

Abstract
Purpose
The provirus integration site for Moloney murine leukemia virus 1 (Pim-1) kinase is overexpressed in various tumors and has been linked to poor prognosis. Its role as proto-oncogene is based on several Pim-1 target proteins involved in pivotal cellular processes. Here, we explore the functional relevance of Pim-1 in colon carcinoma.
Experimental Design
RNAi-based knockdown approaches, as well as a specific small molecule inhibitor, were used to inhibit Pim-1 in colon carcinoma cells. The effects were analyzed regarding proliferation, apoptosis, sensitization toward cytostatic treatment, and overall antitumor effect in vitro and in mouse tumor models in vivo.
Results
We demonstrate antiproliferative, proapoptotic, and overall antitumor effects of Pim-1 inhibition. The sensitization to 5-fluorouracil (5-FU) treatment upon Pim-1 knockdown offers new possibilities for combinatorial treatment approaches. Importantly, this also antagonizes a 5-FU-triggered Pim-1 up-regulation, which is mediated by decreased levels of miR-15b, a microRNA we newly identify to regulate Pim-1. The analysis of the molecular effects of Pim-1 inhibition reveals a complex regulatory network, with therapeutic Pim-1 repression leading to major changes in oncogenic signal transduction with regard to p21Cip1/WAF1, STAT3, c-jun-N-terminal kinase (JNK), c-Myc, and survivin and in the levels of apoptosis-related proteins Puma, Bax, and Bcl-xL.
Conclusions
We demonstrate that Pim-1 plays a pivotal role in several tumor-relevant signaling pathways and establish the functional relevance of Pim-1 in colon carcinoma. Our results also substantiate the RNAi-mediated Pim-1 knockdown based on polymeric polyethylenimine/small interfering RNA nanoparticles as a promising therapeutic approach.
Introduction
Provirus integration site for Moloney murine leukemia virus 1 (Pim-1) kinase belongs to a family of constitutively active serine/threonine kinases [1,2]. The expression of Pim-1 is controlled on the transcriptional level by several interleukins and growth factors through the Jak/STAT pathway involving STAT3 and/or STAT5 [3,4]. Additionally, its expression can be increased upon cellular stress like hypoxia [5] or shear stress [6]. Only recently, we have identified Pim-1 as target of miR-33a, indicating a posttranscriptional regulation of Pim-1 by microRNAs (miRNAs) [7,8]. On the protein level, the stability of Pim-1 is regulated by Hsp90, Hsp70, and the ubiquitin-proteasome pathway [9].
Several target proteins of Pim-1 involved in apoptosis, cell cycle regulation, signal transduction pathways, and transcriptional regulation have been identified, which are overall linked to cell survival (see, e.g., [10,11]). Furthermore, Pim-1 has been shown to act synergistically with the oncogenic transcription factor c-Myc by phosphorylation/stabilization of c-Myc [12] and by interaction with c-Myc on the chromatin level, leading to elevated transcription of c-Myc target genes [13]. The overexpression of Pim-1 can thus substantially contribute to malignant transformation of cells during tumorigenesis ([14]; for review, see [15]), establishing Pim-1 as a proto-oncogene. In several tumor entities, e.g., B-cell lymphoma, prostate cancer, colorectal cancer, or pancreatic cancer, an overexpression of Pim-1 has been described and linked to poor prognosis (for review, see [16]). In hematopoietic malignancies and prostate cancer, Pim-1 is known to promote tumor onset and progression [16,17], and Pim-1 knockdown or inhibition led to antitumor effects [18,19]. In colon carcinoma, Pim-1 is overexpressed [18,19], but the functional relevance of Pim-1 has not been determined.
Colon carcinoma is the third most common form of cancer and the second leading cause of cancer-related death in the Western world. Despite a favorable prognosis when detected at early stages, the presence of metastatic disease is associated with limited survival, indicating the need for novel molecular targets and strategies to specifically block oncogenic pathways.
In this study, we establish the functional relevance of Pim-1 in colon carcinoma. Using RNAi-based knockdown approaches as well as a specific low molecular weight inhibitor, we demonstrate antitumor effects in vitro and in vivo, analyze the cross talk between Pim-1 and the established cytostatic 5-fluorouracil (5-FU), and determine the effects of Pim-1 inhibition on downstream signal transduction pathways relevant for proliferation or apoptosis.
Materials and Methods
Cell Culture and Transfection
Colon carcinoma cell lines HCT-116, LS174T, and HT29 were purchased from the American Type Culture Collection (Manassas, VA) and authenticated by the vendor. Cells were maintained in a humidified incubator under standard conditions (37°C, 5% CO2) in Iscove's modified Dulbecco's medium (IMDM; PAA, Cölbe, Germany) supplemented with 10% fetal calf serum (FCS).
SiRNAs were purchased from Thermo Scientific (Schwerte, Germany); miR-15b sense and antisense strands were purchased from Eurofins MWG Operon (Ebersberg, Germany). Cells were transfected with small interfering RNA (siRNA) targeting Pim-1 [siPim-1: 5′-GGA ACA ACA UUU ACA ACU CdTdT (sense) and 5′-GAG UUG UAA AUG UUG UUC CdTdT (antisense)] or negative control siRNA, targeting luciferase mRNA [siCtrl: 5′-CUU ACG CUG AGU ACU UCG AdTdT (sense) and 5′-UCG AAG UAC UCA GCG UAA GdTdT (antisense)], or targeting enhanced green fluorescent protein (eGFP) [siEGFP: 5′ GCA GCA CGA CUU CUU CAA GdTdT 3′ (sense) and 5′CUU GAA GAA GUC GUG CUG CdTdT 3′(antisense)], or with miR-15b (mature miR-15b sequence, miRBase accession MI0000438). For annealing of the miRNA, sense and antisense strands were dissolved equimolarly in annealing buffer, heated to 95°C for 5 minutes, and cooled down slowly to room temperature.
For transfection, 2 x 102 (HCT-116 and HT29) or 1 x 103 (LS174T) cells were seeded in a 96-well plate, 7 x 104 cells in a 24-well plate, or 3.5 x 105 cells in a 6-well plate, respectively, and incubated under standard conditions unless stated otherwise. SiRNA (20 nM) was transfected using either INTERFERin siRNA transfection reagent or jetPRIME transfection reagent (Peqlab, Erlangen, Germany) according to the manufacturer's protocol and incubated for the indicated time periods. Transfection of plasmid DNA was performed using FuGENE HD Transfection Reagent (Roche, Penzberg, Germany) and 125 ng of plasmid per well (24-well plate) according to the manufacturer's protocol and incubated for the indicated periods of time. For co-transfection of plasmid with miRNA or siRNA, co-complexation was performed using polyethylenimine (PEI) F25-LMW. Briefly, for one well (24-well plate), 125 ng of plasmid and 700 ng of siRNA or miRNA were dissolved in 25 µl of 10 mM Hepes/150 mM NaCl buffer (pH 7.4). PEI F25-LMW (4.2 µg) was diluted in 25 µl of the same buffer and, after incubation for 10 minutes, added to the nucleic acid solution. Complexes were allowed to form during a 30-minute incubation at room temperature and were then added to the cells.
Plasmids and Luciferase Assay
Plasmids encoding luciferase in front of the Pim-1 3′untranslated region (3′UTR) or Pim-1 3′UTR with a mutated miR-15b seed region are based on the pGL3 control luciferase reporter vector (Promega, Mannheim, Germany) and were cloned as described previously [7]. To generate the mutated miR-15b seed region, two point mutations (C→G) were inserted by polymerase chain reaction (PCR)-directed mutagenesis and the result was verified by DNA sequencing. HCT116 cells were seeded in a 24-well plate at a density of 3 x 104 cells/well and co-transfected with plasmid (pGL3 Pim-1 3′UTR or pGL3 Pim-1 3′UTR mut) and miR-15b seed, miR-15b, siPim-1, or, as negative control, siEGFP using PEI F25-LMW as described above. After 72 hours, luciferase activity was determined. Cells were lysed in 24-well plates with 100 µl of lysis buffer (Promega). Lysate (10 µl) was then mixed with 25 µl of luciferase substrate (pjk, Kleinblittersdorf, Germany) and immediately measured using an FB 12 Luminometer (Berthold, Bad Wildbach, Germany).
Treatment of Cells with the Pim-1 Inhibitor KH-CARB13 and Cytostatics
A 20 mg/ml stock solution of the Pim-1 kinase inhibitor KHCARB13 (PubChem CID: 54613583; compound 20 in [20]) was prepared in DMSO and then further diluted in phosphate-buffered saline (PBS) or medium; 1 x 103 HCT-116 or 3 x 103 LS174T cells/well were seeded in a 96-well plate and incubated under standard conditions. For treatment, the medium was aspirated and replaced by 100 µl of IMDM/10% FCS medium containing KH-CARB13 at the indicated concentrations, and cells were incubated at 37°C for 72 hours.
5-FU, oxaliplatin (L-OHP), and docetaxel (DTX) stock solutions in 0.9% NaCl were obtained from the University Hospital Leipzig. The active irinotecan metabolite 7-ethyl-10-hydroxycamptothecin (SN38) was purchased from Sigma-Aldrich (Taufkirchen, Germany) and dissolved in DMSO, before further dilution in PBS. For the analysis of 5-FU-mediated Pim-1 up-regulation, 3.5 x 105 HCT-116 cells/well were seeded in a six-well plate and incubated under standard conditions. Then, the medium was replaced by 2 ml of IMDM/10% FCS containing 1 or 3 µM 5-FU, and cells were incubated at 37°C for 24 hours. For cell viability assays, 5 x 102 HCT-116 cells were seeded in a 96-well plate, transfected as described above, and incubated at 37°C for 24 hours. After aspiration of medium, 100 µl of IMDM/10% FCS medium containing 5-FU, L-OHP, SN38, or DTX at indicated concentrations was added, and cells were incubated for 48 hours (DTX) or 72 hours (5-FU, L-OHP, SN38). For luciferase reporter assays, 3 x 104 HCT-116 cells were seeded in a 24-well plate and transfected as described above. After 48 hours, the medium was replaced by 500 µl of 1 or 3 µM 5-FU diluted in IMDM/10% FCS, and cells were incubated at 37°C for 24 hours. To determine repression of miR-15b by 5-FU treatment, 1.2 x 105 HCT-116 cells were seeded in a six-well plate. After 24 hours, medium was replaced by 2 ml of IMDM/10% FCS containing 3 µM 5-FU, and cells were incubated for 24 hours at 37°C.
RNA Preparation and mRNA and miRNA Detection by Quantitative PCR
Total cellular RNA was prepared by phenol/chloroform extraction using 250 µl of TRI Reagent (Sigma-Aldrich) according to the manufacturer's protocol. miRNA was extracted using mirVana miRNA Isolation Kit (Applied Biosystems, Darmstadt, Germany) according to the manufacturer's protocol. cDNA was transcribed from 1 µgof RNA using the RevertAid H Minus First Strand cDNA Synthesis Kit (Fermentas, St Leon-Roth, Germany) as described previously [8]. For miRNA reverse transcription, specific stem-looped primers were used as described previously [21]. cDNA was diluted 1:10 with nuclease-free water. Quantitative PCR was performed in a LightCycler 2.0 (Roche) using the Absolute QPCR SYBR Green Capillary Mix (Thermo Scientific). All procedures were conducted according to the manufacturers' protocols with 4.5 µl of cDNA (diluted 1:10), 1 µl of primers (5 µM), and 5 µl of SYBR Green master mix. A preincubation for 15 minutes at 95°C was followed by 55 amplification cycles: 10 seconds at 95°C, 10 seconds at 55°C, and 10 seconds at 72°C. The melting curve for PCR product analysis was determined by rapid cooling down from 95 to 65°C and incubation at 65°C for 15 seconds before heating to 95°C. To normalize for equal mRNA/cDNA amounts, PCRs with target-specific and actin-specific primer sets were always run in parallel for each sample, and target levels were determined by the formula 2CP(target)/2CP(actin) with CP as the cycle number at the crossing point (0.3).
Western Blot Analysis
Cells were seeded and transfected in six-well plates as described above. Seventy-two hours after transfection, the medium was removed and cells were washed once with PBS. Two hundred microliters of lysis buffer [1 mM EDTA, 1% NP-40 in PBS, protease inhibitor cocktail (EDTA-free; Roche), Ser/Thr- and Tyr-phosphatase inhibitors (Sigma-Aldrich)] was added per well and plates were incubated on ice for 15 minutes. The suspension was transferred to Eppendorf tubes and sonicated for 20 seconds followed by centrifugation (13,000 rpm, 4°C, 20 minutes). The supernatant was transferred to a new Eppendorf tube and protein concentration was determined using the Bio-Rad DC Protein Assay (Bio-Rad, Munich, Germany) according to the manufacturer's protocol. Loading buffer (4x) was added [0.25 mM Tris-HCl (pH 6.8), 20% glycerol, 10% β-mercaptoethanol, 8% sodium dodecyl sulfate, and 0.08% bromophenol blue] to yield a 1x concentration. Forty micrograms of protein was loaded onto 10% polyacrylamide gels, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transferred onto a 0.2-µm (survivin) or 0.45-µm (all other proteins) Immobilon-P Transfer polyvinylidene difluoride (PVDF) Membrane (Millipore, Billerica, MA). Membranes were blocked with 5% (wt/vol) nonfat dry milk in TBST [10 mM Tris-HCl (pH 7.6), 150 mM NaCl, 0.1% Tween 20], washed in TBST, and incubated with primary antibodies diluted in 3%nonfat dry milk in TBST: anti-phospho-p21Cip1/WAF1 (antibodies online, Aachen, Germany), anti-survivin (Epitomics, Burlingame, CA), anti-Pim-1 (Epitomics), anti-α-tubulin (Sigma-Aldrich), anti-actin (Santa Cruz Biotechnology, Heidelberg, Germany), anti-c-Myc (Santa Cruz Biotechnology), anti-Bax (Cell Signaling Technology, Danvers, MA), anti-Puma (Cell Signaling Technology), or anti-Bcl-xL (Cell Signaling Technology). The other antibodies were diluted in 5% BSA in TBST: anti-phospho-p38 (Thr180/Tyr182) mitogen-activated protein kinase (MAPK), anti-phospho-stress-activated protein kinase/c-jun N-terminal kinase (SAPK/JNK) (Thr183/Tyr185), anti-phospho-p44/42 MAPK [extracellular signal-regulated kinase 1/2 (Erk1/2) Thr202/Tyr204], anti-phospho-STAT3 (Tyr705), or anti-phospho-STAT3 (Ser727; all from Cell Signaling Technology). Blots were incubated overnight at 4°C, washed in TBST, and incubated for 1 hour with HRP-coupled goat anti-rabbit IgG (Cell Signaling Technology) or HRP-coupled goat anti-mouse IgG (Santa Cruz Biotechnology) in TBST containing 3% nonfat dry milk. After washing again, bound antibodies were visualized by enhanced chemiluminescence (ECL Kit; Thermo Scientific). Scanned bands were quantitated using ImageJ (National Institutes of Health, Bethesda, MD).
Proliferation and Soft Agar Assay
For the assessment of anchorage-dependent proliferation, cells were seeded and transfected in 96-well plates as described above. At the time points indicated, the number of viable cells was determined using a colorimetric assay. Briefly, the medium was aspirated and 50 µl of a 1:10 dilution of Cell proliferation Reagent WST-1 (Roche Molecular Biochemicals, Mannheim, Germany) in serum-free medium was added to the cells before incubation for 1 hour at 37°C. The absorbance at 450 nm was measured using a Dynex MRX microplate reader (Pegasus Scientific Inc, Rockville, MD).
For studying anchorage-independent proliferation, soft agar assays were performed as described previously [22]. Briefly, cells were seeded in six-well plates and transfected as described above. Twenty-four hours later, cells were trypsinized and counted. Cells (2 x 105) in 0.35% agar (Carl Roth, Karlsruhe, Germany) were layered on top of 1 ml of a solidified 0.6% agar in a six-well plate. The bottom layer contained 7.5% FCS and the top layer contained 8.5% FCS, respectively. After incubating for 1 week, colonies >50 µm in diameter were counted by at least two blinded investigators.
Apoptosis Assay
To determine the rate of apoptosis, the Caspase-Glo 3/7 Assay (Promega) was used. HCT-116 cells (5 x 102) were seeded in a 96-well plate, transfected as described above, and incubated at 37°C for 72 hours or for 24 hours when followed by a 72-hour treatment with 5 µM 5-FU. The medium was aspirated and 50 µl of caspase substrate, diluted 1:5 in serum-free medium, was added to the cells. After incubation for 1 hour at room temperature in the dark, luminescence was measured using a Fluostar Optima reader (BMG Labtec, Jena, Germany). To normalize for differences in cell densities, a WST-1 assay was performed in parallel on the same plate.
Mouse Xenograft Models
Athymic nude mice (Hsd:Athymic Nude-Foxn1nu, 6–8 weeks of age) were obtained from Harlan Winkelmann (Borchen, Germany) and kept at 23°C in a humidified atmosphere and a 12-hour light/dark cycle, with standard rodent chow and water ad libitum.Experiments were performed according to the national regulations and approved by the Regierungspräsidium Giessen (Giessen, Germany). For tumor establishment, 1.5 x 106 HCT-116 cells in 150 µl of PBS were injected subcutaneously (s.c.) into both flanks of the mice. When the solid tumors reached a volume of 40 to 50 mm3, mice were randomized into specific treatment, negative control treatment, and non-treatment groups (n = 12 tumors per group). SiRNAs were complexed with PEI F25-LMW essentially as described previously [22]. Briefly, 10 µg of siRNA was dissolved in 75 µl of 10 mM Hepes/150 mM NaCl (pH 7.4) and incubated for 10 minutes. Fifty micrograms of PEI F25-LMW was dissolved in the same buffer and, after incubation of 10 minutes, mixed with the siRNA solution. The complexes were aliquoted and stored frozen [22]. Before use, the complexes were thawed and allowed to incubate for 1 hour at room temperature. For local treatment, 3 µg of PEI F25-LMW-complexed siRNA was administered every 2 to 3 days by intratumoral (i.t.) injection. For systemic treatment, 10 µgofPEI F25-LMW-complexed siRNA was administered every 2 to 3 days by intraperitoneal (i.p.) injection, described previously as optimal [22]. Where applicable, 70 or 40 mg/kg 5-FU in PBS was injected i.p. every 3 to 4 days. Tumor volumes were monitored every 2 to 3 days. Mice were sacrificed 1 day after the last treatment, and tumors were removed. Pieces of the tumor tissue where either fixed immediately with 10% paraformaldehyde for paraffin embedding or snap frozen for RNA preparation.
Immunohistochemistry
Paraffin-embedded sections were immunohistochemically stained for Pim-1 essentially as described previously [22]. Briefly, sections were deparaffinized with xylene and rehydrated with graded alcohols. Antigen retrieval was accomplished by incubation in 1 mM EDTA (pH 8.0) at 90 to 95°C temperature for 15 minutes. Endogenous peroxidases were inactivated with 0.3% hydrogen peroxide at 4°C for 30 minutes. Sections were blocked with 10% normal goat serum in phosphate-buffered saline with Tween 20 (PBST)/2% BSA for 1 hour at room temperature before incubation with rabbit monoclonal anti-Pim-1 antibodies (Epitomics) in PBST overnight at 4°C in a wet chamber. After washing in PBST, a 1:1000 solution of biotinylated horse anti-rabbit IgG (Vector Laboratories, Burlingame, CA) in PBST was applied for 1 hour. For visualization, sections were incubated with a streptavidinbiotin-peroxidase complex (ABC Kit; Vector Laboratories) for 30 minutes before washing and incubation with 3,3′-diaminobenzidine. In the presence of immunoreactivity, a brownish color was obtained on the section and the overall staining intensities were ranked from 0 (no staining) to 4 (strong staining).
Statistics
Statistical analyses were performed by Student's t test or one-way analysis of variance/Tukey multiple comparison post-test, and significance levels are *P < .03, **P < .01, ***P < .001, and #not significant. Values are shown as means ± SEM.
Results
siRNA-mediated Knockdown of Pim-1 in Colon Carcinoma Cell Lines
Three Pim-1-specific siRNAs were analyzed in HCT-116 colon carcinoma cells. While all siRNAs revealed a >50% knockdown efficacy compared to the negative control siRNA against an irrelevant gene (luciferase), the Pim-1-specific siRNA 1491 led to the most profound reduction of Pim-1 mRNA levels and was selected for subsequent experiments (Figure W1A). In all three colon carcinoma cell lines, siRNA transfection resulted in a 70% to 80% knockdown of Pim-1 mRNA (Figure 1A), which was paralleled by low residual protein levels as determined by Western blot analysis (Figure 1B).
Figure 1.
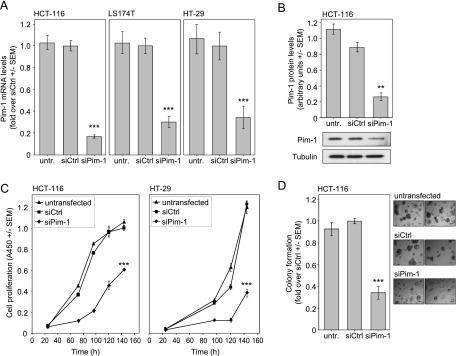
Pim-1 inhibition exerts antiproliferative and proapoptotic effects. (A, B) siRNA-mediated Pim-1 knockdown in colon carcinoma cell lines, as determined on Pim-1 mRNA (A) and protein (B) levels. A representative example of a Western blot is shown. (C, D) Pim-1 knockdown leads to antiproliferative effects in anchorage-dependent growth, as determined by WST-1 assay (C), and (D) anchorage-independent soft agar colony formation. The right panel shows two representative images of colonies.
Antiproliferative and Proapoptotic Effects upon Pim-1 Knockdown
For the assessment of the functional relevance of Pim-1 expression in colon carcinoma cells, we initially analyzed two key features of tumor cells: accelerated proliferation and evasion from apoptosis. Upon siPim-1 transfection, a marked reduction of the anchorage-dependent cell proliferation was detected in all cell lines tested (Figure 1C and data not shown). The comparison of different siRNAs in two cell lines further revealed that the most profound Pim-1 knockdown also resulted in the most prominent inhibition of proliferation (Figure W1B). The antiproliferative effects of the siRNA-mediated Pim-1 knockdown were confirmed in soft agar assays, with a >50% reduction in HCT-116 cell colony formation and the formation of smaller colonies compared to negative control-transfected or wild-type cells (Figure 1D). To test whether this antiproliferative effect is at least in part due to elevated apoptosis, we analyzed the activation of caspase-3/7, representing an early event in apoptosis. Notably, in HCT-116 cells, caspase-3/7 showed a statistically significant ∼1.5-fold increased activity upon Pim-1 knockdown (Figure 2A), demonstrating an antiapoptotic role of Pim-1. To confirm our findings independently of RNAi-mediated knockdown strategies, the newly described Pim-1 kinase inhibitor KH-CARB13 was employed [20]. Increasing concentrations of KH-CARB13 led to a dose-dependent decrease in the viability of LS174T and HCT-116 cells (Figure 2B). Concomitantly, KH-CARB13 led to a moderate but still statistically significant increase in caspase-3/7 activity (Figure 2C), confirming the results obtained after siRNA-mediated Pim-1 knockdown. Taken together, our results establish a proliferative and antiapoptotic role of Pim-1 in colon carcinoma cells.
Figure 2.
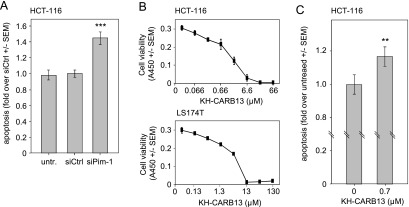
Pim-1 knockdown leads to induction of apoptosis. (A) Elevated caspase-3/7 activity after siPim-1 delivery. (B) Pim-1 inhibitor KH-CARB13 leads to dose-dependent reduction of cell viability in HCT-116 and LS174T colon carcinoma cells and (C) increased apoptosis.
Antitumor Effect upon Local Delivery of Pim-1 siRNA
To test the functional relevance of Pim-1 more rigorously in an in vivo situation, we explored a therapeutic, siRNA-mediated knockdown of Pim-1 in s.c. tumor xenografts. To this end, HCT-116 cells were injected s.c. in athymic nude mice, and upon establishment of solid tumors, mice were randomized into treatment and control groups. Specific or negative control siRNAs were complexed in polymeric nanoparticles based on low molecular weight PEI (PEI F25-LMW) as described previously [22], and 3 µg of siRNA was injected i.t. three times a week for a period of 14 days. In the case of untreated tumors, a rapid ∼12-fold increase of tumor volume was observed, while the i.t. injection of PEI/negative control siRNA complexes led to a slight, statistically nonsignificant inhibition of tumor growth, probably due to some mechanic disruption of the tumor tissue by the injection as observed previously [8]. Notably, however, upon delivery of PEI-complexed Pim-1-specific siRNA, tumor volumes were significantly reduced by ∼50% (P < .01) compared to negative control- treated tumors (∼65% reduction compared to untreated tumors; Figure 3A). In agreement with the previously shown PEI-mediated delivery of siRNAs into tumor cells [22], the immunohistochemical staining of the tumor tissues upon termination of the experiments revealed a decrease in Pim-1 expression in PEI/siPim-1-treated tumors compared to the control groups (Figure 3B), thus confirming that the observed antitumor effects were based on Pim-1 targeting.
Figure 3.

Antitumor effect of PEI/siPim-1 treatment in s.c. HCT-116 colon carcinoma xenograft mouse models. (A) Intratumoral injection of PEI/siPim-1 complexes results in decreased tumor growth compared to treatment with negative control (PEI/siCtrl) or untreated mice. (B) Immunohistochemical staining for Pim-1 shows decreased Pim-1 expression in tumors treated with siPim-1. (C) Mice treated systemically with PEI/siPim-1 complex are more sensitive toward 5-FU therapy. Upon treatment with 70 (left) or 40 mg/kg 5-FU (right), more profound and statistically significant antitumor effects are observed in the PEI/siPim-1 + 5-FU group compared to mice treated with 5-FU + PEI-complexed negative control siRNA. (D) Left: Immunohistochemical staining of the tumor tissues for Pim-1 reveals elevated levels of Pim-1 in 5-FU treatment groups. Right: Exposure of HCT-116 cells to 5-FU in vitro leads to an early increase in Pim-1 mRNA levels (left) and elevated cell growth (right).
Pim-1 Knockdown Sensitizes Colon Carcinoma Cells to 5-FU
While we show that a Pim-1 knockdown exerts antitumor effects, a clinically more relevant scenario may rely on a combination of Pim-1 targeting and established chemotherapy and the systemic rather than i.t. application of siRNAs. We therefore chose a combinatorial approach of systemic PEI/siPim-1 delivery and 5-FU treatment. 5-FU was selected because it is part of established treatment regimens (FOLFOX, FOLFIRI) and in vitro experiments suggested possible synergistic effects (see below), and it was used here as single cytostatic to precisely analyze its combination with Pim-1 knockdown. Upon establishment of HCT-116 tumor xenografts, mice in the treatment groups were i.p. injected three times a week with 10 µg of PEI/siPim-1 or PEI/siCtrl, alone or in combination with 70 mg/kg 5-FU in PBS (i.p., twice a week). Tumors grew rapidly and while, as expected, no difference in tumor size was observed upon PEI/siCtrl treatment, the same was true for PEI/siPim-1 treatment (Figure 3C, left). In contrast, within 1 week of 5-FU treatment (for correct comparison, combined with the PEI-complexed negative control siRNAs), ∼40% reduced tumor volumes were observed. More importantly, however, when combining PEI/siPim-1 and 5-FU treatment, tumor volumes reached only ∼30% of mouse groups treated with PEI/siCtrl or PEI/siPim-1 alone and statistically significant tumor growth inhibition was achieved (Figure 3C, left). This indicates a synergistic effect of PEI/siRNA-mediated Pim-1 knockdown and 5-FU treatment. However, after 1 week, mice that received 5-FU treatment died after losing weight, swelling of the mouth, and showing behavioral alterations. This prompted us to repeat the experiment using a lower dosage of 40 mg/kg 5-FU in PBS (i.p., twice a week). Throughout the whole treatment period, the mouse body weights remained stable and no behavioral alterations or other signs of discomfort were observed (data not shown). Again, untreated tumors grew rapidly, and despite prolonged tumor treatment for 12 days, no difference in tumor volumes was observed between PEI/siCtrl and PEI/siPim-1 treatment (Figure 3C, right). This confirmed that an siRNA-mediated Pim-1 knockdown is insufficient to induce antitumor effects in this tumor model. The treatment of mice with 5-FU (again, for appropriate comparison in combination with the PEI-complexed negative control siRNAs) led to an ∼20% decrease in tumor growth. More importantly, only the combination of PEI/siPim-1 and 5-FU treatment led to a statistically significant decrease in tumor volumes to ∼60% of mouse groups treated with PEI/siCtrl or PEI/siPim-1 alone, confirming the synergistic effect of PEI/siRNA-mediated Pim-1 knockdown and 5-FU treatment (Figure 3C, right).
Surprisingly, the subsequent immunohistochemical staining of the tumor tissues for Pim-1 revealed slightly elevated Pim-1 levels in all 5-FU treatment groups, which possibly obscured the RNAi knockdown effects (Figure 3D, left). To further explore this finding, we exposed HCT-116 cells in vitro to 5-FU and indeed detected an increase of Pim-1 mRNA levels in a concentration-dependent manner (Figure 3D, right) that peaked 24 hours after the start of treatment. In agreement with our findings regarding a proliferative and antiapoptotic role of Pim-1 (see above), we found the cells to grow more densely for the first 24 hours of 5-FU exposure (Figure 3D, right) before the onset of the expected cytotoxic 5-FU effects. Keeping in mind that mice were sacrificed 24 hours after the last 5-FU treatment, these in vitro results are in line with the observed elevation in Pim-1 levels in the dissected tumors.
The effect of Pim-1 levels on the sensitivity of colon carcinoma cells toward single chemotherapeutics that are part of established combinations in colon carcinoma therapy was then characterized more closely in vitro. Indeed, after 72 hours the double treatment of HCT-116 cells with siPim-1 and 5-FU revealed synergistic effects on tumor cell cytotoxicity at low 5-FU concentrations, as determined by a shift of the upper part of the 5-FU dose-response curve to the left, i.e., toward lower 5-FU concentrations, and a more than three-fold decrease in the IC50 (Figure 4A). In contrast, no differences were observed upon treatment of the cells with other cytostatics relevant in colon carcinoma therapy schemes, namely, oxaliplatin or the active metabolite of irinotecan, SN38 (Figure W2). The same was true for the cytotoxic antimicrotubule agent docetaxel. The increase in cellular 5-FU sensitivity upon Pim-1 knockdown thus confirmed the in vivo results (see Figure 3C). Apoptosis assays revealed that it is at least in part due to a slightly more profound increase in 5-FU-mediated apoptosis upon siPim-1 treatment (∼1.7 vs 1.4-fold; compare 5-FU treatment bars in Figure 4B).
Figure 4.

Additive effect of Pim-1 knockdown and 5-FU in HCT-116 colon carcinoma cells. (A) Knockdown of Pim-1 through siRNA results in sensitization of HCT-116 cells to 5-FU exposure 72 hours after the start of treatment [see left shift of the upper part of the dose-response curve and (insert) the calculated IC50 values]. (B) 5-FU-mediated induction of caspase-3/7 is increased by siRNA-mediated knockdown of Pim-1, reaching statistical significance. (C) Pim-1 is directly regulated by miR-15b (compare center bars in each panel; siPim, positive control). (D) The up-regulation of Pim-1 by 5-FU is mediated by miR-15b. Left: 5-FU treatment leads to the down-regulation of miR-15b. Right: The Pim-1 3′UTR is a target of miR-15b, as determined by the activity of a luciferase reporter gene-Pim-1 3′UTR fusion construct. Luciferase activity is increased upon 5-FU treatment (center bars), while this effect is lost upon the introduction of mutations in the miR-15b seed sequence (right bars).
5-FU Effects on Pim-1 Expression Are Mediated by miR-15b
To address the 5-FU-mediated up-regulation of Pim-1 expression in more detail, we speculated that miRNAs may be involved. In silico analyses (TargetScan 6.1) identified the 3′UTR of Pim-1 as a putative target of miR-15b. Indeed, in a reporter gene assay with the intact Pim-1 3′UTR, the transfection with miR-15b markedly reduced luciferase activity to 60% of the siEGFP negative control, whereas no effect was observed in the case of the Pim-1 3′UTR with mutated miR-15b seed (Figure 4C, left vs right panel). This finding points to a direct regulation of Pim-1 by miR-15b. SiPim-1, which binds Pim-1 3′UTR in another position than miR-15b, was able to reduce reporter gene activity of both constructs drastically. Zhou et al. had reported previously that miR-15b is downregulated by 5-FU [23]. Indeed, a 40% reduction in miR-15b levels was found in our experiments (Figure 4D, left). The hypothesis that this 5-FU-mediated miR-15b down-regulation is responsible for Pim-1 elevation was tested using the reporter gene constructs. Transfection of the constructs and subsequent treatment with 5-FU revealed, in accordance with our previous results, a 5-FU concentration-dependent increase in luciferase activity (Figure 4D, right, center bars). In contrast, no increase in luciferase activity was observed in the case of the mutant miR-15b seed, supporting that the 5-FU-mediated up-regulation of Pim-1 is indeed critically dependent on miR-15b.
Downstream Effects of Pim-1 Knockdown
To unravel the molecular mechanisms behind the antitumor effects of Pim-1 knockdown in colon carcinoma cells, we analyzed several proteins that have either been described as downstream effector molecules of Pim-1 kinase activity or that are involved in cell survival, apoptosis pathways, or migration. The cell cycle regulator p21Cip1/WAF1 is a known target of Pim-1 in leukemia cells [24]. Indeed, upon Pim-1 knockdown, levels of p21Cip1/WAF1 phosphorylated at the Pim-1 target site Thr145 were markedly reduced by ∼75% compared to the negative controls (Figure 5A, left, and Western blot, upper panel). In contrast, no change in mRNA expression of p21Cip1/WAF1 was observed (Figure 5A, center panel) indicating that Pim-1 acts posttranslationally through phosphorylation rather than affecting p21Cip1/WAF1 expression.
Figure 5.
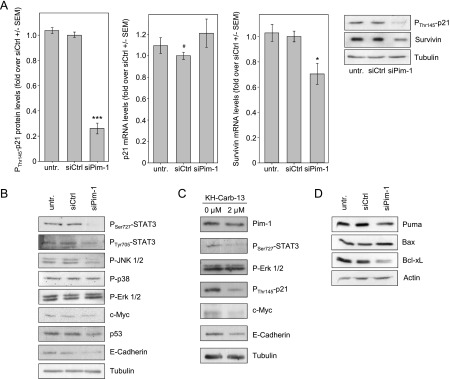
Downstream effects of siRNA-mediated Pim-1 knockdown or Pim-1 inhibition in HCT-116 colon carcinoma cells. (A) Upon Pim-1 knockdown, levels of Thr145-phosphorylated p21 are markedly decreased (left), while overall p21 expression remains unchanged as determined on mRNA levels (center). Downstream of Pim-1, survivin mRNA, and protein levels are decreased (right). Western blots show data from a representative experiment. Since the same lysates were used, one loading control is shown. (B) Analysis of various proteins involved in signal transduction pathways related to cell survival and adhesion. Differences in expression levels are observed upon Pim-1 knockdown (right bands) compared to negative controls. For details, see text. (C) Effects of KH-CARB-13-mediated Pim-1 inhibitor on downstream signaling molecules confirm knockdown results. (D) Alterations in the levels of proapoptotic and antiapoptotic proteins upon Pim-1 knockdown.
The antiapoptotic protein survivin has been described previously to be overexpressed in a variety of tumor entities and to be associated with poor prognosis [25]. Upon Pim-1 knockdown, survivin was reduced on the mRNA and protein level, respectively (Figure 5A, right panel and Western blot). STAT3 has been described previously to be involved in the regulation of Pim-1 [3,4] and of survivin expression [26]. Upon Pim-1 knockdown, a down-regulation of STAT3 phosphorylation at Tyr705 was detected, which is in contrast to Chang et al. who found that STAT3 phosphorylation in prostate and pancreatic carcinomas was mediated by Pim-3 rather than Pim-1 [27]. Notably, in our experiments, a reduction of STAT3 phosphorylation at Ser727 was observed as well. This decrease was even more pronounced than the Tyr705 phosphorylation (Figure 5B) and has not been described before. Moreover, Pim-1 knockdown also led to some other changes (Figure 5B). Seventy-two hours after siPim-1 transfection, phospho-JNK1 and phospho-JNK2 levels were decreased, whereas P-p38 and P-Erk1/2 levels remained unchanged. We furthermore analyzed the oncogenic transcription factor c-Myc and the tumor suppressor p53, whose stability had been identified previously in mantle cell lymphoma to be affected by Pim-1 indirectly through p53 E3 ubiquitin ligase Mdm2 [28]. While we observed an only slight decrease in c-Myc levels 72 hours after siPim-1 transfection, a more profound decrease in p53 expression was detected. Finally, we tested E-cadherin because it is a marker for anchorage-dependent growth and has been shown to be strongly downregulated in an HCT-116 p21Cip1/WAF1 knockout cell line [29]. Indeed, we observed markedly decreased E-cadherin levels upon Pim-1 knockdown, thus linking p21Cip1/WAF1 phosphorylation, rather than p21Cip1/WAF1 expression, to E-cadherin levels. Key findings from the siRNA-mediated Pim-1 knockdown experiments were confirmed by the Pim-1 inhibitor KH-CARB-13 (Figure 5C). Upon incubation of cells with 2 µM inhibitor, a down-regulation of P-STAT3 (with a concomitant decrease in Pim-1 levels), P-p21, c-Myc, and E-cadherin was observed.
Since our results show that a Pim-1 knockdown leads to the induction of apoptosis, we wondered whether apoptosis-related proteins other than the proapoptotic protein Bad, which has already been established as a Pim-1 phosphorylation target, may be affected by Pim-1. Interestingly, Western blot analysis revealed the proapoptotic protein Puma, whose gene is a direct target of p53, to be downregulated upon Pim-1 inhibition, while levels of proapoptotic Bax were elevated (Figure 5D). In line with the induction of apoptosis, the antiapoptotic Bcl-xL was found to be decreased in Pim-1 knockdown cells (Figure 5D).
Discussion
In cardiomyocytes, Pim-1 has important functions in facilitating cell survival under stress [30], whereas its overexpression in tumor cells contributes to malignant transformation, tumor progression, and poor prognosis. While this has been established in hematopoietic malignancies and prostate cancer, little is known about the functional relevance of Pim-1 and the underlying molecular mechanisms in colon carcinoma.
We show here that Pim-1 is embedded in a complex regulatory network of mitogenic and antiapoptotic signals (Figure 6). While STAT3 induces Pim-1 expression, Pim-1 itself can regulate STAT3 activity, thus forming a positive autocrine loop. More specifically, by direct interaction, Pim-1 can activate Etk/BMX tyrosine kinase [31], which in turn leads to STAT3 activation by phosphorylation at Tyr705 [32]. In the tumor context, this leads to the constitutive activation of STAT3, and indeed, we found reduced STAT3 P-Tyr705 levels upon RNAi-mediated knockdown of Pim-1 expression in colon carcinoma. Our results are confirmed by the inhibitor KH-CARB1, which has been described only recently and is used here for the first time in the context of solid tumors. While it has been shown to be largely specific for Pim-1, further improvement with regard to specificity and IC50 will remain a matter of further investigation (F. Bracher, personal communication). In addition to Tyr705 phosphorylation of STAT3, we reveal here that STAT3 P-Ser727 levels are regulated by Pim-1 as well. The consequences of Ser727 phosphorylation on STAT3 activity and DNA binding capacity are discussed controversially with regard to transcriptional activation or inactivation. However, in invasive colorectal cancer, activated STAT3 and STAT3-inducible genes are found to be overexpressed and are associated with increased proliferation and lymph node metastasis [33]. Additionally, simultaneous Tyr705 and Ser727 phosphorylation of STAT3 was found to be essential for oncogenic transformation mediated by the GTPase RhoA [34]. Taken together, this emphasizes the relevance of Pim-1 for STAT3 activation. Beyond Etk/BMX, the MAPK family member JNK1 may represent another possible linker between Pim-1 and STAT3 by mediating STAT3 Ser727 and/or Tyr705 phosphorylation [35]. In agreement with the above-mentioned autocrine loop, we demonstrate that JNK1 and JNK2 phosphorylation was repressed upon Pim-1 knockdown. In addition to STAT3, this may be relevant since there is increasing evidence that in colon carcinoma JNK1 and JNK2 contribute to oncogenesis.
Figure 6.
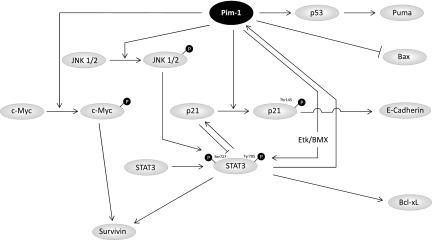
Schematic depiction of the downstream pathways of Pim-1.
p21Cip1/WAF1 is another target of Pim-1. It is phosphorylated by Pim-1 at Thr145 [24], thus leading to the disruption of its binding to proliferating cell nuclear antigen, which in turn is then free to function as processivity factor in DNA replication. Furthermore, P-Thr145 leads to retention of p21Cip1/WAF1 in the cytoplasm or its translocation to the cytoplasm [24]. Nuclear p21Cip1/WAF1 can negatively regulate cell cycle progression and inhibit the transcriptional activity of STAT3 [36], and the Pim-1-mediated shift of p21Cip1/WAF1 toward a cytosolic localization consequently reverses these effects. Concomitantly, we have shown previously that Pim-1 knockdown leads to cell cycle inhibition in colon carcinoma cells [7]. Notably, we also observed a suppression of E-cadherin expression upon Pim-1 knockdown. While this has also been described for p21Cip1/WAF1 knockout cells [29], our data suggest that the Pim-1-mediated subcellular shift of p21Cip1/WAF1 may already be sufficient for the regulation of E-cadherin expression. E-cadherin is a marker for the epithelial phenotype, and loss of E-cadherin is associated with epithelial-mesenchymal transition leading to the disruption of cell-cell contacts, increased migration, and consequently metastatic dissemination. However, Pim-1 knockdown in HCT-116 cells did not result in a loss of cell-cell contacts comparable to HCT-116 p21Cip1/WAF1 knockout cells (data not shown). This is in line with the finding that the loss of E-cadherin alone is insufficient for inducing a change in the cellular phenotype [37], indicating that other molecular effects, e.g., decreased CXCR4 expression [38], may be involved as well. While this further supports the therapeutic benefit of Pim-1 inhibition strategies, their precise impact on migration of colon carcinoma cells and alterations in the expression patterns of transcription factors, chemokine receptors, adhesion molecules, or matrix metalloproteases yet remains to be elucidated.
Our results also establish a connection between Pim-1 and a member of the inhibitor of apoptosis family of proteins, survivin. Although barely detectable in adult tissue, survivin is highly expressed in various tumor entities including colon carcinoma and is associated with elevated cell survival and proliferation, higher tumor grade, poor prognosis, as well as resistance to chemotherapy and radiation (for review, see [25]). The substantial reduction of survivin expression upon Pim-1 knockdown can be readily explained by the inactivation of STAT3, since survivin is a target protein of STAT3 [39]. Furthermore, the c-myc gene is a target of STAT3 [40] and was found to elevate survivin expression when overexpressed in breast cancer [41]. In fact, besides being stabilized by Pim-1 phosphorylation [12], oncogenic c-Myc also synergizes with Pim-1 on the chromatin level, resulting in elevated c-Myc target gene expression [13]. In lymphoma and prostate carcinoma, this synergy has been reported to promote tumor onset and development [42,43], and the direct effect of Pim-1 inhibition on c-Myc as well as the attenuation of c-Myc-induced effects further supports the relevance of Pim-1 as a target molecule.
Our study suggests that low molecular weight inhibitors of Pim-1 are promising drugs in colon carcinoma, while RNAi-based strategies offer the potential to target members of the Pim kinase family with highest specificity. We have shown previously that polymeric nanoparticles based on branched low molecular weight PEI, PEI F25-LMW, offer an efficient and nontoxic delivery platform [22], and the antitumor effects in our mouse tumor model upon i.t. administration confirm the bioactivity of PEI/siPim-1 complexes (Figure 3A). Previous biodistribution studies have also shown that i.p. injected PEI/siRNA complexes reach the s.c. tumors, where full-length siRNAs accumulate [22]; yet, in the HCT-116 xenograft model, no antitumor effects of PEI/siPim-1 single therapy were observed upon systemic (i.p.) injection. Still, tumors were significantly sensitized toward 5-FU treatment, which can be readily explained by the down-regulation of p53 upon Pim-1 knockdown. Indeed, although being a tumor suppressor, p53 overexpression was found to predict poor sensitivity to high-dose 5-FU chemotherapy in colorectal cancer [44]. Even more important for the increase in 5-FU sensitivity upon silencing of Pim-1 may be the Bcl-xL/Bax ratio. Pim-1 knockdown resulted in antiapoptotic Bcl-xL to be downregulated and proapoptotic Bax to be elevated, and a smaller Bcl-xL/Bax ratio has been shown to be associated with increased 5-FU sensitivity in colon carcinoma cells, independent of p53 status [45]. Interestingly, beyond additive or synergistic effects as discussed above, the Pim-1 knockdown may also antagonize the (unwanted) up-regulation of Pim-1 upon 5-FU treatment at early time points (Figure 3). This effect, newly described here, may also have obscured the analysis of PEI/siRNA knockdown efficacies in vivo (note that tumors were taken 24 hours after the last treatment, thus at the peak time of 5-FU-mediated Pim-1 induction). Thus, our data provide the basis for a rational combination therapy based on Pim-1 inhibition and chemotherapy that contains 5-FU.
Certain miRNAs have been shown to regulate Pim-1 expression, including miR-1 [46] and miR33a [7]. In this study, we identify the tumor suppressor miRNA miR-15b to directly target Pim-1. MiR-15b is highly conserved among species and shares its seed sequence with miR15a and miR-16. Its functional relevance in tumor biology has been established by the identification of several target genes involved in apoptosis (e.g., Bcl-2 [47]) and cell cycle (e.g., cyclin E1 [48]), and it negatively regulates chemotherapy-induced epithelial-mesenchymal transition [49]. Here, we describe novel functionalities of miR-15b in the context of Pim-1 inhibition and 5-FU therapy. Notably, members of the miR-15/16 family are often underexpressed in tumors and have already been explored in miRNA replacement therapy. In addition to the reintroduction of the natural Pim-1 regulatory miR-33a described recently [7,8], our results now suggest miR-15b as a novel therapeutic miRNA.
Beyond siRNAs, miRNA replacement therapy extends the set of inhibitory small RNA molecules. siRNAs are intended to target a single mRNA by completely complementary base pairing and subsequent target mRNA degradation, thus exhibiting a strong gene knockdown effect on a single target gene. In contrast, due to the limited sequence complementarity of miRNAs to their targets and their inhibitory effect on mRNA translation, one miRNA may well have several 100 target genes and inhibitory effects mediated by miRNAs are generally broader and milder. Consequently, although molecularly less specific, the replacement of a tumor suppressor miRNA may inhibit the expression of several oncogenes at once and thus rather address the concept of tumorigenesis as a pathway disease. In the case of miR-15b, this may yield (therapeutically beneficial) effects beyond Pim-1 inhibition, although effects unrelated to the inhibition of Pim-1 or other established target genes would have to be monitored carefully.
Supplementary Material
Acknowledgments
We are grateful to Andrea Wüstenhagen for expert assistance with the experiments. HCT-116 p21 -/- cells were obtained from B. Vogelstein.
Abbreviations
- PEI
polyethylenimine
- 5-FU
5-fluorouracil
- siRNA
small interfering RNA
- miRNA
microRNA
Footnotes
This work was supported by grants from the German Cancer Aid (Deutsche Krebshilfe, grants 106992 and 109260 to A.G., R.K.H., and A.A.) and the Deutsche Forschungsgemeinschaft (Forschergruppe ‘Nanohale’ AI 24/6-1 to A.A.).
This article refers to supplementary materials, which are designated by Figures W1 and W2 and are available online at www.neoplasia.com.
References
- 1.Amaravadi R, Thompson CB. The survival kinases Akt and Pim as potential pharmacological targets. J Clin Invest. 2005;115:2618–2624. doi: 10.1172/JCI26273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qian KC, Wang L, Hickey ER, Studts J, Barringer K, Peng C, Kronkaitis A, Li J, White A, Mische S, et al. Structural basis of constitutive activity and a unique nucleotide binding mode of human Pim-1 kinase. J Biol Chem. 2005;280:6130–6137. doi: 10.1074/jbc.M409123200. [DOI] [PubMed] [Google Scholar]
- 3.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bachmann M, Moroy T. The serine/threonine kinase Pim-1. Int J Biochem Cell Biol. 2005;37:726–730. doi: 10.1016/j.biocel.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 5.Chen J, Kobayashi M, Darmanin S, Qiao Y, Gully C, Zhao R, Kondo S, Wang H, Wang H, Yeung SC, et al. Hypoxia-mediated up-regulation of Pim-1 contributes to solid tumor formation. Am J Pathol. 2009;175:400–411. doi: 10.2353/ajpath.2009.080972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tu ML, Wang HQ, Sun XD, Chen LJ, Peng XC, Yuan YH, Li RM, Ruan XZ, Li DS, Xu YJ, et al. Pim-1 is up-regulated by shear stress and is involved in shear stress-induced proliferation of rat mesenchymal stem cells. Life Sci. 2011;88:233–238. doi: 10.1016/j.lfs.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Thomas M, Lange-Grunweller K, Weirauch U, Gutsch D, Aigner A, Grunweller A, Hartmann RK. The proto-oncogene Pim-1 is a target of miR-33a. Oncogene. 2012;31:918–928. doi: 10.1038/onc.2011.278. [DOI] [PubMed] [Google Scholar]
- 8.Ibrahim AF, Weirauch U, Thomas M, Grunweller A, Hartmann RK, Aigner A. MicroRNA replacement therapy for miR-145 and miR-33a is efficacious in a model of colon carcinoma. Cancer Res. 2011;71:5214–5224. doi: 10.1158/0008-5472.CAN-10-4645. [DOI] [PubMed] [Google Scholar]
- 9.Shay KP, Wang Z, Xing PX, McKenzie IF, Magnuson NS. Pim-1 kinase stability is regulated by heat shock proteins and the ubiquitin-proteasome pathway. Mol Cancer Res. 2005;3:170–181. doi: 10.1158/1541-7786.MCR-04-0192. [DOI] [PubMed] [Google Scholar]
- 10.Aho TL, Sandholm J, Peltola KJ, Mankonen HP, Lilly M, Koskinen PJ. Pim-1 kinase promotes inactivation of the pro-apoptotic bad protein by phosphorylating it on the Ser112 gatekeeper site. FEBS Lett. 2004;571:43–49. doi: 10.1016/j.febslet.2004.06.050. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Wang Z, Magnuson NS. Pim-1 kinase-dependent phosphorylation of p21Cip1/WAF1 regulates its stability and cellular localization in H1299 cells. Mol Cancer Res. 2007;5:909–922. doi: 10.1158/1541-7786.MCR-06-0388. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Wang Z, Li X, Magnuson NS. Pim kinase-dependent inhibition of c-Myc degradation. Oncogene. 2008;27:4809–4819. doi: 10.1038/onc.2008.123. [DOI] [PubMed] [Google Scholar]
- 13.Zippo A, De Robertis A, Serafini R, Oliviero S. PIM1-dependent phosphorylation of histone H3 at serine 10 is required for MYC-dependent transcriptional activation and oncogenic transformation. Nat Cell Biol. 2007;9:932–944. doi: 10.1038/ncb1618. [DOI] [PubMed] [Google Scholar]
- 14.Cibull TL, Jones TD, Li L, Eble JN, Ann Baldridge L, Malott SR, Luo Y, Cheng L. Overexpression of Pim-1 during progression of prostatic adenocarcinoma. J Clin Pathol. 2006;59:285–288. doi: 10.1136/jcp.2005.027672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nawijn MC, Alendar A, Berns A. For better or for worse: the role of Pim oncogenes in tumorigenesis. Nat Rev Cancer. 2011;11:23–34. doi: 10.1038/nrc2986. [DOI] [PubMed] [Google Scholar]
- 16.Brault L, Gasser C, Bracher F, Huber K, Knapp S, Schwaller J. PIM serine/threonine kinases in the pathogenesis and therapy of hematologic malignancies and solid cancers. Haematologica. 2010;95:1004–1015. doi: 10.3324/haematol.2009.017079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shah N, Pang B, Yeoh KG, Thorn S, Chen CS, Lilly MB, Salto-Tellez M. Potential roles for the PIM1 kinase in human cancer—a molecular and therapeutic appraisal. Eur J Cancer. 2008;44:2144–2151. doi: 10.1016/j.ejca.2008.06.044. [DOI] [PubMed] [Google Scholar]
- 18.Hu XF, Li J, Vandervalk S, Wang Z, Magnuson NS, Xing PX. PIM-1-specific mAb suppresses human and mouse tumor growth by decreasing PIM-1 levels, reducing Akt phosphorylation, and activating apoptosis. J Clin Invest. 2009;119:362–375. doi: 10.1172/JCI33216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang T, Zhang X, Ding K, Yang K, Zhang Z, Xu Y. PIM-1 gene RNA interference induces growth inhibition and apoptosis of prostate cancer cells and suppresses tumor progression in vivo. J Surg Oncol. 2010;101:513–519. doi: 10.1002/jso.21524. [DOI] [PubMed] [Google Scholar]
- 20.Huber K, Brault L, Fedorov O, Gasser C, Filippakopoulos P, Bullock AN, Fabbro D, Trappe J, Schwaller J, Knapp S, et al. 7, 8-Dichloro-1-oxo-β-carbolines as a versatile scaffold for the development of potent and selective kinase inhibitors with unusual binding modes. J Med Chem. 2012;55:403–413. doi: 10.1021/jm201286z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hobel S, Koburger I, John M, Czubayko F, Hadwiger P, Vornlocher HP, Aigner A. Polyethylenimine/small interfering RNA-mediated knockdown of vascular endothelial growth factor in vivo exerts anti-tumor effects synergistically with Bevacizumab. J Gene Med. 2010;12:287–300. doi: 10.1002/jgm.1431. [DOI] [PubMed] [Google Scholar]
- 23.Zhou J, Zhou Y, Yin B, Hao W, Zhao L, Ju W, Bai C. 5-Fluorouracil and oxaliplatin modify the expression profiles of microRNAs in human colon cancer cells in vitro. Oncol Rep. 2010;23:121–128. [PubMed] [Google Scholar]
- 24.Wang Z, Bhattacharya N, Mixter PF, Wei W, Sedivy J, Magnuson NS. Phosphorylation of the cell cycle inhibitor p21Cip1/WAF1 by Pim-1 kinase. Biochim Biophys Acta. 2002;1593:45–55. doi: 10.1016/s0167-4889(02)00347-6. [DOI] [PubMed] [Google Scholar]
- 25.Kelly RJ, Lopez-Chavez A, Citrin D, Janik JE, Morris JC. Impacting tumor cell-fate by targeting the inhibitor of apoptosis protein survivin. Mol Cancer. 2011;10:35. doi: 10.1186/1476-4598-10-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang H, Yu J, Guo H, Song H, Chen S. Upregulation of survivin by leptin/STAT3 signaling in MCF-7 cells. Biochem Biophys Res Commun. 2008;368:1–5. doi: 10.1016/j.bbrc.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 27.Chang M, Kanwar N, Feng E, Siu A, Liu X, Ma D, Jongstra J. PIM kinase inhibitors downregulate STAT3Tyr705 phosphorylation. Mol Cancer Ther. 2010;9:2478–2487. doi: 10.1158/1535-7163.MCT-10-0321. [DOI] [PubMed] [Google Scholar]
- 28.Hogan C, Hutchison C, Marcar L, Milne D, Saville M, Goodlad J, Kernohan N, Meek D. Elevated levels of oncogenic protein kinase Pim-1 induce the p53 pathway in cultured cells and correlate with increased Mdm2 in mantle cell lymphoma. J Biol Chem. 2008;283:18012–18023. doi: 10.1074/jbc.M709695200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mueller S, Cadenas E, Schonthal AH. p21WAF1 regulates anchorage-independent growth of HCT116 colon carcinoma cells via E-cadherin expression. Cancer Res. 2000;60:156–163. [PubMed] [Google Scholar]
- 30.Borillo GA, Mason M, Quijada P, Volkers M, Cottage C, McGregor M, Din S, Fischer K, Gude N, Avitabile D, et al. Pim-1 kinase protects mitochondrial integrity in cardiomyocytes. Circ Res. 2010;106:1265–1274. doi: 10.1161/CIRCRESAHA.109.212035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie Y, Xu K, Dai B, Guo Z, Jiang T, Chen H, Qiu Y. The 44 kDa Pim-1 kinase directly interacts with tyrosine kinase Etk/BMX and protects human prostate cancer cells from apoptosis induced by chemotherapeutic drugs. Oncogene. 2006;25:70–78. doi: 10.1038/sj.onc.1209058. [DOI] [PubMed] [Google Scholar]
- 32.Tsai YT, Su YH, Fang SS, Huang TN, Qiu Y, Jou YS, Shih HM, Kung HJ, Chen RH. Etk, a Btk family tyrosine kinase, mediates cellular transformation by linking Src to STAT3 activation. Mol Cell Biol. 2000;20:2043–2054. doi: 10.1128/mcb.20.6.2043-2054.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lassmann S, Schuster I, Walch A, Gobel H, Jutting U, Makowiec F, Hopt U, Werner M. STAT3 mRNA and protein expression in colorectal cancer: effects on STAT3-inducible targets linked to cell survival and proliferation. J Clin Pathol. 2007;60:173–179. doi: 10.1136/jcp.2005.035113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aznar S, Valeron PF, del Rincon SV, Perez LF, Perona R, Lacal JC. Simultaneous tyrosine and serine phosphorylation of STAT3 transcription factor is involved in Rho A GTPase oncogenic transformation. Mol Biol Cell. 2001;12:3282–3294. doi: 10.1091/mbc.12.10.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim JH, Lee SC, Ro J, Kang HS, Kim HS, Yoon S. Jnk signaling pathway-mediated regulation of Stat3 activation is linked to the development of doxorubicin resistance in cancer cell lines. Biochem Pharmacol. 2010;79:373–380. doi: 10.1016/j.bcp.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 36.Coqueret O, Gascan H. Functional interaction of STAT3 transcription factor with the cell cycle inhibitor p21WAF1/CIP1/SDI1. J Biol Chem. 2000;275:18794–18800. doi: 10.1074/jbc.M001601200. [DOI] [PubMed] [Google Scholar]
- 37.Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68:3645–3654. doi: 10.1158/0008-5472.CAN-07-2938. [DOI] [PubMed] [Google Scholar]
- 38.Grundler R, Brault L, Gasser C, Bullock AN, Dechow T, Woetzel S, Pogacic V, Villa A, Ehret S, Berridge G, et al. Dissection of PIM serine/threonine kinases in FLT3-ITD-induced leukemogenesis reveals PIM1 as regulator of CXCL12-CXCR4-mediated homing and migration. J Exp Med. 2009;206:1957–1970. doi: 10.1084/jem.20082074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanda N, Seno H, Konda Y, Marusawa H, Kanai M, Nakajima T, Kawashima T, Nanakin A, Sawabu T, Uenoyama Y, et al. STAT3 is constitutively activated and supports cell survival in association with survivin expression in gastric cancer cells. Oncogene. 2004;23:4921–4929. doi: 10.1038/sj.onc.1207606. [DOI] [PubMed] [Google Scholar]
- 40.Kiuchi N, Nakajima K, Ichiba M, Fukada T, Narimatsu M, Mizuno K, Hibi M, Hirano T. STAT3 is required for the gp130-mediated full activation of the c-myc gene. J Exp Med. 1999;189:63–73. doi: 10.1084/jem.189.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cosgrave N, Hill AD, Young LS. Growth factor-dependent regulation of survivin by c-myc in human breast cancer. J Mole Endocrinol. 2006;37:377–390. doi: 10.1677/jme.1.02118. [DOI] [PubMed] [Google Scholar]
- 42.van Lohuizen M, Verbeek S, Krimpenfort P, Domen J, Saris C, Radaszkiewicz T, Berns A. Predisposition to lymphomagenesis in pim-1 transgenic mice: cooperation with c-myc and N-myc in murine leukemia virus-induced tumors. Cell. 1989;56:673–682. doi: 10.1016/0092-8674(89)90589-8. [DOI] [PubMed] [Google Scholar]
- 43.Wang J, Kim J, Roh M, Franco OE, Hayward SW, Wills ML, Abdulkadir SA. Pim1 kinase synergizes with c-MYC to induce advanced prostate carcinoma. Oncogene. 2010;29:2477–2487. doi: 10.1038/onc.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang JT, Huang KC, Cheng YM, Hsu HC, Cheng AL, Hsu CH, Yeh KH, Wang SM, Chang KJ. P53 overexpression predicts poor chemosensitivity to high-dose 5-fluorouracil plus leucovorin chemotherapy for stage IV colorectal cancers after palliative bowel resection. Int J Cancer. 2002;97:451–457. doi: 10.1002/ijc.1637. [DOI] [PubMed] [Google Scholar]
- 45.Violette S, Poulain L, Dussaulx E, Pepin D, Faussat AM, Chambaz J, Lacorte JM, Staedel C, Lesuffleur T. Resistance of colon cancer cells to long-term 5-fluorouracil exposure is correlated to the relative level of Bcl-2 and Bcl-XL in addition to Bax and p53 status. Int J Cancer. 2002;98:498–504. doi: 10.1002/ijc.10146. [DOI] [PubMed] [Google Scholar]
- 46.Nasser MW, Datta J, Nuovo G, Kutay H, Motiwala T, Majumder S, Wang B, Suster S, Jacob ST, Ghoshal K. Down-regulation of micro-RNA-1 (miR-1) in lung cancer. Suppression of tumorigenic property of lung cancer cells and their sensitization to doxorubicin-induced apoptosis by miR-1. J Biol Chem. 2008;283:33394–33405. doi: 10.1074/jbc.M804788200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Xia L, Zhang D, Du R, Pan Y, Zhao L, Sun S, Hong L, Liu J, Fan D. miR-15b and miR-16 modulate multidrug resistance by targeting BCL2 in human gastric cancer cells. Int J Cancer. 2008;123:372–379. doi: 10.1002/ijc.23501. [DOI] [PubMed] [Google Scholar]
- 48.Xia H, Qi Y, Ng SS, Chen X, Chen S, Fang M, Li D, Zhao Y, Ge R, Li G, et al. MicroRNA-15b regulates cell cycle progression by targeting cyclins in glioma cells. Biochem Biophys Res Commun. 2009;380:205–210. doi: 10.1016/j.bbrc.2008.12.169. [DOI] [PubMed] [Google Scholar]
- 49.Sun L, Yao Y, Liu B, Lin Z, Lin L, Yang M, Zhang W, Chen W, Pan C, Liu Q, et al. MiR-200b and miR-15b regulate chemotherapy-induced epithelial-mesenchymal transition in human tongue cancer cells by targeting BMI1. Oncogene. 2012;31:432–445. doi: 10.1038/onc.2011.263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.