight effect sizes and consistent LD patterns within a gene. To ultimately protect against false positive reports of association we encourage further replication in other large study samples with similar cognitive testing.
Our results show association between two genes in the IL1 family and baseline cognitive performance among older adults, providing some genetic evidence that the pro-inflammatory cytokine IL1 may affect cognition, likely via a dementing process. Our study had excellent gene coverage, continuous measures of cognitive test performance collected over time, and provided a large sample size in Caucasians. We saw differences by ethnic background that may be important for future replication and interpretation. Finally, education levels serving as a proxy for cognitive reserve may modify associations found with cognition and genes that lie in inflammatory pathways, so that any future replication efforts should take level of education into consideration.
Supplementary Material
Acknowledgments
This study was supported by contracts N01-HC-85079 through N01-HC-85086, N01-HC-35129, N01-HC-15103, N01 HC-55222, N01-HC-75150, N01-HC-45133, grant number U01 HL080295 from the National, Heart, Lung and Blood Institute, and grant AG15928 from the National Institute on Aging, with additional contribution from the National Institute of Neurological Disorders and Stroke. A full list of principal CHS investigators and institutions can be found at http://www.chs-nhlbi.org/pi.htm. The Johns Hopkins Older Adults Indepedence Center (P30-AG021334) provided funding genotyping and analytic support. We would also like to thank Alexis Rea and Gina Davis for their assistance with the Biotrove genotyping for this project.
Footnotes
Supplementary materials related to this article can be found online at doi:10.1016/j.exger.2011.09.005.
References
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiol aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert MS. Cognitive and neurobiologic markers of early Alzheimer disease. Proc Natl Acad Sci U S A. 1996;93:13547–13551. doi: 10.1073/pnas.93.24.13547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert MS. Memory decline: the boundary between aging and age-related disease. Ann Neurol. 2002;51:282–284. [PubMed] [Google Scholar]
- Arbelaez JJ, Ariyo AA, Crum RM, Fried LP, Ford DE. Depressive symptoms, inflammation, and ischemic stroke in older adults: a prospective analysis in the cardiovascular health study. J Am Geriatr Soc. 2007;55 (11):1825–1830. doi: 10.1111/j.1532-5415.2007.01393.x. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. (Oxford, England) [DOI] [PubMed] [Google Scholar]
- Baune BT, Ponath G, Rothermundt M, Riess O, Funke H, Berger K. Association between genetic variants of IL-1beta, IL-6 and TNF-alpha cytokines and cognitive performance in the elderly general population of the MEMO-study. Psychoneuroendocrinology. 2008;33:68–76. doi: 10.1016/j.psyneuen.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Brennan R, Jorgensen L, Gordon S, Loades K, Hackett C, Russell J. The development of a PCR-based marker linked to resistance to the blackcurrant gall mite (Cecidophyopsis ribis Acari: Eriophyidae) TAG Theor Appl Genet. 2009;118:205–211. doi: 10.1007/s00122-008-0889-x. [DOI] [PubMed] [Google Scholar]
- Cechetto DF, Hachinski V, Whitehead SN. Vascular risk factors and Alzheimer’s disease. Expert Rev Neurother. 2008;8:743–750. doi: 10.1586/14737175.8.5.743. [DOI] [PubMed] [Google Scholar]
- de Bakker PI, Yelensky R, Pe’er I, Gabriel SB, Daly MJ, Altshuler D. Efficiency and power in genetic association studies. Nat Genet. 2005;37:1217–1223. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- Déniz-Naranjo MC, Muñoz-Fernandez C, Alemany-Rodríguez MJ, Pérez-Vieitez MC, Aladro-Benito Y, Irurita-Latasa J, Sánchez-García F. Cytokine IL-1 beta but not IL-1 alpha promoter polymorphism is associated with Alzheimer disease in a population from the Canary Islands, Spain. Eur J Neurol. 2008;15 (10):1080–1084. doi: 10.1111/j.1468-1331.2008.02252.x. [DOI] [PubMed] [Google Scholar]
- Elkins JS, Longstreth WT, Jr, Manolio TA, Newman AB, Bhadelia RA, Johnston SC. Education and the cognitive decline associated with MRI-defined brain infarct. Neurology. 2006;67:435–440. doi: 10.1212/01.wnl.0000228246.89109.98. [DOI] [PubMed] [Google Scholar]
- Engelhart MJ, Geerlings MI, Meijer J, Kiliaan A, Ruitenberg A, van Swieten JC, Stijnen T, Hofman A, Witteman JC, Breteler MM. Inflammatory proteins in plasma and the risk of dementia: the rotterdam study. Arch Neurol. 2004;61:668–672. doi: 10.1001/archneur.61.5.668. [DOI] [PubMed] [Google Scholar]
- Fried LP, Borhani NO, Enright P, Furberg CD, Gardin JM, Kronmal RA, Kuller LH, Manolio TA, Mittelmark MB, Newman A, et al. The Cardiovascular Health Study: design and rationale. Ann Epidemiol. 1991;1:263–276. doi: 10.1016/1047-2797(91)90005-w. [DOI] [PubMed] [Google Scholar]
- Giedraitis V, Kilander L, Degerman-Gunnarsson M, Sundelöf J, Axelsson T, Syvänen AC, Lannfelt L, Glaser A. Genetic analysis of Alzheimer’s disease in the Uppsala longitudinal study of adult men. Dement Geriatr Cogn Disord. 2009;27 (1):59–68. doi: 10.1159/000191203. [DOI] [PubMed] [Google Scholar]
- Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer’s disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain pathology. 1998;8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. (Zurich, Switzerland) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi LM, Casadei VM, Ferri C, Veglia F, Licastro F, Annoni G, Biunno I, De Bellis G, Sorbi S, Mariani C, Canal N, Griffin WS, Franceschi M. Association of early-onset Alzheimer’s disease with an interleukin-1alpha gene polymorphism. Ann Neurol. 2000;47 (3):361–365. [PubMed] [Google Scholar]
- Harrison P, Pointon JJ, Chapman K, Roddam A, Wordsworth BP. Interleukin-1 promoter region polymorphism role in rheumatoid arthritis: a meta-analysis of IL-1B-511A/G variant reveals association with rheumatoid arthritis. Rheumatology. 2008;47:1768–1770. doi: 10.1093/rheumatology/ken374. (Oxford, England) [DOI] [PubMed] [Google Scholar]
- Hedley R, Hallmayer J, Groth DM, Brooks WS, Gandy SE, Martins RN. Association of interleukin-1 polymorphisms with Alzheimer’s disease in Australia. Ann Neurol. 2002;51:795–797. doi: 10.1002/ana.10196. [DOI] [PubMed] [Google Scholar]
- Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Jones N, Stretton A, Thomas C, Richards A, Ivanov D, Widdowson C, Chapman J, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Beaumont H, Warden D, Wilcock G, Love S, Kehoe PG, Hooper NM, Vardy ER, Hardy J, Mead S, Fox NC, Rossor M, Collinge J, Maier W, Jessen F, Rüther E, Schürmann B, Heun R, Kölsch H, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frölich L, Hampel H, Gallacher J, Hüll M, Rujescu D, Giegling I, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Mühleisen TW, Nöthen MM, Moebus S, Jöckel KH, Klopp N, Wichmann HE, Pankratz VS, Sando SB, Aasly JO, Barcikowska M, Wszolek ZK, Dickson DW, Graff-Radford NR, Petersen RC, van Duijn CM, Breteler MM, Ikram MA, DeStefano AL, Fitzpatrick AL, Lopez O, Launer LJ, Seshadri S, Berr C, Campion D, Epelbaum J, Dartigues JF, Tzourio C, Alpérovitch A, Lathrop M, Feulner TM, Friedrich P, Riehle C, Krawczak M, Schreiber S, Mayhaus M, Nicolhaus S, Wagenpfeil S, Steinberg S, Stefansson H, Stefansson K, Snaedal J, Björnsson S, Jonsson PV, Chouraki V, Genier-Boley B, Hiltunen M, Soininen H, Combarros O, Zelenika D, Delepine M, Bullido MJ, Pasquier F, Mateo I, Frank-Garcia A, Porcellini E, Hanon O, Coto E, lvarez V, Bosco P, Siciliano G, Mancuso M, Panza F, Solfrizzi V, Nacmias B, Sorbi S, Bossù P, Piccardi P, Arosio B, Annoni G, Seripa D, Pilotto A, Scarpini E, Galimberti D, Brice A, Hannequin D, Licastro F, Jones L, Holmans PA, Jonsson T, Riemenschneider M, Morgan K, Younkin SG, Owen MJ, O’Donovan M, Amouyel P, Williams J Alzheimer’s Disease Neuroimaging Initiative; CHARGE consortium; EADI1 consortium. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43 (5):429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulkkonen J, Laippala P, Hurme M. A rare allele combination of the interleukin-1 gene complex is associated with high interleukin-1 beta plasma levels in healthy individuals. Eur Cytokine Netw. 2000;11 (2):251–255. [PubMed] [Google Scholar]
- Joy S, Fein D, Kaplan E, Freedman M. Speed and memory in WAIS-R-NI digit symbol performance among healthy older adults. J Int Neuropsychol Soc. 2000;6:770–780. doi: 10.1017/s1355617700677044. [DOI] [PubMed] [Google Scholar]
- Klimkowicz-Mrowiec A, Marona M, Wołkow P, Maruszak A, Styczynska M, Barcikowska M, Zekanowski C, Szczudlik A, Slowik A. Interleukin-1 gene −511 CT polymorphism and the risk of Alzheimer’s disease in a Polish population. Dement Geriatr Cogn Disord. 2009;28 (5):461–464. doi: 10.1159/000259460. [DOI] [PubMed] [Google Scholar]
- Knopman DS, Mosley TH, Catellier DJ, Coker LH. Fourteen-year longitudinal study of vascular risk factors, APOE genotype, and cognition: the ARIC MRI study. Alzheimers Dement. 2009;5:207–214. doi: 10.1016/j.jalz.2009.01.027. [DOI] [PubMed] [Google Scholar]
- Kronfol Z. Cytokines and Mental Health. Kluwer Academic Publishers; Boston: 2003. [Google Scholar]
- Kuller LH, Shemanski L, Manolio T, Haan M, Fried L, Bryan N, Burke GL, Tracy R, Bhadelia R. Relationship between ApoE, MRI findings, and cognitive function in the Cardiovascular Health Study. Stroke j cerebral circulation. 1998;29:388–398. doi: 10.1161/01.str.29.2.388. [DOI] [PubMed] [Google Scholar]
- Lee KA, Ki CS, Kim HJ, Sohn KM, Kim JW, Kang WK, Rhee JC, Song SY, Sohn TS. Novel interleukin 1beta polymorphism increased the risk of gastric cancer in a Korean population. J Gastroenterol. 2004;39:429–433. doi: 10.1007/s00535-003-1315-4. [DOI] [PubMed] [Google Scholar]
- Licastro F, Chiappelli M. Brain immune responses cognitive decline and dementia: relationship with phenotype expression and genetic background. Mech ageing dev. 2003;124:539–548. doi: 10.1016/s0047-6374(03)00034-4. [DOI] [PubMed] [Google Scholar]
- Licastro F, Pedrini S, Ferri C, Casadei V, Govoni M, Pession A, Sciacca FL, Veglia F, Annoni G, Bonafe M, Olivieri F, Franceschi C, Grimaldi LM. Gene polymorphism affecting alpha1-antichymotrypsin and interleukin-1 plasma levels increases Alzheimer’s disease risk. Ann Neurol. 2000;48:388–391. [PubMed] [Google Scholar]
- Licinio J, Wong ML. Pathways and mechanisms for cytokine signaling of the central nervous system. J Clin Invest. 1997;100:2941–2947. doi: 10.1172/JCI119846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez OL, Becker JT, Kaufer DI, Hamilton RL, Sweet RA, Klunk W, DeKosky ST. Research evaluation and prospective diagnosis of dementia with Lewy bodies. Arch Neurol. 2002;59:43–46. doi: 10.1001/archneur.59.1.43. [DOI] [PubMed] [Google Scholar]
- Lyketsos CG, Lopez O, Jones B, Fitzpatrick AL, Breitner J, DeKosky S. Prevalence of neuropsychiatric symptoms in dementia and mild cognitive impairment: results from the cardiovascular health study. Jama. 2002;288:1475–1483. doi: 10.1001/jama.288.12.1475. [DOI] [PubMed] [Google Scholar]
- McDowell TL, Symons JA, Ploski R, Forre O, Duff GW. A genetic association between juvenile rheumatoid arthritis and a novel interleukin-1 alpha polymorphism. Arthritis and Rheumatism. 1995;38:221–228. doi: 10.1002/art.1780380210. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Inflammation and the degenerative diseases of aging. Ann N Y Acad Sci. 2004;1035:104–116. doi: 10.1196/annals.1332.007. [DOI] [PubMed] [Google Scholar]
- Morley JE. The top 10 hot topics in aging. j gerontology. 2004;59:24–33. doi: 10.1093/gerona/59.1.m24. [DOI] [PubMed] [Google Scholar]
- Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JS, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, St George-Hyslop P, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, DeCarli C, DeKosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Parisi JE, Perl DP, Peskind E, Petersen RC, Poon WW, Quinn JF, Rajbhandary RA, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosenberg RN, Sano M, Schneider LS, Seeley W, Shelanski ML, Slifer MA, Smith CD, Sonnen JA, Spina S, Stern RA, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Woltjer RL, Cantwell LB, Dombroski BA, Beekly D, Lunetta KL, Martin ER, Kamboh MI, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43 (5):436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll JA, Mrak RE, Graham DI, Stewart J, Wilcock G, MacGowan S, Esiri MM, Murray LS, Dewar D, Love S, Moss T, Griffin WS. Association of interleukin-1 gene polymorphisms with Alzheimer’s disease. Ann Neurol. 2000;47 (3):365–368. [PMC free article] [PubMed] [Google Scholar]
- Pociot F, Molvig J, Wogensen L, Worsaae H, Nerup J. A TaqI polymorphism in the human interleukin-1 beta (IL-1 beta) gene correlates with IL-1 beta secretion in vitro. Eur J Clin Investig. 1992;22:396–402. doi: 10.1111/j.1365-2362.1992.tb01480.x. [DOI] [PubMed] [Google Scholar]
- Rachal Pugh C, Fleshner M, Watkins LR, Maier SF, Rudy JW. The immune system and memory consolidation: a role for the cytokine IL-1beta. Neurosci Biobehav Rev. 2001;25:29–41. doi: 10.1016/s0149-7634(00)00048-8. [DOI] [PubMed] [Google Scholar]
- Romero JR, Beiser A, Seshadri S, Benjamin EJ, Polak JF, Vasan RS, Au R, DeCarli C, Wolf PA. Carotid artery atherosclerosis, MRI indices of brain ischemia, aging, and cognitive impairment: the Framingham study. Stroke j cerebral circulation. 2009;40:1590–1596. doi: 10.1161/STROKEAHA.108.535245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross FM, Allan SM, Rothwell NJ, Verkhratsky A. A dual role for interleukin-1 in LTP in mouse hippocampal slices. J Neuroimmunol. 2003;144:61–67. doi: 10.1016/j.jneuroim.2003.08.030. [DOI] [PubMed] [Google Scholar]
- Rothwell NJ. Annual review prize lecture cytokines — killers in the brain? J Physiol. 1999;514 (Pt 1):3–17. doi: 10.1111/j.1469-7793.1999.003af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozzini R, Ferrucci L, Losonczy K, Havlik RJ, Guralnik JM. Protective effect of chronic NSAID use on cognitive decline in older persons. J Am Geriatr Soc. 1996;44:1025–1029. doi: 10.1111/j.1532-5415.1996.tb02932.x. [DOI] [PubMed] [Google Scholar]
- Salthouse TA. The role of memory in the age decline in digit-symbol substitution performance. J Gerontol. 1978;33:232–238. doi: 10.1093/geronj/33.2.232. [DOI] [PubMed] [Google Scholar]
- Stern Y. The concept of cognitive reserve: a catalyst for research. Journal of clinical and experimental neuropsycholog. 2003;25:589–593. doi: 10.1076/jcen.25.5.589.14571. [DOI] [PubMed] [Google Scholar]
- Szekely CA, Thorne JE, Zandi PP, Ek M, Messias E, Breitner JC, Goodman SN. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease: a systematic review. Neuroepidemiology. 2004;23:159–169. doi: 10.1159/000078501. [DOI] [PubMed] [Google Scholar]
- Teng EL, Chui HC. The Modified Mini-Mental State (3MS) examination. J clin psychiatry. 1987;48:314–318. [PubMed] [Google Scholar]
- Thomas AJ, Davis S, Morris C, Jackson E, Harrison R, O’Brien JT. Increase in interleukin-1beta in late-life depression. Am J Psychiatry. 2005;162:175–177. doi: 10.1176/appi.ajp.162.1.175. [DOI] [PubMed] [Google Scholar]
- Trompet S, de Craen AJ, Slagboom P, Shepherd J, Blauw GJ, Murphy MB, Bollen EL, Buckley BM, Ford I, Gaw A, Macfarlane PW, Packard CJ, Stott DJ, Jukema JW, Westendorp RG. Genetic variation in the interleukin-1 beta-converting enzyme associates with cognitive function. The PROSPER study. Brain. 2008;131:1069–1077. doi: 10.1093/brain/awn023. [DOI] [PubMed] [Google Scholar]
- Veyrieras JB, Kudaravalli S, Kim SY, Dermitzakis ET, Gilad Y, Stephens M, Pritchard JK. High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet. 2008;4:e1000214. doi: 10.1371/journal.pgen.1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitkovic L, Bockaert J, Jacque C. “Inflammatory” cytokines: neuromodulators in normal brain? J Neurochem. 2000;74:457–471. doi: 10.1046/j.1471-4159.2000.740457.x. [DOI] [PubMed] [Google Scholar]
- Weaver JD, Huang MH, Albert M, Harris T, Rowe JW, Seeman TE. Interleukin-6 and risk of cognitive decline: MacArthur studies of successful aging. Neurology. 2002;59:371–378. doi: 10.1212/wnl.59.3.371. [DOI] [PubMed] [Google Scholar]
- Wehr H, Bednarska-Makaruk M, Lojkowska W, Graban A, Hoffman-Zacharska D, Kuczynska-Zardzewialy A, Mrugala J, Rodo M, Bochynska A, Sulek A, Ryglewicz D. Differences in risk factors for dementia with neurodegenerative traits and for vascular dementia. Dement Geriatr Cogn Disord. 2006;22:1–7. doi: 10.1159/000092845. [DOI] [PubMed] [Google Scholar]
- Whalley LJ, Deary IJ, Appleton CL, Starr JM. Cognitive reserve and the neurobiology of cognitive aging. Ageing research reviews. 2004;3:369–382. doi: 10.1016/j.arr.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Finch CE, Cohen HJ. Cytokines and cognition—the case for a head-to-toe inflammatory paradigm. J Am Geriatr Soc. 2002;50:2041–2056. doi: 10.1046/j.1532-5415.2002.50619.x. [DOI] [PubMed] [Google Scholar]
- Yaffe K, Haan M, Byers A, Tangen C, Kuller L. Estrogen use, APOE, and cognitive decline: evidence of gene-environment interaction. Neurology. 2000;54:1949–1954. doi: 10.1212/wnl.54.10.1949. [DOI] [PubMed] [Google Scholar]
- Yaffe K, Lindquist K, Penninx BW, Simonsick EM, Pahor M, Kritchevsky S, Launer L, Kuller L, Rubin S, Harris T. Inflammatory markers and cognition in well-functioning African-American and white elders. Neurology. 2003;61:76–80. doi: 10.1212/01.wnl.0000073620.42047.d7. [DOI] [PubMed] [Google Scholar]
- Zandi PP, Breitner JC. Do NSAIDs prevent Alzheimer’s disease? And, if so, why? The epidemiological evidence. Neurobiol aging. 2001;22:811–817. doi: 10.1016/s0197-4580(01)00297-4. [DOI] [PubMed] [Google Scholar]
- Zheng C, Huang DR, Bergenbrant S, Sundblad A, Osterborg A, Bjorkholm M, Holm G, Yi Q. Interleukin 6, tumour necrosis factor alpha, interleukin 1beta and interleukin 1 receptor antagonist promoter or coding gene polymorphisms in multiple myeloma. Br J Haematol. 2000;109:39–45. doi: 10.1046/j.1365-2141.2000.01963.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.