

Abstract
Castration-resistant prostate cancers (CRPCs) that relapse after androgen deprivation therapies (ADTs) are responsible for the majority of mortalities from prostate cancer (PCa). While mechanisms enabling recurrent activity of androgen receptor (AR) are certainly involved in the development of CRPC, there may be factors that contribute to the process including acquired neuroendocrine (NE) cell-like behaviors working through alternate (non-AR) cell signaling systems or AR-dependent mechanisms. In this study, we explore the potential relationship between the AR axis and a novel putative marker of NE differentiation, the human male protocadherin-PC (PCDH-PC), in vitro and in human situations. We found evidence for an NE transdifferentiation process and PCDH-PC expression as an early-onset adaptive mechanism following ADT and elucidate AR as a key regulator of PCDH-PC expression. PCDH-PC overexpression, in turn, attenuates the ligand-dependent activity of the AR, enabling certain prostate tumor clones to assume a more NE phenotype and promoting their survival under diverse stress conditions. Acquisition of an NE phenotype by PCa cells positively correlated with resistance to cytotoxic agents including docetaxel, a taxane chemotherapy approved for the treatment of patients with metastatic CRPC. Furthermore, knockdown of PCDH-PC in cells that have undergone an NE transdifferentiation partially sensitized cells to docetaxel. Together, these results reveal a reciprocal regulation between the AR axis and PCDH-PC signals, observed both in vitro and in vivo, with potential implications in coordinating NE transdifferentiation processes and progression of PCa toward hormonal and chemoresistance.
Introduction
Prostate cancer (PCa) is the most commonly diagnosed malignancy among men in Western nations [1]. It is well recognized that androgens working through the androgen receptor (AR), play a key role in PCa disease initiation and progression [2] and are known to stimulate the PCa cell growth and diminish their rate of apoptosis. This is the basis for the use of androgen deprivation therapy (ADT) in the form of medical or surgical castration as standard frontline therapy for patient with advanced disease [3]. Despite the fact that ADT has been proven to extend life span in accordance with its effect of limiting the growth of “androgen-sensitive” PCa cells and inducing cell death of “androgendependent” PCa cells, one important aspect of PCa is that the majority of cases eventually develop resistance to ADT and castration-resistant prostate cancer (CRPC) emerges. Although there are a number of approved and promising therapies for metastatic CRPC, including taxane chemotherapies (i.e., docetaxel, cabazitaxel) and potent AR-targeted agents (i.e., abiraterone, MDV3100) [4], all patients develop resistance, and as such, metastatic CRPC accounts for most PCa-related deaths.
A key mechanism involved in progression of PCa from a hormone-sensitive to castration-resistant state includes acquisition of molecular alterations of the androgen/AR axis, such that PCa cells retain active AR even in the setting of castrate levels of circulating testosterone [5]. However, an alternative mechanism that dominates in some cases of CRPC involves transformation toward an androgen-independent state, in which certain PCa cells offset their sensitivity to androgens by altering their apoptotic pathways such that active androgen/AR signaling is no longer mandatory for their survival. These androgen-independent cell populations may either arise from progenitor or neuroendocrine (NE)-like cells in the primary prostate tumor or from prostate adenocarcinoma cells that transdifferentiate to NE-like cells. It has been more than a decade since the concept first emerged from in vitro studies suggesting the latter, that under certain circumstances, including hormonal manipulation, PCa cells have the potential to transdifferentiate to acquire NE characteristics [6–10]. Despite evidence of upregulated NE differentiation in patients receiving ADT [11,12], the origin of NE cells in the prostate remains uncertain. Moreover, the relative lack of knowledge regarding the chain of events and the mechanistic paradigm underlying the trans-differentiation process supports the need for further investigations.
We previously reported that overexpression of protocadherin-PC (PCDH-PC, also referred to as PCDH11Y), a gene primarily identified for its antiapoptotic properties that encodes from the Y-chromosome at Yp11.2 [13,14], can drive NE transdifferentiation in LNCaP [15], a cell line originally established from a lymph node metastatic lesion of human PCa characterized by its androgen-dependent growth [16]. Here, by exploring the potential relationship between the androgen/AR axis and PCDH-PC, we investigated the possibility that PCa progression toward androgen independence is indeed characterized by a putative subpopulation of cancer cells that undergo an NE transdifferentiation. We also explore the extent to which the emergence of these populations is influenced by current therapies for advanced CRPC.
Materials and Methods
Cell Culture and Chemicals
The human PCa cell lines LNCaP and 22Rv1 were obtained from ATCC (Manassas, VA), authenticated at this site, and maintained in recommended medium. For androgen-reduced conditions, cells were cultured in phenol red-free RPMI supplemented with 10% dextran charcoal-stripped FBS (CS-FBS). The LNCaP-PCDH-PC cells were previously described [17]. Steroids and chemotherapeutic agents were obtained from Sigma-Aldrich (St Quentin Fallavier, France). Bicalutamide was obtained from LKT Laboratories (St Paul, MN).
Human Prostate Tissue Samples
The prostate samples have been collected as part of an Institutional Review Board-approved protocol at Henri Mondor Hospital. Specimens consisted of formalin-fixed paraffin-embedded (FFPE) tissues from hormone-naïve PCa (HNPC; n = 222), neoadjuvant hormone therapy-treated PCa (HTPC; n = 32) obtained from radical prostatectomy specimens, and CRPC specimens (n = 60), of which 54 were collected at the time of the transurethral resection of the prostate for obstructive CRPC and 6 isolated from rapid autopsy specimens with metastatic lesions. The study also included a few specimens derived from normal prostates of young donors.
Immunohistochemistry and Immunofluorescence
Paraffin-embedded tissues were sectioned at 5-µm thickness and deparaffinized, and endogenous peroxidase activity was inactivated in a solution containing 3% hydrogen peroxide (H2O2) for 10 minutes. Sections were then cleared in running water followed by phosphate-buffered saline. Antigen unmasking was performed by heat retrieval with citrate buffer (pH 6; Dako, Trappes, France). The primary antibodies used are listed in Table W1. Antibodies purified from HB 0337 SSA hybridoma and raised against PCDH-PC are available upon request to Prof. F. Vacherot (vacherot@u-pec.fr). Biotin-labeled antibodies (Jackson ImmunoResearch, New Market, United Kingdom) were used as secondary antibodies. Antigen-antibody reactions were revealed using the streptavidin method with DAB as substrate. All slides were read by a genitourinary pathologist (Y.A.) and the intensity of staining was scored as null (0), weak (1), moderate (2), and strong (3). In this analysis, a case was considered positive only when the score was 2 or more in at least 10% of cancer cells, whereas cases with less than 10% staining or scored below 2 were considered as negative. For dual immunofluorescence staining, samples were processed as above but using, as secondary antibodies, anti-mouse Alexa Fluor 488 (Life Technologies, Grand Island, NY) and biotinylated anti-rabbit antibodies (Jackson ImmunoResearch) with subsequent incubation with Streptavidin-Fluoprobes 647H (Interchim, Montluçon, France). Slides were mounted using Vectashield mounting medium (Vector Laboratories, Burlingame, CA) and inspected by confocal microscopy.
Transient Transfection and Luciferase Reporter Assays
Transient transfection assays and measures of luciferase and β-galactosidase (β-Gal) activities were performed as previously described [15] with minor modifications. The PSA-61-luc plasmid was described previously [18] and used as reporter of AR activity. Briefly, cells (6 x 105 per well) were plated onto 24-well plates and cotransfected the next day using Lipofectamine 2000 (Life Technologies) mixed with up to 400 ng of pcDNA3-PCDH-PC vector or empty pcDNA3 along with 500 ng of a PSA-61-luc and 50 ng of a Lac-Z luciferase plasmid as a transfection control, so that all wells received ∼1 µg of DNA. On the next day, cells were treated with dihydrotestosterone (DHT) for 24 hours after which cell lysates were prepared and processed for luciferase activity and β-Gal activity using the Luciferase Reporter Assay and β-Gal Reporter Gene Assay Kits (Roche Diagnostics, Meylan, France), respectively. Measures have been performed using Wallac VICTOR3 1420 Multilabel Counter (Perkin-Elmer, Courtaboeuf, France).
PCDH-PC Knockdown
All siRNAs were from Thermo Scientific (Waltham, MA). Knockdown of PCDH-PC in 22Rv1 cells was performed using ON-TARGETplus SMARTpool Human PCDH11Y (L-013624); 100 nM ON-TARGETplus Non-Targeting Pool (D-001810) or siRNAs against PCDH-PC were transfected in 22Rv1 cells as indicated using Lipofectamine 2000. Knockdown of PCDH-PC in LNCaP-NE-like cells was carried out using Accell SMARTpool Human PCDH11Y (E-013624). Accell Non-Targeting Pool D-001910 and Accell Green Non-Targeting siRNA were also used. LNCaP-NE-like cells were incubated in Accell siRNA Delivery Media mixed with either 1 µM of Non-Targeting siRNAs or siRNAs against PCDH-PC according to the manufacturer's instructions. On the next day, media were changed and cells were subsequently cultured in the indicated medium.
Cell Growth and Cell Viability
Cell growth was monitored by cell counting and the population doubling time (DT) was estimated (in hours) by using the following formula: DT = h * ln(2)/ln(C2/C1), where C1 and C2 are the cell concentrations at the beginning and the end of the chosen period of time. Cell viability was assessed by the tetrazolium bromide (MTT) assay [19] or WST-1 assay (Roche Diagnostics) as described previously [20].
Western Blot Analysis
Protein lysates were prepared and processed as described previously [21].
cDNA Synthesis and Real-Time Polymerase Chain Reaction
RNA was extracted using the TRIzol reagent (Life Technologies), subjected to DNase treatment (DNA-Free Kit; Applied Biosystems, Foster City, CA) according to the manufacturer's instructions. One microgram of total RNA was then reverse transcribed using SuperScript II (Life Technologies). Quantitative polymerase chain reaction (qPCR) was performed using SYBR Green dye on a StepOnePlus Real-Time PCR System (Applied Biosystems). Unless indicated, the amount of each target gene relative to the housekeeping gene RPLP0 or HMBS was determined for each sample using 2-ΔΔCT method. Primer sequences are provided in Table W2.
Statistical Analysis
For qualitative data, χ2 test and Fisher exact test were applied. For in vitro studies, comparisons between groups were performed using the Student's t test. All statistical tests used a two-tailed α = 0.05 level of significance and were performed using GraphPad Prism (GraphPad Software, La Jolla, CA).
Results
Phenotypic Changes in the PCa Cell Line LNCaP upon Androgen Depletion
LNCaP cells are commonly used in vitro to model the response to ADT of PCa in patients following hormone manipulation [22]. Thus, we first searched for perturbation in PCDH-PC expression and various markers in LNCaP cells maintained in androgen-depleted medium for an extended period. This included known androgen-upregulated gene products KLK3 (PSA) and KLK2, previously described androgen-repressed genes, the neuron-specific enolase (NSE) [6], neuronal class III β-tubulin (TUBB3) [7], and the hedgehog ligand SHH [23], as well as various genes assumed to be critical in PCa progression comprising Bcl-2, Akt, TP53, MYC, and AR [5,24]. Western blot (WB) and quantitative reverse transcription-polymerase chain reaction (qRT-PCR) analyses showed that when cells are switched to androgen-deficient medium, NSE and TUBB3, two prominent markers of NE differentiation, are induced along with PCDH-PC, which shows a peak expression (∼125-fold increase) at 2 weeks (Figures 1, A and B, and W1A). SHH was also augmented (Figure W1B). This period was associated with a decreased of cell growth accompanied by the emergence of neuritelike outgrowths from the cells (Figure 1C). We likewise observed a down-regulation of PSA and KLK2 levels, two AR target genes, during the first weeks of androgen depletion, as expected. We also noted some increase in phosphorylated Akt and a decrease in expression of p53 and MYC (Figures 1, A and B, and W1A). Intriguingly, PCDH-PC expression was found to be gradually decreased with time in conjunction with reappearance of an epithelial-like morphology and a loss of neurite outgrowth (Figure 1C). After 3 months of culturing in androgen-depleted medium, PSA and KLK2 were again detected, suggestive of AR activity (Figures 1, A and B, and W1A). This was concomitant with the down-modulation of PCDH-PC, NSE, and TUBB3 and increased expression of active phosphorylated Akt, p53, and MYC. Together, these observations further qualified PCDH-PC as a novel in vitro marker of NE differentiation in PCa cells and indicate that its expression may fluctuate in concordance with AR activity. After more than 11 months of culturing, the obtained LNCaP derivative grows perfectly in androgen-depleted media and expresses significant levels of AR and PSA. The growth rate was comparable to cultures of parental LNCaP cells grown in normal media (Figure W1C). For subsequent studies, these cells will be referred to as LNCaP-androgen-independent (LNCaP-AI).
Figure 1.
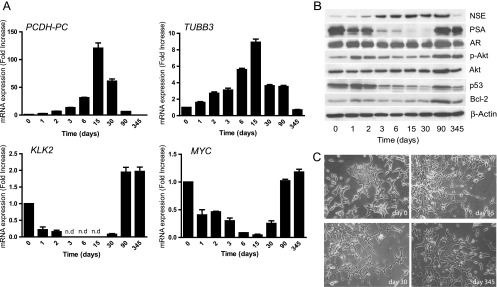
Phenotypic changes in LNCaP cells upon long-term androgen deprivation. At day 0, monolayer cultures of LNCaP cells were grown in 10% CS-FBS-containing medium. (A) qRT-PCR analysis for mRNA expression of PCDH-PC, TUBB3, KLK2, and MYC. (B) Western blot analysis for indicated proteins. β-Actin is used as a loading control. Densitometry of some western blot bands is provided in Figure W1A. (C) Morphology of cultured LNCaP cells maintained in medium containing 10% FBS (day 0) or 10% CS-FBS-containing medium for 15, 30, or 345 days. Photomicrographs are taken at x10 objective magnification under inverted light microscopy.
The Androgen/AR Axis Regulates PCDH-PC Expression
We then sought to determine the extent to which the androgen/AR axis regulates PCDH-PC expression. LNCaP were treated during 24 hours with increasing concentrations of the androgen DHT, and KLK3 (PSA) and PCDH-PC mRNA levels were measured by qRT-PCR. The increased level of KLK3, an AR-targeted gene, was used as a positive control of the AR activity in the presence of DHT. In DHT-treated cells, we observed a four-fold reduction in PCDH-PC mRNA levels in conjunction with increased KLK3 expression (Figure 2A). The temporal effects of androgen were further tested in an experiment where the cells were maintained in androgen-depleted media for 72 hours and then DHT was added back for 6, 12, and 24 hours. In such conditions, inhibition of PCDH-PC expression was detectable as early as 6 hours following DHT supplementation, suggesting that the androgen/AR axis directly mediates PCDH-PC expression (Figure 2B).
Figure 2.

Androgenic regulation of the PCDH-PC gene. (A) Cultures of LNCaP cells were grown 24 hours in 10% CS-FBS media supplemented or not with incremental doses of DHT. RT-PCR analysis for PCDH-PC (left) and KLK3 (right) levels in DHT-treated over vehicle-treated cells. (B) LNCaP cells were grown for 72 hours in 5% CS-FBS media and then refreshed with media supplemented with 100 nM DHT, and PCDH-PC and KLK3 levels were inspected at the indicated times. (C) LNCaP cells were grown in 10% FBS in the presence or absence of bicalutamide (10 µM) for 10 days, and mRNA levels for PCDH-PC and KLK3 were examined. (D) Histograms showing normalized levels of KLK2 (left), KLK3 (middle), and PCDH-PC (right) from LNCaP-AI cultures treated with bicalutamide for 8 days. (E) Time course expression of NSE (left), TUBB3 (middle), and PCDH-PC (right) in LNCaP-AI cells cultivated at 2.5 nM docetaxel. Bars represent means ± SEM of two independent experiments done in triplicate. (F) Morphology of LNCaP-AI cells maintained in medium containing 10% CS-FBS (left panel), supplemented with docetaxel for 15 days (middle) or 30 days (right). Scale bar, 200 µm.
Moreover, PCDH-PC expression was similarly reduced when cells were chronically exposed to androgens (Figure W2A), estrogen, or progesterone, which are two alternative ligands of mutated AR in this line [25]. We then asked whether a functional AR is required to mediate the repressive effect of androgens on PCDH-PC expression. LNCaP cells were incubated in the presence of the antiandrogen bicalutamide [26]. A 10-day treatment resulted in augmenting by seven-fold PCDHPC expression (Figure 2C) while expectedly reducing KLK3 expression. Changes in cell morphology were also visible upon the treatment (Figure W2B). We next applied bicalutamide treatment to the LNCaP-AI derivative. We observed a dose-dependent relative decrease in KLK3 and KLK2 expression compared to untreated cells with a concurrent increase in PCDH-PC expression (Figure 2D). To ascertain our assumption that PCDH-PC is repressed by AR activity, we next treated the LNCaP-AI cells with docetaxel. Docetaxel is the standard-of-care first-line chemotherapy for men with metastatic CRPC. In PCa cells, recent studies showed that short-term treatment with docetaxel impeded AR activity [27]. Here, we exposed LNCaP-AI cells to 2.5 nM docetaxel for a prolonged period and examined the expression of PCDH-PC and NE markers over time. After 15 days, we found that the cell populations surviving this chronic exposure to docetaxel had greater levels of NE markers NSE (∼2- to 4-fold increase), TUBB3 (∼2- to 5-fold increase), and PCDH-PC (∼25- to 125-fold increase) compared to untreated cells (Figure 2E). The morphology of the cells also changed substantially with the formation of neurite outgrowths (Figure 2F). These data suggest that NE-like cancer cells likely emerged through transdifferentiation following the chronic exposure to docetaxel.
PCDH-PC Is a Negative Mediator of Ligand-Dependent AR Transcriptional Activity
We earlier found that transient overexpression of PCDH-PC, under certain circumstances, can perturb AR protein stability in LNCaP cells through a complex mechanism that involves Akt activation and increase proteasomal activity toward AR [28]. However, the potential links between AR activity, PCDH-PC expression, and phenotypic changes in LNCaP cells have not been investigated. Here, we tested the possibility that PCDH-PC expression could disrupt androgen signaling. We transiently overexpressed PCDH-PC using cultures of LNCaP cells. Increased expression of PCDH-PC was verified by qRT-PCR (Figure W3A); Western blot analysis showed a marked down-regulation of PSA in PCDH-PC-transfected cells while expectedly increased NSE and phospho-Akt levels (Figure 3A). There was also significant enrichment for inactivated phospho-glycogen synthase kinase-3 beta (GSK-3β; Ser9). The AR level was not perturbed, suggesting that PCDH-PC expression disrupted androgen signaling by inhibition of AR activity in our conditions. To further explore this inhibitory effect, we performed luciferase reporter assays on these latter cells following transfection of incremental amounts of the PCDH-PC expression construct. These analyses demonstrated a dose-dependent decrease of the PSA promoter transactivation (Figure 3B). We then investigated long-term effects of PCDH-PC expression by analyzing PSA expression in LNCaP derivatives stably transfected with PCDH-PC. In normal culture conditions, these cells showed more neurites and a decrease in cell growth compared to control cells (Figure 3C). PCDH-PC mRNA and protein levels in LNCaP-pcDNA3 and LNCaP-PCDH-PC are depicted in Figure W3, B and C. Stable transfectants exhibited reduced AR activity compared to vectortransfected LNCaP cells (Figure 3D). These cells have enhanced levels of endogenous NSE, phospho-Akt, and phospho-GSK-3β, comparable AR expression, but lower levels of PSA protein compared to the vector-transfected or LNCaP-AI cells (Figure 3E). Interestingly, inhibition of phosphatidylinositol 3-kinase (PI3K)/Akt signal using the PI3K inhibitor LY294002 compromised NE features in these cells (Figure 3F). We next investigated whether knockdown of PCDH-PC could affect the AR activity in the 22Rv1 PCa cells [29], which endogenously express PCDH-PC. 22Rv1 cells are androgen-independent given that they can grow in the absence of androgens. However, they remain AR dependent expressing several AR target genes including KLK3 and KLK2. When 22Rv1 cells were maintained in the presence of androgens, ablation of PCDH-PC with PCDH-PC-targeted siRNAs did not significantly affect KLK3 expression (Figure 3G). By contrast, this led to KLK2 levels that were approximately 12-fold higher. It was earlier demonstrated that 22Rv1 is androgen responsive for KLK2 but weakly for KLK3 expression [30]. We confirmed this information in an experiment where cells were exposed to 10 nMDHTfor 24 hours (Figure 3H). Thus, we conceived that PCDH-PC is a potential repressor of ligand-dependent AR activity in this line. To pursue this possibility, we transiently transfected 22Rv1 cells with a PCDH-PC expression construct or control vector and measured KLK2 and KLK3 in either control (ethanol) or DHT-treated cells. Overexpression of PCDH-PC resulted in a significant decrease in KLK2 expression compared to minor changes for KLK3 (Figure 3I), and the effect was perceived only in the presence of DHT. Together, these results strongly suggest that PCDH-PC overexpression inhibits ligand-dependent activity of AR in PCa cells, with no or marginal effects on its ligand-independent activity.
Figure 3.

PCDH-PC expression reduces ligand-bound AR activity. (A) Western blot analysis 48 hours following transient transfection of PCDH-PC cDNA or the control vector. (B) PSA promoter activity was assessed in transfected LNCaP cells by measuring luciferase activity 24 hours after DHT treatment in cellular extracts normalized to β-Gal activity. (C) Stable transfectants of vector- and PCDH-PC-transfected LNCaP (LNCaP-PCDH-PC) were examined for differences in cell morphology and cell growth (DT), and PSA promoter activity (D) as in B. (E) Western blot made against proteins from LNCaP-pcDNA3, LNCaP-AI, and LNCaP-PCDH-PC cells showing reduced PSA and increased levels of NSE in the LNCaP-PCDH-PC cells. (F) LNCaP-PCDH-PC cells were treated for 3 days with either the PI3K inhibitor LY294002 (10 µM) or vehicle (DMSO). A Western blot was performed and probed as above. (G) 22Rv1 cells transfected either with siRNAs raised against PCDH-PC mRNA or non-targeting siRNA were analyzed for mRNA expression of PCDH-PC, KLK3, and KLK2. Down-regulation of PCDH-PC is accompanied by elevation of KLK2 mRNA but had minor effects on KLK3. (H) 22Rv1 cells were treated with vehicle (EtOH) or DHT (10 nM) for 24 hours, and endogenous levels of KLK3 and KLK2 were examined. (I) 22Rv1 cells pretransfected with PCDH-PC plasmid were treated with vehicle (EtOH) or DHT (10 nM) for 24 hours, and PCDH-PC, KLK3, and KLK2 levels were compared by qPCR.
PCDH-PC Expression during PCa Progression
By immunohistochemistry, we then explored the distribution of PCDH-PC protein in normal and pathologic specimens. In tissues derived from normal prostate, luminal epithelial cells were consistently found to be negative for PCDH-PC and pronounced expression of this protein was observed in lonely cells scattered within the epithelium (Figure 4A, i). Occasionally, a faint staining was detected in the basal cell layer (Figure W4). A series of HNPC specimens was examined using tissue microarrays. This analysis revealed moderate to high expression of PCDH-PC in at most 11% (25 of 222) of evaluable cases (Table 1). There was no significant correlation with clinicopathologic data (Table W3). Evaluation of PCDH-PC expression in CRPC samples indicated a much higher proportion of positive cases (that is, 61%, 33 of 54 CRPC; Figure 4A, ii and Table 1). It is noteworthy that PCDH-PC protein was also detectable in cancer cells of metastatic CRPC lesions present in the brain and the lymph nodes of patients (Figure 4A, iii–iv). Despite only six cases were analyzed, this suggested that deregulated expression of PCDH-PC in CRPC is not restricted to recurrent lesions localized to the prostate.
Figure 4.

(A) Expression of PCDH-PC in human prostatic tissues. Anti-PCDH-PC identifies single normal cells in the prostatic epithelium of a healthy subject (i), in PCa cells in prostate tissue of CRPC (ii), in brain containing PCa metastases (iii), and in a lymph node metastasis (iv) of CRPC. (v) Positive PCDH-PC staining in cancer cells of a section of the surgical piece from a patient who had received 3 months of neoadjuvant ADT. (vi) Representative biopsy core from the same patient before neoadjuvant ADT showing negativity for PCDH-PC. (B, C) Expression of PCDH-PC correlates with NE characteristics in human PCa. Representative consecutive sections stained with antibodies to PCDH-PC, CgA, and PSA of primary PCa from a patient treated by neoadjuvant ADT. Immunohistochemical stains reveal mixed populations of cancer cells suggesting a common origin.
Table 1.
PCDH-PC Expression before and after ADT.
| Prostate Carcinoma | No. of PCDH-PC-Negative Samples (%) | No. of PCDH-PC-Positive Samples (%) | |
| HNPC | 197 (88.8) | 25 (11.2) | |
| HTPC | 18 (56.3) | 14 (43.7) | |
| CRPC | 21 (38.9) | 33 (61.1) | |
| Pearson χ2 test | P < .0001 | ||
| Fisher exact test | |||
| HNPC/HTPC | P < .0001 | ||
| HTPC/CRPC | P = .178 | ||
| HNPC/CRPC | P < .0001 |
We then evaluated a series of prostatectomy specimens of PCa obtained from patients treated for 3 to 6 months with neoadjuvant hormone therapy (HTPC). Of the 32 cases of HTPC evaluated, 14 (43.7%) were recorded as positive for PCDH-PC (Table 1). Especially, intense expression was consistently detected in clusters comprising of 5 to 100 cells (Figure 4A, v). For the overall HTPC group, PCDH-PC was found to be significantly higher when compared with the HNPC group as evaluated by Fisher exact test (P < .0001). To test further the hypothesis that ADT is causative for increased expression of PCDH-PC in these specimens, we examined the hormone-naïve tissues of these patients by examining their initial prostatic biopsies. Matched biopsy specimens were available in seven cases. In six of these index cases, we found no evidence of PCDH-PC expression after analyzing cancer foci of several biopsy specimens (Figure 4A, vi), and one other case showed strong positivity for PCDH-PC but in dispersed isolated cells rather than in clusters. These results demonstrate that high PCDH-PC expression is rare in men with still hormonally untreated PCa but substantially increases in response to hormonal manipulation.
PCDH-PC Expression Associates with NE Features in Human Prostate Tissues
Given the apparent link between PCDH-PC and NE features in vitro, we explored the value of PCDH-PC as a novel candidate marker for NE transdifferentiation in human PCa specimens. Examination of the hormone-treated samples for CgA and PSA expressions consistently revealed that cancer cells expressing PCDH-PC are present in tumor foci showing a large majority of CgA-expressing cells but with reduced expression of PSA (Figures 4, B and C, 5A, and W5A). Dual immunofluorescence procedure also revealed that in these tumor areas, not all cells exhibited the same NE characteristics such that varied levels of NE markers were observed in the cells (Figure 5B). In adjacent benign epithelia, we detected a few isolated cells staining positive for both CgA and PCDH-PC likely representing nonmalignant NE cells (Figure W5B).
Figure 5.
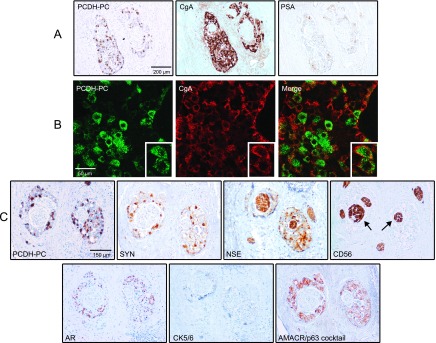
(A) Immunohistochemical analysis further validating the inverse correlation between PCDH-PC/CgA stainings and PSA expression in tumor foci of a hormonally treated case. (B) Dual immunofluorescence in the previous index case identifies cancer cells coexpressing PCDH-PC and CgA. The cells can express varied levels of the two proteins. (C) A positive PCDH-PC cancer focus was analyzed for expression of synaptophysin (SYN), NSE, N-CAM (CD56), AR, basal cytokeratins 5/6, AMACR, and p63. Note the areas positive for NSE and CD56 (arrows) but negative for the other markers representing nontumoral nerves present in the prostate tissue.
On further analysis of cancer foci positive for PCDH-PC, we found positivity for the AR as well as for NSE and synaptophysin, two established NE markers, but we consistently failed to detect staining for CD56 (NCAM1; Figure 5C), another NE marker. Of note, cancer areas within the different tissues analyzed (PCDH-PC positive and negative) were consistently negative for the Ki67 antigen (not shown). Moreover, PCDH-PC-expressing cells were negative for the basal cytokeratins 5/6 and p63 but positive for α-methylacyl-CoA racemase (AMACR; Figure 5C), a highly specific marker of PCa epithelia, thus supporting a PCa origin [31].
Collectively, these observations strongly suggest PCDH-PC as a novel early marker for transition from epithelial to NE phenotype in PCa treated by ADT. Intriguingly however, at the castration state of prostate adenocarcinoma, the relationship between PCDH-PC expression and NE (as assessed by CgA staining) appeared to be lost, and although PCDH-PC immunostaining of PCa cells sometimes coincides with staining for NE markers such as NSE (Figure W5C), in many cases the PCDH-PC-positive contingents examined did not show coincidental staining (not shown).
NE-like PCa Cells Are Resistant to Chemotherapeutic Agents
Several pieces of evidence suggest that PCa NE-like cells are resistant to multiple therapeutics agents [32,33]. Here, we assessed further the chemoresistance spectrum of LNCaP-NE-like cells. After culturing LNCaP cells for 15 days in androgen-depleted medium, the cells exhibit an NE-like phenotype and reduced growth (Figure 6A) concomitant with a loss of their epithelial characteristics. Sensitivity with respect to diverse agents was evaluated 96 hours after treatment of LNCaP-NE-like, LNCaP, or LNCaP-AI cells. Treatments included two taxanes, docetaxel and paclitaxel, as well as 12-O-tetradecanoylphorbol-13-acetate (TPA) and camptothecin, two well-known inducers of apoptosis in LNCaP cells [34,35]. At the indicated doses, LNCaP-NE-like cells were overwhelmingly resistant to these drugs compared to LNCaP or LNCaP-AI cells (Figure 6B). LNCaP-NE-like cells also showed enhanced resistance to various cytotoxic agents commonly used in management of various malignancies (Figure W6A). We next wanted to gauge the dependence of LNCaP-NE-like cells with respect to PCDH-PC expression for their viability. To this end, LNCaP-NE-like cells were treated for 24 hours with Accell Green Non-Targeting siRNAs used to control effective uptake of the siRNAs (Figure W6B), pools of Accell Non-Targeting siRNAs, or Accell siRNAs raised against PCDH-PC transcripts, then cultured for 8 days in hormone-deprived medium supplemented or not with docetaxel (10 nM). PCDH-PC silencing was found to be efficient in these conditions (Figure 6C). In the presence of docetaxel, LNCaP-NE cells that had been preincubated with the PCDH-PC siRNAs showed a significant decrease in cell viability (relative to cells exposed to NT siRNA in the presence or absence of docetaxel), whereas in the absence of docetaxel, PCDH-PC siRNA treatment had limited effect (Figure 6D). Moreover, the effect was not seen when similar treatments were applied to the chemosensitive PC3 PCa lineage (Figure 6E), which lacks PCDH-PC or LNCaP-AI that expresses low amounts of PCDH-PC (Figure 6F). Subsequent analyses showed that attenuating PCDH-PC expression similarly sensitized LNCaP-NE-like cells to TPA and camptothecin (Figure W6, C and D). These data argue for a chemoprotective role for PCDH-PC in LNCaP-NE-like cells.
Figure 6.

Acquired NE phenotype correlates with chemoresistance in LNCaP cells. (A) LNCaP, LNCaP-NE like, and LNCaP-AI derivatives were examined for differences in cell growth. (B) Viability assay of LNCaP (white bars), LNCaP-NE-like (gray bars), and LNCaP-AI (black bars) cells at 96 hours after treatment with docetaxel, paclitaxel, camptothecin, or phorbol ester (TPA) relative to untreated cells. (C) Verification of efficient PCDH-PC knockdown by qRT-PCR in LNCaP-NE-like cells pretreated 24 hours with either Accell Non-Targeting or PCDH11Y siRNAs and then maintained in the presence or absence of docetaxel for 48 hours. (D) Cell viability as assessed by WST-1 assay using siRNAs treated with LNCaP-NE-like cells alone or subsequently treated with docetaxel for 8 days. (E) As in C except using PC3 cells and 96-hour docetaxel treatment. (F) As in D except using LNCaP-AI cells. Bars represent means ± SEM of quintuplets from one experiment representative of three independent experiments.
Discussion
The androgen/AR axis remains active in the majority of CRPCs. However, as prostate tumors develop resistance to treatment, NE differentiation has been proposed as a mechanism for hormonal escape or AR independence [4,10–12,36–38]. Yet, the impact of NE differentiation on the clinical outcome, the mechanisms by which NE differentiation emerges after ADT, and the consequence of targeting these cell populations remain uncertain. The current study significantly expands our understanding of NE differentiation in PCa and qualifies PCDH-PC as a surrogate marker for human PCa cell subpopulations experiencing NE transdifferentiation under hormonal treatment.
With respect to progression toward a castration-resistant phenotype, results obtained from LNCaP cultures grown in androgen-reduced medium support a model in which AR function is attenuated in a first phase following ADT, concomitantly with the acquisition of NE features by PCa cells. In situ, we found evidence that high PCDH-PC expression also parallels CgA and other NE markers in clusters of tumor cells from neoadjuvant hormonally treated PCa. The fact that normal NE cells are considered as post-mitotic [39], coupled with data showing that the proliferating rate of PCa cells is relatively low in primary prostate tumors [40], strongly suggests that NE-like clusters revealed in this study originated from the NE transdifferentiation of preexisting epithelial-looking PCa cells. Thus, we propose that in clinical setting, overexpression of PCDH-PC and concomitant induction of NE trans-differentiation by a fraction of PCa cells in early response to hormonal treatment reflects one route for PCa cells to adapt and survive in a low androgen environment.
In a second step, AR may be reactivated [5,41,42] to promote proliferation in conjunction with partial or total loss of NE features along with reappearance of significant amounts of PSA as observed in LNCaP-AI cells. Further studies are warranted to decrypt the mechanisms involved in reactivation of AR in these cells.
Enigmatically, the relationship between PCDH-PC and NE differentiation was not evident in CRPC specimens. This could reflect the multifaceted role of PCDH-PC in the more advanced stages of PCa with functions that may occur independently of NE differentiation. Alternatively, this could be indicative of various subtypes of NE differentiation (from well differentiated to poorly differentiated) in tumors with varied proliferative activity and expressing various levels of NE markers [43,44]. In that respect, it will be important to examine the role of PCDH-PC in the setting of small cell carcinoma of prostate, a rare poorly differentiated NE PCa associated with poor prognosis and poor response to therapies [45]. It is also tempting to speculate that AR plays a crucial role in this potential molecular switch as AR is consistently implicated in the growth of castrate-resistant tumors [41,46]. We have shown here that PCDH-PC expression inhibits AR activity. However, this inhibition appeared to be incomplete in the sense that it is likely restricted to the ligand-dependent activity of AR. Although we already know that PI3K/Akt activity may be an important mediator of this effect, the precise mechanism through which PCDH-PC regulates the ligand-dependent AR activity has yet to be fully determined.
If confirmed, this regulation could also indicate that among castrate-resistant tumors, those overexpressing PCDH-PC might progress to the favor of tumor clones dependent on a ligand-independent activity of AR [46–48].
Our experimental data consistently revealed that androgen exposure inhibits PCDH-PC expression in LNCaP cells, although it is unlikely that androgens completely switch off PCDH-PC expression. Likewise, the contribution of other recurrent alterations found in PCa, such as TMPRSS2-ERG gene fusion or loss of PTEN, known to perturb AR signaling, should be considered [49,50].
Another interesting observation is that the NE status of LNCaP cells correlates with resistance to a wide range of chemotherapeutic agents including docetaxel, the current standard for metastatic CRPC. One could suggest that those resistances are likely linked to the reduced growth rate of LNCaP-NE-like cells. Indeed, from a clinical perspective, the observation that NE transdifferentiation could confer a multidrug-resistant phenotype allowing a cell to remain arrested until it can reacquire the ability to proliferate could make that process a formidable tumor promoter at any stage of PCa progression. Interestingly, by targeting NE-like PCa cells using RNA interference against PCDH-PC, it was possible to sensitize cells to chemohormonal treatment. Together with prior work identifying PCDH-PC as an antiapoptotic factor in PCa cells [13], this qualifies PCDH-PC as a general survival factor in PCa cells and provides a biologic rationale for further assessment of targeting malignant NE-like cells.
Although not emphasized here, in neoadjuvant hormonally treated tumors, we found many instances with NE-like PCa (PCDH-PC+, CgA+, PSA-) cells adjacent to malignant epithelial-like (PCDH-PC-, CgA-, PSA+) cells, thus continuing to use the androgen/AR axis despite ADT (Figure 4). Clearly, the manifestation of these mixed populations gives reason to further examine whether these phenotypically distinct cell populations may cooperate to promote transition toward castration resistance [8,51], which would either help support or refute a rationale of treating both adenocarcinoma and NE components.
In summary, our study provides support for the likelihood of transdifferentiation model of PCa cells to explain the emergence of NE differentiation in human PCa following ADT. We substantiate PCDH-PC, a human male-specific protocadherin, as a critical factor in this process that appears to be regulated by cross modulation between PCDH-PC and AR. Along this line, our data revealed novel paradigms linking the AR axis and NE transdifferentiation in PCa cells with apparent implications for the emergence of chemohormonal resistance.
Supplemental Experimental Procedures
Generation of Monoclonal Antibodies Specific for PCDH-PC
BALBc mice were immunized with a recombinant PCDH-PC expressed from an Escherichia coli (BL21-CodonPlus competent cells; Stratagene, Paris, France) transformed with a pET3a-PCDH-PC expression construct. After four repeats of immunization, mice splenocytes were fused using polyethylene glycol with X63 myeloma cells and grown in hypoxanthine-aminopterin-thymidine selection medium to generate hybridomas. After limit dilution, supernatants from hybridoma clones were screened for efficient detection of PCDH-PC using an ELISA procedure wherein recombinant PCDH-PC was coated on Immulon flat-bottom microtiter plates. Immunocytochemistry detection of PCDH-PC in the LNCaP-PCDH-PC was performed and compared to vector-transfected LNCaP cells to verify the specificity of the antibodies (Figure W3C). One hybridoma cell line designed “HB 0337 SSA” was selected for in situ detection of human PCDH-PC and deposited under No. I-3561 to the Collection Nationale de Cultures de Microorganismes, Institut Pasteur (Paris, France).
Acknowledgments
We are grateful to Ralph Buttyan (Vancouver Prostate Centre) for advice and critical reading of this work. We are also grateful to Fannie Semprez, Luis Queires, Renée Delbé, and Noëmie Lemenand for technical support critical to this work.
Abbreviations
- ADT
androgen deprivation therapy
- AR
androgen receptor
- CRPC
castration-resistant prostate cancer
- DHT
dihydrotestosterone
- NE
neuroendocrine
- PCDH-PC
protocadherin-PC
Footnotes
This work was supported by INSERM and Université Paris-Est Créteil, through grants from the Association pour la Recherche sur les Tumeurs de la Prostate (to S.T.), by the Fondation ARC pour la Recherche sur le Cancer (to F.V.), and by the Association Française d'Urologie (to A.D.L.T.). This work has been supported by institutional and charity grants only and there are no financial or other interest in the publication of this paper that might be considered a conflict of interest.
This article refers to supplementary materials, which are designated by Tables W1 to W3 and Figures W1 to W6 and are available online at www.neoplasia.com.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Isaacs JT, Isaacs WB. Androgen receptor outwits prostate cancer drugs. Nat Med. 2004;10:26–27. doi: 10.1038/nm0104-26. [DOI] [PubMed] [Google Scholar]
- 3.Denmeade SR, Isaacs JT. A history of prostate cancer treatment. Nat Rev Cancer. 2002;2:389–396. doi: 10.1038/nrc801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beltran H, Beer TM, Carducci MA, de Bono J, Gleave M, Hussain M, Kelly WK, Saad F, Sternberg C, Tagawa ST, et al. New therapies for castration-resistant prostate cancer: efficacy and safety. Eur Urol. 2011;60:279–290. doi: 10.1016/j.eururo.2011.04.038. [DOI] [PubMed] [Google Scholar]
- 5.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- 6.Burchardt T, Burchardt M, Chen MW, Cao Y, de la Taille A, Shabsigh A, Hayek O, Dorai T, Buttyan R. Transdifferentiation of prostate cancer cells to a neuroendocrine cell phenotype in vitro and in vivo. J Urol. 1999;162:1800–1805. [PubMed] [Google Scholar]
- 7.Wright ME, Tsai MJ, Aebersold R. Androgen receptor represses the neuroendocrine transdifferentiation process in prostate cancer cells. Mol Endocrinol. 2003;17:1726–1737. doi: 10.1210/me.2003-0031. [DOI] [PubMed] [Google Scholar]
- 8.Yuan TC, Veeramani S, Lin FF, Kondrikou D, Zelivianski S, Igawa T, Karan D, Batra SK, Lin MF. Androgen deprivation induces human prostate epithelial neuroendocrine differentiation of androgen-sensitive LNCaP cells. Endocr Relat Cancer. 2006;13:151–167. doi: 10.1677/erc.1.01043. [DOI] [PubMed] [Google Scholar]
- 9.Sauer CG, Roemer A, Grobholz R. Genetic analysis of neuroendocrine tumor cells in prostatic carcinoma. Prostate. 2006;66:227–234. doi: 10.1002/pros.20338. [DOI] [PubMed] [Google Scholar]
- 10.Cindolo L, Cantile M, Vacherot F, Terry S, de la Taille A. Neuroendocrine differentiation in prostate cancer: from lab to bedside. Urol Int. 2007;79:287–296. doi: 10.1159/000109711. [DOI] [PubMed] [Google Scholar]
- 11.Jiborn T, Bjartell A, Abrahamsson PA. Neuroendocrine differentiation in prostatic carcinoma during hormonal treatment. Urology. 1998;51:585–589. doi: 10.1016/s0090-4295(97)00684-5. [DOI] [PubMed] [Google Scholar]
- 12.di Sant'Agnese PA. Neuroendocrine differentiation in human prostatic carcinoma. Hum Pathol. 1992;23:287–296. doi: 10.1016/0046-8177(92)90110-o. [DOI] [PubMed] [Google Scholar]
- 13.Chen MW, Vacherot F, DeLaTaille A, Gil-Diez-De-Medina S, Shen R, Friedman RA, Burchardt M, Chopin DK, Buttyan R. The emergence of protocadherin-PC expression during the acquisition of apoptosis-resistance by prostate cancer cells. Oncogene. 2002;21:7861–7871. doi: 10.1038/sj.onc.1205991. [DOI] [PubMed] [Google Scholar]
- 14.Blanco P, Sargent CA, Boucher CA, Mitchell M, Affara NA. Conservation of PCDHX in mammals; expression of human X/Y genes predominantly in brain. Mamm Genome. 2000;11:906–914. doi: 10.1007/s003350010177. [DOI] [PubMed] [Google Scholar]
- 15.Yang X, Chen MW, Terry S, Vacherot F, Chopin DK, Bemis DL, Kitajewski J, Benson MC, Guo Y, Buttyan R. A human- and male-specific protocadherin that acts through the wnt signaling pathway to induce neuroendocrine transdifferentiation of prostate cancer cells. Cancer Res. 2005;65:5263–5271. doi: 10.1158/0008-5472.CAN-05-0162. [DOI] [PubMed] [Google Scholar]
- 16.Horoszewicz JS, Leong SS, Kawinski E, Karr JP, Rosenthal H, Chu TM, Mirand EA, Murphy GP. LNCaP model of human prostatic carcinoma. Cancer Res. 1983;43:1809–1818. [PubMed] [Google Scholar]
- 17.Terry S, Queires L, Gil-Diez-de-Medina S, Chen MW, de la Taille A, Allory Y, Tran PL, Abbou CC, Buttyan R, Vacherot F. Protocadherin-PC promotes androgen-independent prostate cancer cell growth. Prostate. 2006;66:1100–1113. doi: 10.1002/pros.20446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cleutjens KB, van der Korput HA, van Eekelen CC, van Rooij HC, Faber PW, Trapman J. An androgen response element in a far upstream enhancer region is essential for high, androgen-regulated activity of the prostate-specific antigen promoter. Mol Endocrinol. 1997;11:148–161. doi: 10.1210/mend.11.2.9883. [DOI] [PubMed] [Google Scholar]
- 19.Romijn JC, Verkoelen CF, Schroeder FH. Application of the MTT assay to human prostate cancer cell lines in vitro: establishment of test conditions and assessment of hormone-stimulated growth and drug-induced cytostatic and cytotoxic effects. Prostate. 1988;12:99–110. doi: 10.1002/pros.2990120112. [DOI] [PubMed] [Google Scholar]
- 20.Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, Wang Y, Sheikh KL, Terry S, Tagawa ST, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011;1:487–495. doi: 10.1158/2159-8290.CD-11-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Terry S, Ploussard G, Allory Y, Nicolaiew N, Boissiere-Michot F, Maille P, Kheuang L, Coppolani E, Ali A, Bibeau F, et al. Increased expression of class III β-tubulin in castration-resistant human prostate cancer. Br J Cancer. 2009;101:951–956. doi: 10.1038/sj.bjc.6605245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Steenbrugge GJ, van Uffelen CJ, Bolt J, Schroder FH. The human prostatic cancer cell line LNCaP and its derived sublines: an in vitro model for the study of androgen sensitivity. J Steroid Biochem Mol Biol. 1991;40:207–214. doi: 10.1016/0960-0760(91)90184-7. [DOI] [PubMed] [Google Scholar]
- 23.Chen M, Tanner M, Levine AC, Levina E, Ohouo P, Buttyan R. Androgenic regulation of hedgehog signaling pathway components in prostate cancer cells. Cell Cycle. 2009;8:149–157. doi: 10.4161/cc.8.1.7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bernard D, Pourtier-Manzanedo A, Gil J, Beach DH. Myc confers androgen-independent prostate cancer cell growth. J Clin Invest. 2003;112:1724–1731. doi: 10.1172/JCI19035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Veldscholte J, Berrevoets CA, Ris-Stalpers C, Kuiper GG, Jenster G, Trapman J, Brinkmann AO, Mulder E. The androgen receptor in LNCaP cells contains a mutation in the ligand binding domain which affects steroid binding characteristics and response to antiandrogens. J Steroid Biochem Mol Biol. 1992;41:665–669. doi: 10.1016/0960-0760(92)90401-4. [DOI] [PubMed] [Google Scholar]
- 26.Masiello D, Cheng S, Bubley GJ, Lu ML, Balk SP. Bicalutamide functions as an androgen receptor antagonist by assembly of a transcriptionally inactive receptor. J Biol Chem. 2002;277:26321–26326. doi: 10.1074/jbc.M203310200. [DOI] [PubMed] [Google Scholar]
- 27.Zhu ML, Horbinski CM, Garzotto M, Qian DZ, Beer TM, Kyprianou N. Tubulin-targeting chemotherapy impairs androgen receptor activity in prostate cancer. Cancer Res. 2010;70:7992–8002. doi: 10.1158/0008-5472.CAN-10-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang X, Chen MW, Terry S, Vacherot F, Bemis DL, Capodice J, Kitajewski J, de la Taille A, Benson MC, Guo Y, et al. Complex regulation of human androgen receptor expression by Wnt signaling in prostate cancer cells. Oncogene. 2006;25:3436–3444. doi: 10.1038/sj.onc.1209366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sramkoski RM, Pretlow TG, II, Giaconia JM, Pretlow TP, Schwartz S, Sy MS, Marengo SR, Rhim JS, Zhang D, Jacobberger JW. A new human prostate carcinoma cell line, 22Rv1. In Vitro Cell Dev Biol Anim. 1999;35:403–409. doi: 10.1007/s11626-999-0115-4. [DOI] [PubMed] [Google Scholar]
- 30.Denmeade SR, Sokoll LJ, Dalrymple S, Rosen DM, Gady AM, Bruzek D, Ricklis RM, Isaacs JT. Dissociation between androgen responsiveness for malignant growth vs. expression of prostate specific differentiation markers PSA, hK2, and PSMA in human prostate cancer models. Prostate. 2003;54:249–257. doi: 10.1002/pros.10199. [DOI] [PubMed] [Google Scholar]
- 31.Rubin MA, Zhou M, Dhanasekaran SM, Varambally S, Barrette TR, Sanda MG, Pienta KJ, Ghosh D, Chinnaiyan AM. α-Methylacyl coenzyme A racemase as a tissue biomarker for prostate cancer. JAMA. 2002;287:1662–1670. doi: 10.1001/jama.287.13.1662. [DOI] [PubMed] [Google Scholar]
- 32.Deng X, Liu H, Huang J, Cheng L, Keller ET, Parsons SJ, Hu CD. Ionizing radiation induces prostate cancer neuroendocrine differentiation through interplay of CREB and ATF2: implications for disease progression. Cancer Res. 2008;68:9663–9670. doi: 10.1158/0008-5472.CAN-08-2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frigo DE, McDonnell DP. Differential effects of prostate cancer therapeutics on neuroendocrine transdifferentiation. Mol Cancer Ther. 2008;7:659–669. doi: 10.1158/1535-7163.MCT-07-0480. [DOI] [PubMed] [Google Scholar]
- 34.Day ML, Zhao X, Wu S, Swanson PE, Humphrey PA. Phorbol ester-induced apoptosis is accompanied by NGFI-A and c-fos activation in androgen-sensitive prostate cancer cells. Cell Growth Differ. 1994;5:735–741. [PubMed] [Google Scholar]
- 35.Akao Y, Kusakabe S, Banno Y, Kito M, Nakagawa Y, Tamiya-Koizumi K, Hattori M, Sawada M, Hirabayasi Y, Ohishi N, et al. Ceramide accumulation is independent of camptothecin-induced apoptosis in prostate cancer LNCaP cells. Biochem Biophys Res Commun. 2002;294:363–370. doi: 10.1016/S0006-291X(02)00462-X. [DOI] [PubMed] [Google Scholar]
- 36.Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, Macvicar GR, Varambally S, Harwood J, Bismar TA, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res. 2004;64:9209–9216. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 37.Hirano D, Okada Y, Minei S, Takimoto Y, Nemoto N. Neuroendocrine differentiation in hormone refractory prostate cancer following androgen deprivation therapy. Eur Urol. 2004;45:586–592. doi: 10.1016/j.eururo.2003.11.032. discussion 592. [DOI] [PubMed] [Google Scholar]
- 38.Wafa LA, Palmer J, Fazli L, Hurtado-Coll A, Bell RH, Nelson CC, Gleave ME, Cox ME, Rennie PS. Comprehensive expression analysis of L-dopa decarboxylase and established neuroendocrine markers in neoadjuvant hormone-treated versus varying Gleason grade prostate tumors. Hum Pathol. 2007;38:161–170. doi: 10.1016/j.humpath.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 39.Bonkhoff H, Stein U, Remberger K. Endocrine-paracrine cell types in the prostate and prostatic adenocarcinoma are postmitotic cells. Hum Pathol. 1995;26:167–170. doi: 10.1016/0046-8177(95)90033-0. [DOI] [PubMed] [Google Scholar]
- 40.Berges RR, Vukanovic J, Epstein JI, CarMichel M, Cisek L, Johnson DE, Veltri RW, Walsh PC, Isaacs JT. Implication of cell kinetic changes during the progression of human prostatic cancer. Clin Cancer Res. 1995;1:473–480. [PMC free article] [PubMed] [Google Scholar]
- 41.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 42.Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25:276–308. doi: 10.1210/er.2002-0032. [DOI] [PubMed] [Google Scholar]
- 43.Klimstra DS, Modlin IR, Adsay NV, Chetty R, Deshpande V, Gonen M, Jensen RT, Kidd M, Kulke MH, Lloyd RV, et al. Pathology reporting of neuroendocrine tumors: application of the Delphic consensus process to the development of a minimum pathology data set. Am J Surg Pathol. 2010;34:300–313. doi: 10.1097/PAS.0b013e3181ce1447. [DOI] [PubMed] [Google Scholar]
- 44.Helpap B, Kollermann J. Immunohistochemical analysis of the proliferative activity of neuroendocrine tumors from various organs. Are there indications for a neuroendocrine tumor-carcinoma sequence? Virchows Arch. 2001;438:86–91. doi: 10.1007/s004280000337. [DOI] [PubMed] [Google Scholar]
- 45.Wang W, Epstein JI. Small cell carcinoma of the prostate. A morphologic and immunohistochemical study of 95 cases. Am J Surg Pathol. 2008;32:65–71. doi: 10.1097/PAS.0b013e318058a96b. [DOI] [PubMed] [Google Scholar]
- 46.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, Page ST, Coleman IM, Nguyen HM, Sun H, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–2730. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–5477. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu J, Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, Wang X, Wu L, Li J, Hu M, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2ERG gene fusions in prostate cancer progression. Cancer Cell. 2010;17:443–454. doi: 10.1016/j.ccr.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19:575–586. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jin RJ, Wang Y, Masumori N, Ishii K, Tsukamoto T, Shappell SB, Hayward SW, Kasper S, Matusik RJ. NE-10 neuroendocrine cancer promotes the LNCaP xenograft growth in castrated mice. Cancer Res. 2004;64:5489–5495. doi: 10.1158/0008-5472.CAN-03-3117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.