

Abstract
We have investigated the importance of interleukin-6 (IL-6) in promoting tumor growth and metastasis. In human primary breast cancers, increased levels of IL-6 were found at the tumor leading edge and positively correlated with advanced stage, suggesting a mechanistic link between tumor cell production of IL-6 and invasion. In support of this hypothesis, we showed that the IL-6/Janus kinase (JAK)/signal transducer and activator of transcription 3 (Stat3) pathway drives tumor progression through the stroma and metastatic niche. Overexpression of IL-6 in tumor cell lines promoted myeloid cell recruitment, angiogenesis, and induced metastases. We demonstrated the therapeutic potential of interrupting this pathway with IL-6 receptor blockade or by inhibiting its downstream effectors JAK1/2 or Stat3. These clinically relevant interventions did not inhibit tumor cell proliferation in vitro but had profound effects in vivo on tumor progression, interfering broadly with tumor-supportive stromal functions, including angiogenesis, fibroblast infiltration, and myeloid suppressor cell recruitment in both the tumor and pre-metastatic niche. This study provides the first evidence for IL-6 expression at the leading edge of invasive human breast tumors and demonstrates mechanistically that IL-6/JAK/Stat3 signaling plays a critical and pharmacologically targetable role in orchestrating the composition of the tumor microenvironment that promotes growth, invasion, and metastasis.
Introduction
The aberrant tyrosine phosphorylation or activation of signal transducer and activator of transcription 3 (pStat3) has been broadly characterized as a regulator of tumorigenesis through its effects in both tumor cells and the tumor microenvironment [1]. In contrast to normal cells in which Stat phosphorylation is tightly regulated, Stat3 is persistently phosphorylated in many cancers through increased production of positive effectors, such as specific cytokines and cytokine receptors, and decreased expression of negative regulators, such as the SOCS proteins and tyrosine phosphatases [2]. We and others have determined that pStat3 is expressed in ∼40% of all breast cancers [3–6], particularly on the leading edge of tumors in association with stromal cells [7]. pStat3 is expressed in the triple-negative (TN) subtype of breast cancer, in part through autocrine expression of the inflammatory cytokine interleukin-6 (IL-6) [5,8–10]. Additionally, paracrine IL-6 can induce autocrine IL-6 expression in adjacent cells, thus creating an IL-6+ niche [8]. The IL-6 family of cytokines is produced by numerous cell types within a tumor (e.g., cancer cells, bone marrow-derived cells, adipocytes, and fibroblasts) and, coupled with the IL-6 receptor (IL-6R) and gp130 receptor, activates a Janus kinase (JAK)-dependent signaling cascade, mediating tyrosine phosphorylation of Stat3.
The mechanisms by which this signaling pathway regulates mammary tumorigenesis and metastatic progression are complex, involving both tumor-intrinsic and tumor-extrinsic roles. For example, targeted reduction of Stat3 in mammary epithelial cells has little effect on their in vitro growth, while the in vivo consequences on tumor growth and metastatic progression are significant and correlate with a reduction in angiogenesis [11–14]. These observations suggest a context or microenvironment-dependent role for the activation of the IL-6/JAK/Stat3 signaling pathway in regulating mammary tumorigenesis. The cells, which constitute the tumor stroma (including endothelial cells, cancer-associated fibroblasts, and bone marrow-derived cells), are recognized as principal determinants of tumor progression [15–17]. Moreover, many of these cells express pStat3 and targeting this transcription factor in bone marrow-derived myeloid cells nearly abrogated the growth of metastatic disease in models of melanoma and bladder cancer demonstrating the importance of myeloid-specific Stat3 activation in metastatic progression [18–21]. Thus, IL-6/JAK/Stat3-driven regulatory programs in tumor cells are hypothesized to orchestrate the formation of a pro-tumorigenic/metastatic microenvironment through the activation of Stat3 in the stroma. However, the significance of this signaling pathway in regulating the interactions among these cell types as well as their function in mammary gland pathogenesis remains unclear.
In this study, we demonstrated that high expression levels of IL-6 on the leading edge of human mammary tumors positively correlated with advanced stage, suggesting a role for this cytokine in promoting metastasis. Increasing IL-6 levels in human breast cancer models induced metastasis, which was associated with the mobilization of tumor-associated suppressive myeloid cells and a robust stromal and endothelial cell infiltrate. Additionally, pStat3, a principal target of IL-6 signaling, was coexpressed with IL-6 in primary human specimens and in murine models of breast cancer in both tumor cells and those comprising the microenvironment including myeloid suppressor cells. Moreover, by knocking out Stat3 in human and in transgenic mammary tumor cells, IL-6 levels were significantly reduced, as were tumor growth and metastasis. A similar phenotype was observed by targeting IL-6 and JAKs using pharmacological interventions. These data demonstrate the formation of an autocrine/paracrine IL-6/JAK/Stat3 feed-forward loop, which participates in tumor proliferation, shaping of the tumor microenvironment, and metastasis. The observed suppression of tumor growth and metastatic potential through the use of clinically relevant inhibitors supports the therapeutic potential of targeting this pathway in breast cancer.
Materials and Methods
Animal Studies
Female BALB/c, FVB, C57/BL6, IL-6-/- C57/BL6, athymic nude, and non-obese diabetic/severe combined immunodeficiency mouse (NOD/SCID) mice (5–8 weeks old) were obtained from NCI (Frederick, MD) and Jackson Laboratory (Bar Harbor, ME). MMTV-PyMT/MMTV-Cre/Stat3flox/flox mice were generated by crossing C57/BL6 MMTV-PyMT transgenic mice (Ming Li, MSKCC) with C57/BL6 MMTV-Cre (Ming Li, MSKCC) and Stat3flox/flox mice (kindly provided by Hua Yu, City of Hope). Animal work was done in accordance with a protocol approved by MSKCC's Institutional Animal Care and Use Committee (IACUC). For mammary fat pad (MFP) tumor injection, cells were harvested by trypsinization, washed twice in phosphate-buffered saline (PBS), and resuspended in PBS (+30% Matrigel). The fourth inguinal mammary glands of mice were revealed and 106 cells were injected directly into the MFP. Microdissected (<1 mm) tumors and/or equal numbers of mammospheres (103) were transplanted into the MFP of 4- to 6-week-old female recipient mice. Mice bearing tumors were treated with JAK inhibitor (JAKi), IL-6R blocking antibody (IL-6RAb), or isotype control and vehicle (see Cell Lines, Antibodies, and Drug Administration section). Tumor growth was monitored weekly by measuring the tumor length (L) and width (W), and volume was calculated as π x L x W2/6. For metastasis assays, MFP tumors were surgically resected upon reaching a volume greater than 300 mm3, and metastatic progression was assessed by bioluminescence imaging (BLI; Xenogen IVIS System) and quantitative polymerase chain reaction [PCR; green fluorescent protein positive (GFP+) tumor cells]. For lung colonization assays, 2 x 105 cells (0.1 ml) were injected into the lateral tail vein. Photon flux (BLI) was measured and normalized to the value obtained after xenografting at day 0.
Cell Lines, Antibodies, and Drug Administration
The tumor cell lines 4T1 and 67NR were provided by Dr Fred Miller (Michigan Cancer Foundation) and the xenograft MARY-X by Dr Mary L. Alpaugh (MSKCC). Met-1 cells were derived from an MMTV-PyMT tumor (FVB background; provided by Dr Kent Hunter, NCI). 4175 and 1833 cell lines were provided by Dr Massague (MSKCC) and express a reporter construct encoding GFP and firefly luciferase for bioluminescent tracking [22]. The 1833-IL-6 overexpression cell line was created by retrovirally introducing pBabe-IL-6 and pBabe control into the 1833 cell line selected with puromycin. Individual clones were isolated and their relative IL-6 levels were determined by ELISA. PyMT control and Stat3-/- PyMT cell lines were generated from tumors derived from PyMT and Stat3-/- PyMT transgenic mice. Tumors were dissected and digested (collagenase III and liberase; minimum essential medium (SMEM) medium, Invitrogen, Carlsbad, CA; 37°C, 3 hours). Isolated cells were cultured in WIT media (low attachment plates). Anti-IL-6R mAb (Tocilizumab) and isotype control were provided by Dr Nori Nishimoto [23] and administered by intraperitoneal injection weekly. AZD1480 was provided by AstraZeneca (Waltham, MA; Dennis Huszar). For in vitro studies, AZD1480 was dissolved in DMSO; for in vivo studies, it was resuspended in 0.5% methylcellulose/0.1% Tween 80 and administered by oral gavage twice daily.
Quantitative Reverse Transcription-PCR
Lung tissue from mice was homogenized and RNA extracted in TRIzol reagent according to the manufacturer's instructions (Invitrogen) and reverse transcribed to cDNA using Superscript III (Invitrogen). Gene expression was quantified by quantitative reverse transcription-PCR [qRT-PCR; SyBR-Green PCR reactions; ABI Prism 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA)]. GFP expression quantification was based on a standard curve (standardized dilutions of GFP; 3 x 102 to 3 x 106 copy numbers), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) or β-actin was used as a housekeeping control transcript. The following primer sequences were used: GFP-FW5′-CTGCTGCCCGACAACCA-3′; GFP-RV5′-TCCAGCAGGACCATGTGATC-3′; β-actin-FW5′-CGGTTGGCCTTAGGGTTCA-3′; β-actin-RV5′-GTGGGCCGCTCTAGGCACCA-3′; GAPDH-FW5′-GAAACCTGCCAAGTATGATGACAT-3′; GAPDH-RV5′-TTGTCATACCAGGAAATGAGCTTG-3′; CSF1-FW5′-CAATGCTAACGCCACCGAGAG-3′; CSF1-RV5′-CGGACACAGGCCTTGTTCTGC-3′; ARG1-FW5′-GGAGACCACAGTCTGGCAGTTG-3′; ARG1-RV5′-AGGACACAGGTTGCCCATGCAG-3′; IL-1β-FW5′-CCAAAAGATGAAGGGCTGCTTC-3′; IL-1β-RV5′-AGCCACAATGAGTGATACTGCC-3′; NOS2-FW5′-CTGCATCGGCAGGATCCAGTG-3′; NOS2-RV5′-TGATGGCCGACCTGATGTTGCC-3′; S100A8-FW5′-CAAGGAAATCACCATGCCCTCTAC-3′; S100A8-RV5′-CGATATTTATATTCTGCACAAACTGAGGAC-3′; S100A9-FW5′-CCTGACACCCTGAGCAAGAAG-3′; S100A9-RV5′-CATTTCTCTTCTCTTTCTTCATAAAGG-3′. Differences in the expression levels of genes were determined by calculating the fold change in expression (2-ddCT).
Immunohistochemistry/Immunofluorescence
Tumors were fixed in 4% paraformaldehyde and embedded in paraffin. Immunohistochemical and immunofluorescence (IF) experiments were performed at the Molecular Cytology Core Facility (MSKCC) using Discovery XT processor (Ventana Medical Systems, Oro Valley, AZ). The primary antibodies used were pStat3 (Tyr705; 0.2 µg/ml; Cell Signaling Technology, Boston MA), smooth muscle actin (SMA; 1 µg/ml; Sigma, St Louis, MO), Meca-32 (3 µg/ml; DSHB, Iowa City, IA), CD45 (0.25 µg/ml; BD, San Jose, CA), Gr1 and anti-mouse CD11b (BD), and anti-human CD33 (Leica BOND). Corresponding biotinylated secondary antibodies were purchased from Vector Laboratories (Burlingame, CA). Tyramide-Alexa 488 and Alexa 568 (Invitrogen) were used for double IF detection.
Immunohistochemical Analysis of IL-6 in Human Breast Cancer Specimens
Forty-six surgical specimens, obtained from patients with ductal/lobular breast carcinoma who underwent quadrantectomy or mastectomy, were collected for immunohistochemical analysis (Table W1). All the procedures were approved by Comitato Etico dell'Azienda Ospedaliera, University of Bologna, and written informed consent was obtained from patients (M.T.). Tissues were histologically classified according to World Health Organization (WHO) criteria and graded following the classification described by Elston and Ellis. The tumors were also typed by nuclear grading and classified as mild, moderate, and severe nuclear atypia (graded 1, 2, and 3, respectively). Tumor size and axillary lymph node involvement were also recorded using pathological tumor-node-metastasis [pTNM; International Union Against Cancer (UICC)] pathologic staging criteria. The cytoplasmic IL-6-immunoreactive population was evaluated [anti-IL-6 antibody, clone 1936; R&D Systems, Minneapolis, MN. Formalin fixed paraffin embedded (FFPE) sections were subjected to antigen retrieval using a Tris-EDTA buffer (pH 9.0) for 20 minutes and incubated with the antibody (1:250) in PBS overnight at room temperature]. For each field, the neoplastic cells were classified according to both positive percentage and staining intensity: percentage = 0 if <1%, 1 if >1% < 25%, 2 if >25% < 50%, 3 if >50% <75%, 4 if >75%; intensity = 1 (weak), 2 (moderate), and 3 (strong). A final classification was obtained by multiplying the two mean values (percentage and intensity) and classifying the results (range, 0–12) as follows: score 0 (value = 0); score 1 (0.1.1); score 2 (>1 < 4); score 3 (>4 < 8); score 4 (>8). pStat3 levels were determined as previously described [7].
Western Blot and ELISA
Whole-cell lysates were prepared as previously described [5], and Western blot analysis was carried out by standard methods. Antibodies used included Stat3, pStat3 (Cell Signaling Technology), tubulin, and β-actin (Santa Cruz Biotechnology, Dallas, TX). IL-6 levels were examined from conditioned media (50–85% confluent cells) or sera using an IL-6 ELISA Kit (R&D Systems) according to the manufacturer's instructions.
Flow Cytometry
Cells were isolated from tumor, spleen, or lung and stained with CD45 (Pe-Cy7 rat anti-mouse, 30-F11; BD), CD11b (APC rat anti-mouse, M1/70; BD), Gr1 (APC-Cy7 rat anti-mouse, RB6-8C5; BD), F4/80 (Brilliant Violet 421, anti-mouse, BM8; Biolegend, San Diego, CA), CD3 (PE rat anti-mouse, 17A2; BD), and CD4 (PerCP rat antimouse, RM4-5; BD). Cell viability was determined using LIVE/DEAD Fixable Yellow Dead Cell Stain Kit (Invitrogen). Data were collected by CyAn ADP Violet Cytometer (Dako Cytomation, Carpinteria, CA) and analyzed with FlowJo (Tree Star, Ashland, OR). CD11b+/Gr1+ and F4/80+ cells were isolated by fluorescence-activated cell sorter (FACS) sorting (BD FACS Aria III).
Arginase and NO Assay
Splenic myeloid-derived suppressor cells (MDSCs) were isolated using MACS cell isolation magnetic beads according to the manufacturer's instructions (Miltenyi Biotech, Auburn, CA). The purity of the resulting cells (>90% CD11b+/Gr1+) was assessed by flow cytometry. Isolated MDSCs were activated by culturing 1 x 106 cells per 200-µl well in Dulbecco's modified Eagle's medium containing 5% FBS plus interferon gamma (IFN-γ; 2 ng/ml) and lipopolysaccharide (LPS; 100 ng/ml) in a 96-well plate for 24 hours. Arginase was quantified by measuring the production of urea as described [24]. Absorbance (urea concentration) was measured spectrophotometrically at 540 nm. NO was measured using Griess Reagent System (Promega, Madison, WI) according to the manufacturer's instructions.
Statistical Analysis
Data are expressed as means ± SEM. The statistical significance of differences was evaluated using an unpaired, nonparametric Student's t test. Significant differences between experimental groups were *P < .05 or **P < .01.
Results
Consequences of IL-6 Expression on Mammary Tumorigenesis
IL-6 and pStat3 are co-expressed in a number of cancer subtypes, including mammary tumors [5,7,10,25,26]. Paracrine IL-6 was shown to promote autocrine expression of IL-6 within cancer cells [8,9], suggesting a positive feed-forward loop, in which IL-6 engages with IL-6R/gp130, resulting in JAK and Stat3 activation, which in turn increases expression of autocrine IL-6 [27–29]. These observations prompted us to examine the relative levels and distribution of IL-6 expression by immunohistochemistry in human primary breast cancers including those with metastatic involvement in matched axillary lymph nodes (Figure 1, A and B). We determined that the highest levels of IL-6 were found on the tumor edge enriched in stromal/immune cells, areas of lymphovascular invasion (LVI), and axillary lymph nodes. In contrast, the central portion of the tumor expressed lower levels of IL-6 (Figure 1A). Furthermore, Figure 1C depicts the positive correlation observed between high IL-6 levels on the tumor edge and the number of lymph nodes affected by metastatic disease (the most important predictor of survival in breast cancer) [30]. These results suggested to us that the relative levels and distribution of IL-6 in breast tumors might play a role in metastatic progression. Additionally, the lack of generalized IL-6 staining supports the idea of an interdependence between tumor and stromal cells (frequently found on the edge of tumors) as an important positive regulator of IL-6 expression in cancers.
Figure 1.

Differential IL-6 expression in human breast cancer. (A) IL-6 expression was examined by IHC in 45 primary breast cancer samples, 18 with matched axillary lymph nodes. Relative IL-6 levels [low (0, 1+) versus moderate/high (2, 3, 4+)] were determined and found to be highest on the tumor edge, areas of LVI, and lymph nodes relative to the central portion of the tumor (P < .05, Fisher exact C2). Normal lymph node inset displays no IL-6 expression. (B) IL-6 scoring. Cytoplasmic IL-6 immunoreactivity was evaluated at x200 (see Materials and Methods section). Representative examples are shown. (C) Higher IL-6 levels on the tumor edge were associated with number of lymph nodes involved with metastatic disease.
We hypothesized that autocrine/paracrine IL-6 expression from tumor cells would lead to activation of Stat3 in both stromal and tumor cells enhancing growth and metastasis. We therefore determined the effects of modulating the levels of IL-6 and IL-6 signaling in human breast cancer models on both primary tumor growth and metastasis. We examined two TN breast cancer-derived cell lines with either a high (4175) or no capacity (1833) to spontaneously metastasize to the lung [22]. The 4175 cells expressed two-fold higher levels of IL-6 and pStat3 compared to the 1833 cell line (Figure 2A). We first determined if increasing the levels of IL-6 in the 1833 cells (∼1800 pg/ml) to those observed in 4175 cells (∼2900 pg/ml) could alter their growth in the lungs and MFP, as well as their capacity to spontaneously metastasize to the lungs. We introduced an IL-6 retroviral expression vector in 1833 cells and selected several clones that expressed two to four (∼4000 pg/ml) times more IL-6 than vector-infected control cells (1833-pB), which correspondingly showed relatively more pStat3 expression (Figure 2, A and B). We compared the in vitro growth of 1833-pB and 1833-IL-6 cells and no differences were observed, suggesting that IL-6 does not regulate proliferation in a tumor-autonomous manner (Figure W1A). We compared the in vivo growth of 1833-pB to 1833-IL-6 cells following tail vein (lung colonization) and MFP injection by in vivo luciferase imaging. While 1833-pB cells never gave rise to lung metastases, 1833-IL-6 cells grew in the lungs as effectively as 4175 cells (Figure 2C and data not shown). Examination of the lungs of mice injected with 1833-IL-6 cells compared to those injected with 1833-pB cells revealed a 20-fold increase in pStat3+ cells and a 5-fold increase in leukocytes (CD45+) before the development of macro-metastatic disease (3 weeks after injection; Figure 2D). The growth of 1833-IL-6 cells in the MFP was significantly greater than that of the 1833-pB cells, demonstrating a proliferative advantage (Figure 2E). A comparison of the MFP tumors revealed higher levels/numbers of pStat3+ cells, leukocytes (CD45+), myeloid cells (CD11b+/Gr1+), and endothelial cells (Meca-32) in the 1833-IL-6 compared to the 1833 tumors (Figure W1, B and C). A significant difference in the development of lung metastases was observed between mice bearing 1833-IL-6 and 1833-pB MFP tumors (as measured by qRT-PCR of GFP+ tumor cells). That is, 1833-IL-6 MFP tumors spontaneously metastasized to the lung, while 1833-pB MFP tumors never gave rise to lung metastases regardless of primary tumor size (Figure 2F). These results demonstrated that a two- to three-fold increase in tumor-derived IL-6 levels can dramatically enhance inflammation in the tumor and pre-metastatic sites and alter the fate of a cancer cell, rendering it capable of proliferating in distant metastatic sites.
Figure 2.

IL-6 expression promotes metastatic progression in the lung. (A) Conditioned media from 1833-pB (vector control), 4175, and 1833-IL-6 cells were analyzed for IL-6 levels by ELISA. (B) Extracts from 1833-pB, 4175, and 1833-IL-6 cells were analyzed for pStat3, Stat3, and tubulin levels. (C) 1833-pB and 1833-IL-6 cells (1 x 105) were injected intravenously into mice, and tumor growth was determined weekly by normalized BLI (n = 10 mice/group). Representative BLI images of mice from each group at week 7 are shown. (D) IHC staining of CD45 and pStat3 expression in lung sections of 1833 vector control and 1833-IL-6-injected mice. Number of CD45+ and pStat3+ cells are indicated/high-powered field (hpf; n = 10 images). (E) 1833 and 1833-IL-6 cells (1 x 106) were injected into the MFP of athymic nude mice, and tumor growth was determined weekly by normalized BLI (n = 10 mice/group). (F) qRT-PCR for GFP (tumor cells: GFP+) was performed on the RNA of lungs from 1833-IL-6 and 1833-pB primary tumor-bearing mice (n = 5/group; *P < .05).
Conversely, we reasoned that blocking this cytokine and its associated signaling pathway would abrogate the metastatic process. Here, we turned to IL-6RAb, which inhibited IL-6 signaling in 1833-IL-6 cells. Specifically, IL-6RAb was added to 1833-IL-6 cells leading to a reduction in pStat3 levels (Figure W2A). We also demonstrated that 1833-IL-6 cells secrete both the soluble IL-6R (sIL-6R) and the IL-6 ligand, which form a complex with the gp130 receptor (both murine and human). This signaling, known as trans-signaling, was effectively blocked by the IL-6RAb (Figure W2B and data not shown). Notably, the IL-6RAb had no effect on the in vitro growth of 1833-IL-6 cells (data not shown).
To determine the effects of IL-6R blockade on tumorigenesis, we colonized lungs and injected MFPs with 1833-IL-6 cells and mice received either the IL-6RAb or isotype control antibody. The growth of colonized cells in the lungs and MFP and the spontaneous metastases were largely abrogated (Figure 3, A–D). The levels of pStat3+ cells, endothelial cells (Meca-32+), leukocytes (CD45+), and CD11b+/Gr1+ cells were reduced in the MFP tumors and lungs of IL-6RAb-treated mice (Figures W2, C–E, and 3, E and F). These observations were also determined in the 4175 cell line (expressing high levels of IL-6 and pStat3) in which IL-6RAb reduced MFP growth, spontaneous metastases, and the growth of established lung metastases with an associated decrease in pStat3+ leukocytes, endothelial cells, and CD11b+/Gr1+ cells (data not shown). These findings demonstrate that two- to three-fold fluctuations in IL-6 levels or IL-6 signaling blockade can result in significant phenotypic consequences.
Figure 3.

IL-6R blockade inhibits growth of tumors and metastases and prevents metastatic progression. (A) 1833-IL-6 cells (1 x 105) were injected into the tail vein (n = 10/group) and were randomized to receive weekly intraperitoneal injections of control IgG (C) or IL-6RAb (IL-6R) given at the time of injection. Tumor growth was determined weekly by normalized BLI. Representative lung images are shown. (B) qRT-PCR for GFP relative to β-actin (tumor cells: GFP+) was performed to quantify tumor lung colonization described in A (n = 5). (C) 1833-IL-6 cells (1 x 106) were injected into the MFP of athymic nude mice and randomized to receive weekly intraperitoneal injections of control IgG (C) or IL-6RAb (IL-6R). Growth was determined weekly by normalized BLI. (D) qRT-PCR for GFP was performed on the lungs of mice (n = 5) in C. Quantification of CD11b+ and Gr1+ cells/hpf in the lungs (E) and MFP tumors (F) of mice described in A and C, respectively (n = 10 sections/group; **P < .01, *P < .05).
Stat3-IL-6 Feed-Forward Loop Regulates Mammary Tumorigenesis
A principal target of IL-6 signaling is the Stat3 transcription factor. Here, we observed, as with IL-6, that phosphorylated pStat3 was expressed primarily on the edge of the tumors and in areas of LVI in association with stromal cells (Figure 4A). These observations suggested that Stat3 may positively regulate the expression of IL-6. Moreover, chromatin immunoprecipitation (ChIP) results demonstrated Stat3 binding to the IL-6 promoter in 4175 breast cancer cells (Figure W3A) [31,32]. These data led us to hypothesize that a positive feed-forward loop may take place between Stat3 transcriptionally regulating IL-6 expression. We investigated whether the selective absence of Stat3 in mammary tumor cells would decrease IL-6 expression/paracrine signaling, which we hypothesize would alter the tumor microenvironment, consequently decreasing tumor growth and metastatic progression. Reduction of Stat3 levels by short hairpin-silencing RNAs to Stat3 (Stat3-shRNA) in 4175 cells had no effects on in vitro growth but potently reduced in vivo growth, which correlated with reduced IL-6, CD45, and Meca-32 levels (Figure W3, B–E and data not shown). Furthermore, 4175 cells silenced for Stat3 display an eight-fold change in IL-6 mRNA (data not shown).
Figure 4.

Stat3 deficiency abrogates tumorigenesis. (A) Representative IHC images examining pStat3 expression within the central portion, edge, and invasive (lymphovascular invasion) regions of human and MMTV-PyMT breast cancers. (B) Tumor pieces (1 mm) from Stat3+/+ (PyMT S3+) and Stat3-/- (PyMT S3-) PyMT-bearing mice were implanted orthotopically in the MFP of syngeneic C57/BL6 hosts, and tumor growth (volume and weight) was determined over 3 weeks (n = 8/group). (C) Representative histologic images of PyMT S3+ and PyMT S3- tumors stained for hematoxylin and eosin (H&E), pStat3, SMA, and Meca-32. (D) Flow cytometric analysis of PyMT S3+ and PyMT S3- tumors for CD11b+/Gr1+ cells (n = 5/group). (E) qRT-PCR analysis to determine the fold change (2-ddCT) in the expression of Stat3-regulated genes in CD11b+/Gr1+ cells isolated from PyMT S3+ and PyMT S3- tumors. (F) Representative co-IF staining for pStat3 and CD33 in primary invasive ductal carcinomas (n = 20). Thirty-three percent of the CD33+ cells were also pStat3+ (**P < .01).
We next investigated the role of Stat3 in the transgenic MMTV-PyMT model of mammary tumorigenesis, which recapitulates many aspects of human breast cancer, from preinvasive lesions to invasive carcinoma and metastatic disease [33]. High levels of nuclear pStat3 were observed on the invasive edge of tumors in association with stromal cells. Conversely, well-differentiated areas of tumor with few stromal cells expressed much lower levels of pStat3 (Figure 4A). Conditional Stat3 (Stat3Flox/Flox) mice were bred with MMTV-PyMT and MMTV-Cre mice, and the onset of mammary tumor formation between the Stat3+ versus Stat3- MMTV-PYMT mice was quite variable (6–8 months) [34]. Due to insufficient numbers (attributable to poor breeding of the C57/BL6 strain), we were unable to determine the phenotypic consequences of Stat3 deficiency on tumor initiation. Additionally, the MMTV-PyMT mice develop innumerable tumor foci in their mammary glands, making it difficult to monitor tumor growth and metastasis progression [33,35]. To examine the growth and progression of single tumor foci over time, we isolated multiple pairs of tumors from littermates (Stat3-/- vs Stat3+/+ PyMT mice), generated cell lines from these tumors, and confirmed that they either expressed or lacked pStat3/Stat3 (Figure W4A). Interestingly, the Stat3+/+ tumors had a robust stromal cell infiltrate compared to Stat3-/- tumors upon initial culturing (data not shown). We observed no differences in their in vitro growth rates, relative numbers of stem-like cells (CD49f/CD24-positive cells), or capacity to form mammospheres (which are enriched in tumor-initiating cells [36]; Figure W4B and data not shown). In vivo growth was assessed by orthotopically transplanting equal numbers of mammospheres or small chunks (1 mm) of tumor (from littermates, as described above) in the MFP of syngeneic (C57/BL6) hosts. Tumor growth was significantly reduced in the Stat3-/- PyMT tumors compared to control (Stat3+/+; Figure 4B). There was no evidence of macroscopic metastases in the mice bearing Stat3-/- tumors, while Stat3+/+ tumor-bearing mice had widelymetastatic disease (Figure W4C and data not shown). Morphologically, the Stat3+/+ tumors were poorly differentiated, displayed greater pleiomorphic features, and increased vascular invasion compared to the Stat3-/- tumors (Figure 4C). Further examination of these tumors revealed a reduction in pStat3 in both tumor and stromal cells, blood vessel number (Meca-32), mesenchymal cell infiltrate (SMA), and MDSCs (CD11b+/Gr1+) compared to the Stat3+/+ tumors by immunohistochemistry (IHC) and FACS analyses (Figure 4, C and D). We also observed an increase in the levels of CD8+ T cells in the Stat3-/- compared to the Stat3+/+ tumors (Figure W4D). Furthermore, the observed reduction in the number of CD11b+/Gr1+ cells was also accompanied by their decreased expression of immunosuppressive cytokines, such as Arg1, Csf1, Il-1β, Nos, S100a8, and S100a9 (Figure 4E).
Given this evidence, we hypothesized that many of the pStat3+ cells in the tumor microenvironment of breast cancer patients would include immature myeloid cells, including MDSCs. We therefore performed co-IF staining for pStat3 and CD33 (a pan-myeloid marker that recognizes MDSCs [37,38]) in 20 primary invasive ductal carcinomas and determined that ∼30% of CD33+ cells were also pStat3+. Moreover, these cells were found primarily within the tumor stroma (Figure 4F). Thus, tumor-derived Stat3 appears to favor the presence of an immune and mesenchymal cell infiltrate that constitutes a tumor-promoting microenvironment.
Additionally, analysis of the lungs of PyMT mice bearing Stat3+/+ versus Stat3-/- tumors (3 weeks after orthotopic injection) revealed a significant reduction in the levels of pStat3+ cells and CD11b+/Gr1+ cells (in the absence of lung metastases by morphologic analysis), suggesting that Stat3-dependent tumor-derived factors mediate the formation of a pro-inflammatory microenvironment or premetastatic niche (i.e., the receptive environment within distant target organs that forms before metastasis of tumor cells; Figure 5A) [39,40].
Figure 5.

Tumor-extrinsic effects of Stat3 deficiency. (A) Representative IHC images of the lungs of tumor-bearing (PyMT S3+ and PyMT S3-) mice stained for pStat3. The proportion (%) of CD11b+/Gr1+ cells in the lungs of these mice was determined by flow cytometry. (B) IL-6 levels in the serum were determined by ELISA and tumors were determined by qRT-PCR in PyMT S3+ and PyMT S3- mice, respectively (n = 5/group). (C) Tumor pieces (1mm) from mice bearing Stat3+/+ (PyMT S3+) cancerswere implanted orthotopically in theMFP of syngeneic (C57/BL6) IL6+/+ and IL-6-/- hosts, and tumor growth (weight) was determined after 3 weeks (n = 4/group). **P < .01, *P < .05.
We hypothesized that IL-6 levels (a regulator of MDSC expansion) would be reduced in Stat3-/- versus Stat3+/+ PyMT tumors [41,42]. Indeed, IL-6 mRNA/protein levels were low in Stat3-/- versus Stat3+/+ tumor-derived cell lines, demonstrating a dependence for Stat3 on autocrine IL-6 expression on Stat3 (data not shown). Moreover, the lack of IL-6 in orthotopically transplanted tumor cells resulted in an overall reduction in IL-6 mRNA levels in the tumor (both tumor stroma and tumor cells) and a reduction in circulating IL-6 levels in Stat3-/- versus Stat3+/+ tumor-bearing mice (Figure 5B). A requirement for stromal IL-6 in tumorigenesis was determined by implanting Stat3+/+ PyMT tumors in IL-6-deficient and wild-type (WT) mice. The lack of host-derived IL-6 suppressed tumor growth even further (Figure 5C). Overall, these data suggest the presence of an IL-6/Stat3 autocrine/paracrine feed-forward loop with profound effects on tumor size and metastatic potential primarily due to alterations in the tumor and pre-metastatic microenvironment.
Effects of JAK Inhibition on the Tumor Microenvironment
JAKs are the mediators of IL-6 signaling that phosphorylate/activate Stat3. Inhibitors of JAKs have been shown to not only reduce Stat3 phosphorylation (pStat3) but also reduce cytokine levels (including IL-6) in patients with myeloproliferative disorders (MPDs) [43]. In this study, we examined the effect of JAK1/2 inhibition on tumor growth and spontaneous metastatic progression in a number of breast cancer models with differential expression levels and distribution of pStat3. We first examined the effects of JAK inhibition in the transgenic MMTV-PyMT model. Treatment of mice bearing multiple spontaneous mammary tumors with JAKi led to a significant reduction in tumor growth and abrogated the appearance of new tumors and the formation of metastasis, compared to vehicle-treated mice (Figure 6A and data not shown). Immunohistochemical staining of the tumor demonstrated a reduction in pStat3 levels in both the tumor and its stroma and a reduction in infiltrating endothelial (Meca-32) and fibroblast/myoepithelial cells (SMA+; Figure 6B).
Figure 6.

JAK inhibition abrogates in vivo but not in vitro growth of transgenic and orthotopic mammary tumors. (A) MMTV-PyMT tumor-bearing transgenic mice (n = 8) were treated with vehicle (C) or JAKi (AZD1480, 30 mg/kg twice daily). The size of tumors was determined weekly. JAKi-treated mice, compared to control mice, did not develop any new mammary tumors. (B) Representative IHC images of tumors from vehicle- (C) and JAKi-treated MMTV-PyMT tumor-bearing mice stained for pStat3, Meca-32, and SMA. (C) Met-1 mammary breast cancer cells were grown in culture and treated with JAKi (1 µM) or vehicle control (C). Extracts from these cells were analyzed for pStat3 and Stat3 by Western blot. (D) Met-1 cells (1 x 105) were injected into the MFPs of FVB mice, and when tumors reached ∼400 mm3, mice were treated with JAKi (30 mg/kg twice daily) or vehicle control (C). Tumor size was determined after 3 weeks (n = 5/group). (E) Representative IHC images of Met-1 tumors from control (C) and JAKi-treated mice for pStat3, Meca-32, and SMA. (F) MARY-X cells were orthotopically injected into the MFPs of nude mice, and when tumors reached 150 mm3, mice were treated with either vehicle control (C) or JAKi (30 mg/kg twice daily). (G) Representative IHC staining patterns for pStat3, Meca-32, and SMA of MFP tumor sections from control (C) or JAKi-treated mice.
In addition to this transgenic model of mammary tumorigenesis, we also examined the role of JAK inhibition in orthotopic mammary tumor models, including the murine PyMT-derived cell line Met-1, the human inflammatory breast cancer xenograft MARY-X, and the 4T1 murine mammary tumor model [44–46]. JAKi treatment of Met-1 cells in vitro (1 µM) suppressed Stat3 phosphorylation status; however, it had no effect on the in vitro proliferation of the cells (Figure 6C) as also reported for other cell lines (14). Similarly to MMTV-PyMT, administration of JAKi significantly reduced tumor growth, which correlated with a reduction in pStat3, Meca-32, and SMA expression (Figure 6, D and E). The MARY-X model of inflammatory breast cancer expresses very low levels of pStat3 in tumor cells but high pStat3 in the surrounding stroma. Treatment of these xenografts with JAKi led to a reduction in tumor size in conjunction with a decrease in pStat3 expression, angiogenesis (Meca-32), and SMA levels in the stroma (Figure 6, F and G). Additionally, we examined the 4T1 murine model of mammary tumorigenesis. As with the other cell lines, JAKi reduced pStat3 levels and had no effect on in vitro growth (Figure 7A), but a significant reduction in lung colonization, tumor growth, and spontaneous lung metastasis following JAKi administration was observed (Figure 7, B and C). IHC staining of 4T1 tumors of mice treated with JAKi revealed a marked reduction in pStat3 levels in both the tumor and stromal cells as well as in Meca-32 and SMA levels, compared to vehicle-treated mice (Figure 7D).
Figure 7.

JAK inhibition has no in vitro growth effects yet inhibits tumor growth and metastasis of 4T1 mammary cancers. (A) 4T1 breast cancer cells were grown in culture treated with JAKi (1 µM) or vehicle control. Calcein AM was performed daily (C). Extracts from these cells were analyzed for pStat3 and Stat3 by Western blot. (B) 4T1 cells were injected into the tail vein of mice (1 x 105), and once BLI values reached ∼5 x 106, mice were randomized to receive vehicle control (C) or JAKi (30 mg/kg twice daily). Normalized BLI signal of the mice (n = 10/group) was determined weekly. (C) 4T1 cells (2 x 105) were injected into the MFP of BALB/c mice, which were randomized to receive vehicle (C) or JAKi (30 mg/kg twice daily). Normalized BLI signals of MFP (n = 10/group) were determined weekly. Tumor weight and spontaneous lung metastasis for either vehicle (C) or JAKi-treated mice were determined after 3 weeks. (D) Representative IHC images of tumors from vehicle (C) and JAKi-treated 4T1 tumor-bearing mice stained for pStat3, CD45, Meca-32, and SMA. **P < .01, *P < .05.
Given our observation that a reduction in IL-6/Stat3 signaling correlated with decreased MDSCs (see Figure 4D), we examined the effect of JAKi on the immune cell infiltrate in tumor-bearing mice. Flow cytometric analysis of the 4T1 MFP tumors revealed an increase in CD3+, CD4+, and CD8+ cells (Figure 8A) and a reduction in the levels of CD11b+/Gr1+ cells in MMTV-PyMT, Met-1, and 4T1 MFP tumors and in pre-metastatic sites as a consequence of JAKi treatment (Figure 8B). Moreover, CD11b+/Gr1+ cells isolated from 4T1 tumors had reduced expression levels of immunosuppressive factors, such as Arg1, Csf1, Il-1β, Nos, S100a8, and S100a9 compared to their control counterparts, as determined by qRT-PCR (Figure 8C). A functional analysis of the isolated CD11b+/Gr1+ cells revealed a reduction in arginase (urea) and nitric oxide production, suggesting that the CD11b+/Gr1+ cells (MDSCs) isolated from JAKi-treated tumor-bearing mice are hypofunctional (Figure 8D). In addition to CD11b+/Gr1+ cells, we also observed a trend to a decrease in the number of the pro-inflammatory tumor-promoting CD11b+/F480+ cells in the tumors of JAKi-treated mice (Figure 8E). However, qRT-PCR analysis of F4/80+ cells isolated from 4T1 tumor-bearing mice revealed a reduction in the expression levels of the tumor-promoting M2 growth factors, including Arg1, Il-10, Fizz1, Ccl2, and Ym1 following JAKi administration (Figure 8F). These data suggest that JAK inhibition promotes an antitumor response in part through the reduction of tumor-promoting immune cells (CD11b+/Gr1+, F4/80+).
Figure 8.

JAK inhibition alters the cellular immune phenotype of the tumor microenvironment. (A) The proportion (%) of CD3+, CD4+, and CD8+/CD45+ cells (n = 5/group) found in the tumors of 4T1 tumor-bearing mice treated with vehicle control (C) or JAKi (30 mg/kg twice daily) was determined by flow cytometry. (B) Flow cytometric analysis for CD11b+/Gr1+ cells in tumors and lungs of mice bearing MMTV-PyMT, Met-1, and 4T1 MFP tumors treated with vehicle control or JAKi (30 mg/kg twice daily). (C) qRT-PCR analysis to determine the fold change (2-ddCT) in the expression of Stat3-regulated genes in CD11b+/Gr1+ cells isolated from 4T1 tumors treated with vehicle (C) or JAKi (30 mg/kg twice daily). (D) The urea and NO2- concentrations observed in the CD11b+/Gr1+ cells isolated from 4T1 tumor-bearing mice treated with vehicle control (C) or JAKi (30 mg/kg) were determined by arginase and NO assays, respectively (n = 6/group). (E) The proportion (%) of CD11b+ and F480+ cells (n = 5/group) found in the tumors of MMTV-PyMT transgenic mice and in Met-1 and 4T1 xenografts from mice treated with vehicle control (C) or JAKi (30 mg/kg) was determined by flow cytometry. (F) qRT-PCR analysis to determine the fold change (2-ddCT) in the expression of immunosuppressive genes (Arg1, Il-10, Ccl2, Fizz1, Ym1, Ifnγ, S100a8, and S100a9) from CD11b+/F480+ cells isolated from 4T1 xenografts from tumor-bearing mice treated with vehicle control or JAKi (30 mg/kg twice daily; **P < .01, *P < .05).
Discussion
IL-6 is a pro-inflammatory cytokine produced primarily by the cells comprising the tumor microenvironment (e.g., fibroblasts, myeloid cells, and lymphoid cells) and has been characterized as a potent activator of Stat3. In this study, we demonstrate that the presence of high IL-6 expression on the invasive edge of breast cancers in association with stromal cells correlates with the degree of involvement of lymph nodes in cancer, a marker of poor prognosis [30]; in contrast, IL-6 levels are comparatively lower in areas devoid of stroma such as the central portion of tumors. These data suggest a role for IL-6 in promoting invasion and metastasis. Additionally, we observed that the heterogeneity in IL-6 expression is mirrored by the heterogeneity in pStat3 expression. The heterogeneity or localized distribution of IL-6/pStat3 staining suggests multiple hypotheses. For example, paracrine sources of IL-6 from tumor cells lead to the activation of pStat3 and IL-6 expression in cancer-associated fibroblasts, endothelial cells, and myeloid cells, which in turn enhances a paracrine/autocrine IL-6/JAK/Stat3 signaling loop within the stroma-rich areas of a tumor [8,9,13]. It would also suggest an interdependence between tumor and stromal cells to positively regulate IL-6 expression in cancers. Additionally, differential expression of the negative regulators of IL-6 and pStat3 expression (e.g., the nuclear hormone receptors, suppressors of cytokine signaling, protein inhibitors of activated Stat3, and tyrosine phosphatases) and the positive regulators of IL-6 [nuclear factor-kappa β (NF-κβ) and CCAAT/enhancer-binding protein β (CEBPβ)] may account for the localized distribution of IL-6/pStat3.
The presence of this regulatory loop is supported by our data demonstrating that genetic disruption of Stat3 in tumor cells led to a reduction in IL-6 expression in the tumor and in the circulation of tumor-bearing mice. As a consequence, pStat3 expression was reduced in tumor-extrinsic stromal cells and in pre-metastatic sites, such as the lungs. Additionally, pharmacological blockade of tumor-derived IL-6/IL-6R signaling led to a reduction in pStat3 in the tumor, including the stroma and pre-metastatic and metastatic sites of disease. Conversely, increasing tumor IL-6 levels led to an increase in pStat3 in tumor and associated stromal cells. Thus, by interfering with IL-6, JAK, or Stat3, we can disrupt the pro-tumorigenic cross talk between tumor and stromal cells.
Interestingly, the in vitro versus in vivo phenotypic consequences of both enhancing or disrupting the IL-6/JAK/Stat3 pathway support the hypothesis that the principal function of this pathway in regulating mammary tumorigenesis is through its effects on the tumor, premetastatic, and metastatic microenvironments. Specifically, increasing IL-6 levels or blocking its activity (IL-6R blocking antibodies, JAK inhibition, or reducing Stat3 levels) had no effects on in vitro growth. In contrast, overexpression of tumor-derived IL-6 induced metastasis and tumor growth, while interfering with this signaling pathway suppressed growth and metastatic progression. These observations are consistent with studies conducted by a number of investigators, which show that the in vitro growth effects of inhibiting JAK/Stat3 signaling are nominal compared to the striking in vivo effects in models of mammary tumorigenesis [11,13,14,47]. Although blockade of the IL-6/JAK/Stat3 pathway prevented tumor growth, there was no evidence of tumor regression, which is consistent with the absence of any direct cytotoxic tumor cell-intrinsic effects upon inhibition of this pathway. While it was suggested that the IL-6/JAK/Stat3 pathway plays a role in the development of tumor-initiating cells [10,48], we saw no evidence to support this in the murine MMTV-PyMT model of mammary tumorigenesis. For example, the absence of Stat3 had no effect on mammosphere formation/growth or on the relative numbers of tumor-initiating cells.
The significance of JAK signaling on the tumor microenvironment is supported by our data demonstrating the growth-suppressive effects of JAK inhibition on a pStat3-/- tumor surrounded by pStat3+/+ stromal cells. Additionally, the effects of IL-6/JAK blockade were more pronounced on metastasis than on primary tumor growth, which we hypothesize is due to the significant contribution of a pStat3+ microenvironment to metastatic progression. For example, reducing Stat3 in myeloid cells alone led to a potent reduction in metastatic growth of melanoma and bladder cancer [18,40]. These observations further support the prominent role of the IL-6 signaling pathway on tumor stroma, including endothelial cells, fibroblasts, and cells of the immune system, which are active participants in vasculogenesis/angiogenesis, inflammatory and immune-suppressive responses, and initiating and promoting tumor progression and metastasis.
The phenotypic consequences of modulating the IL-6/JAK/Stat3 pathway described above correlated with changes in the abundance of cancer-associated fibroblasts, endothelial cells, lymphocytes, and myeloid cells, as well as with changes in their activity. In this study, we observed that both IL-6 and pStat3 levels were highest on the invasive edge of the mammary tumors, which coincided with an abundance of fibroblasts, endothelial cells, and MDSCs (CD33+ in human specimens and CD11b+/Gr1+ cells in murine models). Overexpression of IL-6 in human orthotopic xenograft models increased the levels of these stromal cells, including endothelial (Meca-32+), fibroblast (SMA+), and myeloid cells (CD11b+ and Gr1+) within the tumor and in the pre-metastatic niche, which were essentially reversed upon treatment with IL-6RAb. Additionally, Stat3-/- tumors compared to Stat3+/+ tumors had few endothelial, fibroblast, and myeloid cells. Finally, treatment of tumor-bearing mice with JAKi led to a decrease in the number of MDSCs recruited to the tumor stroma and to the pre-metastatic sites. The link between MDSCs and Stat3 expression in the promotion of metastasis is well established, since the accumulation of MDSCs plays a key role in the formation of the pre-metastatic niche [18,40].
Moreover, the presence of MDSCs within the tumor and metastatic stroma favors the creation of an immunosuppressive environment by triggering T cell anergy, inhibiting host-mediated antitumor responses [49,50]. For example, Stat3 inactivation in myeloid or T cells augmented the effector functions of adoptively transferred T cells inducing tumor regression [18,19]. In this study, we show that targeting the IL-6/JAK/Stat3 pathway resulted in reduced expression levels of Stat3-regulated genes, such as S100a8, S100a9, and IL-1β, in MDSCs isolated from S3-/- or JAKi-treated tumor-bearing mice. Additionally, a concomitant increase in T cell numbers (CD3+, CD4+, CD8+) was observed, suggesting the possible initiation of an antitumor response. This is further supported by a decrease in the proportion of the proinflammatory tumor-promoting macrophages (CD11b+, F4/80+) upon JAKi administration and a reduction in the expression levels of the immunosuppressive M2 markers Il-10, Ccl2, Arg1, Fizz1, and Ym1.
Our results reveal the presence of a feed-forward loop elicited by the tumor that involves IL-6 expression, which correlated with poor prognosis, and subsequent phosphorylation of Stat3 in cells that constitute the tumor, pre-metastatic, and metastatic microenvironments (Figure 9). This IL-6/Stat3-regulated interplay within the tumor stroma determines tumor proliferation, angiogenesis, inflammatory status, and metastatic potential. For example, the observed low levels and hypofunctional properties of MDSCs in association with reduced tumor burden (including immunocompromised xenografts) have a direct effect on angiogenesis as well as on the activity of other tumor-associated cells [20]. However, the lack of regression with IL-6R blockade or JAKi treatment encourages the identification of combination approaches that can enhance an antitumor response, such as the addition of agents that enhance T cell responses such as antibodies blocking programmed cell death-1 or cytotoxic T-lymphocyte antigen 4 and those that target cells for antibody-dependent cellular cytotoxicity or in conjunction with cytotoxic agents [51]. Agents that target tumor stroma, especially macrophages and myeloid cells, are presently being investigated in clinical trials based on encouraging preclinical studies in breast cancer models [52]. We suggest that anti-IL-6/JAK/Stat3 therapies will function as potent antineoplastic agents in those malignancies or conditions, in which the stroma plays a significant role in regulating tumor growth and metastasis.
Figure 9.
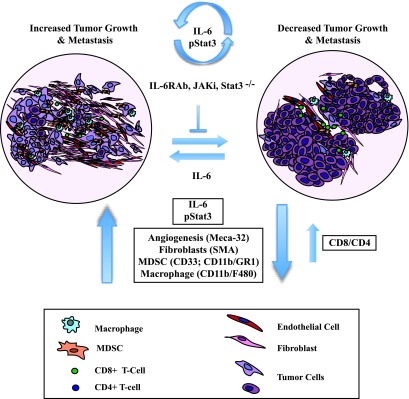
The IL-6/JAK/Stat3 feed-forward loop drives mammary tumorigenesis. A schematic diagram demonstrating that the selective targeting of the IL-6/JAK/Stat3 signaling pathway (IL-6mAb, JAKi, Stat3-/-) has tumor-extrinsic effects on angiogenesis (Meca-32+), fibroblasts (SMA+), and the immune microenvironment [specifically, a decrease in the levels of pro-inflammatory CD11b+/F480+ and CD11b+/Gr1+ cells and an increase in T (CD4+, CD8+) cell responses]. These effects are IL-6-dependent and result in a decrease in tumor size and metastatic potential.
Supplemental Materials and Methods
4175 cell line was provided by Dr Massague (MSKCC) and express a reporter construct encoding GFP and firefly luciferase for bioluminescent tracking [1].
Stat3-shRNA lentiviral and scrambled control shRNA constructs were previously described [2]. Murine Stat3 retroviral construct was introduced into the 4175ShS3 cells to assess for off-target effects of the Stat3-ShRNA.
4175 cells were cross-linked with 1% formaldehyde before being harvested and sonicated. ChIP was performed as previously described [3]. PCR was done with IL-6 promoter primers (sense, 5′-AAACTTCGTGCATGACTTCAGC-3′; antisense, 5′-CCAGTCCTATATTTATTGGGGG′). The supernatant before IP was used as an input control.
Tumors were fixed in 4% paraformaldehyde and embedded in paraffin. Tissue sections were stained for IL-6 using anti-IL-6 (1:200; Abcam, Cambridge, MA) using previously described methods [2]. Imaging was carried out on a Leica Inverted Confocal Microscope. All other methods are described in the manuscript.
PyMT control and Stat3-/- PyMT cell lines were generated from tumors derived from PyMT and Stat3-/- PyMT transgenic mice. Tumors were dissected and digested (collagenase III and liberase; SMEM medium; Invitrogen; 37°C, 3 hours). Isolated cells were cultured in WIT media (low attachment plates).
Supplementary Material
Acknowledgments
AstraZeneca provided AZD1480. We thank Hua Yu (City of Hope), Fred Miller (Michigan Cancer Foundation), and Joan Massague (MSKCC) for kindly providing animals and cell lines for this study. We also thank the members of our laboratories for helpful discussions and the members of the MSKCC Animal Imaging, Antitumor Assessment, and Molecular Cytology core facilities (particularly Huiyong Zhao, Juan Qiu, Afsar Barlas, Sho Fujisawa, and Volodia D. Gueorguiev).
Abbreviations
- IL-6
interleukin-6
- JAK
Janus kinase
- LVI
lymphovascular invasion
- MFP
mammary fat pad
- MDSCs
myeloid-derived suppressor cells
- Stat3
signal transducer and activator of transcription 3
- TN
triple negative
Footnotes
Our work was supported by grants from the National Institutes of Health [U54: CA148967 (J.B. and G.A.-B.) and R01: CA87637 (J.B.)], Charles and Marjorie Holloway Foundation (J.B.), Sussman Family Fund (J.B.), Lerner Foundation (J.B.), AstraZeneca (J.B.), Breast Cancer Alliance (J.B.), Manhasset Women's Coalition Against Breast Cancer (J.B.), NYS Women's Bowling Association (J.B.), American Hellenic Educational Progressive Association 5th District (E.B. and D.L.), Department of Defense (Postdoctoral Award W81XWH-10-1-1013) and Fondazione Carisbo di Bologna (P.S.), Children's Cancer and Blood Foundation (D.L.), The Manning Foundation (D.L.), The Hartwell Foundation (D.L.), Pediatric Oncology Experimental Therapeutics Investigators Consortium (D.L.), Stavros S. Niarchos Foundation (D.L.), Champalimaud Foundation (D.L.), The Nancy C. and Daniel P. Paduano Foundation (D.L.), The Mary Kay Foundation (D.L.), The Malcolm Hewitt Wiener Foundation (D.L.), The George Best Costacos Foundation (D.L.), National Cancer Institute [R01CA 098234-01 and U54-CA143836 PSOC training grant (D.L.)], Susan G. Komen for the Cure (D.L.), and The Beth C. Tortolani Foundation (J.B. and D.L.). J.B. has consulted for Roche, Medimmune, and Bristol-Myers Squibb and has received research support from AstraZeneca. No potential conflicts of interest were disclosed by the other authors.
This article refers to supplementary materials, which are designated by Table W1 and Figures W1 to W4 and are available online at www.neoplasia.com.
References
- 1.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu H, Jove R. The STATs of cancer—new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 3.Dolled-Filhart M, Camp RL, Kowalski DP, Smith BL, Rimm DL. Tissue microarray analysis of signal transducers and activators of transcription 3 (Stat3) and phospho-Stat3 (Tyr705) in node-negative breast cancer shows nuclear localization is associated with a better prognosis. Clin Cancer. 2003;Res 9:594–600. [PubMed] [Google Scholar]
- 4.Walker SR, Nelson EA, Zou L, Chaudhury M, Signoretti S, Richardson A, Frank DA. Reciprocal effects of STAT5 and STAT3 in breast cancer. Mol Cancer Res. 2009;7:966–976. doi: 10.1158/1541-7786.MCR-08-0238. [DOI] [PubMed] [Google Scholar]
- 5.Berishaj M, Gao SP, Ahmed S, Leslie K, Al-Ahmadie H, Gerald WL, Bornmann W, Bromberg JF. Stat3 is tyrosine-phosphorylated through the interleukin-6/glycoprotein 130/Janus kinase pathway in breast cancer. Breast Cancer Res. 2007;9:R32. doi: 10.1186/bcr1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sonnenblick A, Shriki A, Galun E, Axelrod JH, Daum H, Rottenberg Y, Hamburger T, Mali B, Peretz T. Tissue microarray-based study of patients with lymph node-positive breast cancer shows tyrosine phosphorylation of signal transducer and activator of transcription 3 (tyrosine705-STAT3) is a marker of good prognosis. Clin Transl Oncol. 2012;14:232–236. doi: 10.1007/s12094-012-0789-z. [DOI] [PubMed] [Google Scholar]
- 7.Azare J, Doane A, Leslie K, Chang Q, Berishaj M, Nnoli J, Mark K, Al-Ahmadie H, Gerald W, Hassimi M, et al. Stat3 mediates expression of autotaxin in breast cancer. PLoS One. 2011;6:e27851. doi: 10.1371/journal.pone.0027851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, Taffurelli M, Ceccarelli C, Santini D, Paterini P, Marcu KB, et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest. 2007;117:3988–4002. doi: 10.1172/JCI32533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sullivan NJ, Sasser AK, Axel AE, Vesuna F, Raman V, Ramirez N, Oberyszyn TM, Hall BM. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene. 2009;28:2940–2947. doi: 10.1038/onc.2009.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, Bloushtain-Qimron N, Kim JJ, Choudhury SA, Maruyama R, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44+CD24- stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011;121:2723–2735. doi: 10.1172/JCI44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ranger JJ, Levy DE, Shahalizadeh S, Hallett M, Muller WJ. Identification of a Stat3-dependent transcription regulatory network involved in metastatic progression. Cancer Res. 2009;69:6823–6830. doi: 10.1158/0008-5472.CAN-09-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barbieri I, Quaglino E, Maritano D, Pannellini T, Riera L, Cavallo F, Forni G, Musiani P, Chiarle R, Poli V. Stat3 is required for anchorage-independent growth and metastasis but not for mammary tumor development downstream of the ErbB-2 oncogene. Mol Carcinog. 2010;49:114–120. doi: 10.1002/mc.20605. [DOI] [PubMed] [Google Scholar]
- 13.Xin H, Herrmann A, Reckamp K, Zhang W, Pal S, Hedvat M, Zhang C, Liang W, Scuto A, Weng S, et al. Antiangiogenic and antimetastatic activity of JAK inhibitor AZD1480. Cancer Res. 2011;71:6601–6610. doi: 10.1158/0008-5472.CAN-11-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, Buettner R, Proia D, Kowolik CM, Xin H, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–497. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 16.Kaplan RN, Psaila B, Lyden D. Bone marrow cells in the ‘premetastatic niche’: within bone and beyond. Cancer Metastasis Rev. 2006;25:521–529. doi: 10.1007/s10555-006-9036-9. [DOI] [PubMed] [Google Scholar]
- 17.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kortylewski M, Swiderski P, Herrmann A, Wang L, Kowolik C, Kujawski M, Lee H, Scuto A, Liu Y, Yang C, et al. In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat Biotechnol. 2009;27:925–932. doi: 10.1038/nbt.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herrmann A, Kortylewski M, Kujawski M, Zhang C, Reckamp K, Armstrong B, Wang L, Kowolik C, Deng J, Figlin R, et al. Targeting Stat3 in the myeloid compartment drastically improves the in vivo antitumor functions of adoptively transferred T cells. Cancer Res. 2010;70:7455–7464. doi: 10.1158/0008-5472.CAN-10-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kujawski M, Kortylewski M, Lee H, Herrmann A, Kay H, Yu H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest. 2008;118:3367–3377. doi: 10.1172/JCI35213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, Niu G, Kay H, Mule J, Kerr WG, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11:1314–1321. doi: 10.1038/nm1325. [DOI] [PubMed] [Google Scholar]
- 22.Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoshizaki K, Nishimoto N, Mihara M, Kishimoto T. Therapy of rheumatoid arthritis by blocking IL-6 signal transduction with a humanized anti-IL-6 receptor antibody. Springer Semin Immunopathol. 1998;20:247–259. doi: 10.1007/BF00832010. [DOI] [PubMed] [Google Scholar]
- 24.Sinha P, Clements VK, Ostrand-Rosenberg S. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol. 2005;174:636–645. doi: 10.4049/jimmunol.174.2.636. [DOI] [PubMed] [Google Scholar]
- 25.Gao SP, Mark KG, Leslie K, Pao W, Motoi N, Gerald WL, Travis WD, Bornmann W, Veach D, Clarkson B, et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest. 2007;117:3846–3856. doi: 10.1172/JCI31871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH, Levy DE, Settleman J, Engelman JA, Bardeesy N. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 2011;71:5020–5029. doi: 10.1158/0008-5472.CAN-11-0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ernst M, Putoczki TL. Stat3: linking inflammation to (gastrointestinal) tumourigenesis. Clin Exp Pharmacol Physiol. 2012;39:711–718. doi: 10.1111/j.1440-1681.2011.05659.x. [DOI] [PubMed] [Google Scholar]
- 28.Lee H, Deng J, Kujawski M, Yang C, Liu Y, Herrmann A, Kortylewski M, Horne D, Somlo G, Forman S, et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat Med. 2010;16:1421–1428. doi: 10.1038/nm.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hartman ZC, Yang XY, Glass O, Lei G, Osada T, Dave SS, Morse MA, Clay TM, Lyerly HK. HER2 overexpression elicits a proinflammatory IL-6 autocrine signaling loop that is critical for tumorigenesis. Cancer Res. 2011;71:4380–4391. doi: 10.1158/0008-5472.CAN-11-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jatoi I, Hilsenbeck SG, Clark GM, Osborne CK. Significance of axillary lymph node metastasis in primary breast cancer. J Clin Oncol. 1999;17:2334–2340. doi: 10.1200/JCO.1999.17.8.2334. [DOI] [PubMed] [Google Scholar]
- 31.Kang JW, Park YS, Lee DH, Kim JH, Kim MS, Bak Y, Hong J, Yoon DY. Intracellular interaction of interleukin (IL)-32α with protein kinase Cε (PKCε) and STAT3 protein augments IL-6 production in THP-1 promonocytic cells. J Biol Chem. 2012;287:35556–35564. doi: 10.1074/jbc.M112.400911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoon S, Woo SU, Kang JH, Kim K, Kwon MH, Park S, Shin HJ, Gwak HS, Chwae YJ. STAT3 transcriptional factor activated by reactive oxygen species induces IL6 in starvation-induced autophagy of cancer cells. Autophagy. 2010;6:1125–1138. doi: 10.4161/auto.6.8.13547. [DOI] [PubMed] [Google Scholar]
- 33.Lin EY, Jones JG, Li P, Zhu L, Whitney KD, Muller WJ, Pollard JW. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol. 2003;163:2113–2126. doi: 10.1016/S0002-9440(10)63568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Winter SF, Hunter KW. Mouse modifier genes in mammary tumorigenesis and metastasis. J Mammary Gland Biol Neoplasia. 2008;13:337–342. doi: 10.1007/s10911-008-9089-1. [DOI] [PubMed] [Google Scholar]
- 35.Kouros-Mehr H, Bechis SK, Slorach EM, Littlepage LE, Egeblad M, Ewald AJ, Pai SY, Ho IC, Werb Z. GATA-3 links tumor differentiation and dissemination in a luminal breast cancer model. Cancer Cell. 2008;13:141–152. doi: 10.1016/j.ccr.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dimri G, Band H, Band V. Mammary epithelial cell transformation: insights from cell culture and mouse models. Breast Cancer Res. 2005;7:171–179. doi: 10.1186/bcr1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaplan RN, Rafii S, Lyden D. Preparing the “soil”: the premetastatic niche. Cancer Res. 2006;66:11089–11093. doi: 10.1158/0008-5472.CAN-06-2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deng J, Liu Y, Lee H, Herrmann A, Zhang W, Zhang C, Shen S, Priceman SJ, Kujawski M, Pal SK, et al. S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Cancer Cell. 2012;21:642–654. doi: 10.1016/j.ccr.2012.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166:678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 42.Smith C, Chang MY, Parker KH, Beury DW, Duhadaway JB, Flick HE, Boulden J, Sutanto-Ward E, Soler AP, Laury-Kleintop LD, et al. IDO is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discov. 2012;2:722–735. doi: 10.1158/2159-8290.CD-12-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, Catalano JV, Deininger M, Miller C, Silver RT, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366:799–807. doi: 10.1056/NEJMoa1110557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Borowsky AD, Namba R, Young LJ, Hunter KW, Hodgson JG, Tepper CG, McGoldrick ET, Muller WJ, Cardiff RD, Gregg JP. Syngeneic mouse mammary carcinoma cell lines: two closely related cell lines with divergent metastatic behavior. Clin Exp Metastasis. 2005;22:47–59. doi: 10.1007/s10585-005-2908-5. [DOI] [PubMed] [Google Scholar]
- 45.Alpaugh ML, Tomlinson JS, Shao ZM, Barsky SH. A novel human xenograft model of inflammatory breast cancer. Cancer Res. 1999;59:5079–5084. [PubMed] [Google Scholar]
- 46.Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992;52:1399–1405. [PubMed] [Google Scholar]
- 47.Ling X, Arlinghaus RB. Knockdown of STAT3 expression by RNA interference inhibits the induction of breast tumors in immunocompetent mice. Cancer Res. 2005;65:2532–2536. doi: 10.1158/0008-5472.CAN-04-2425. [DOI] [PubMed] [Google Scholar]
- 48.Korkaya H, Liu S, Wicha MS. Regulation of cancer stemcells by cytokine networks: attacking cancer's inflammatory roots. Clin Cancer Res. 2011;17:6125–6129. doi: 10.1158/1078-0432.CCR-10-2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, Schreiber H. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007;67:425. doi: 10.1158/0008-5472.CAN-06-3037. author reply 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kusmartsev S, Gabrilovich DI. Inhibition of myeloid cell differentiation in cancer: the role of reactive oxygen species. J Leukoc Biol. 2003;74:186–196. doi: 10.1189/jlb.0103010. [DOI] [PubMed] [Google Scholar]
- 51.Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12:237–251. doi: 10.1038/nrc3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Denardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Supplemental References
- 1.Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leslie K, Gao SP, Berishaj M, Podsypanina K, Ho H, Ivashkiv L, Bromberg J. Differential interleukin-6/Stat3 signaling as a function of cellular context mediates Ras-induced transformation. Breast Cancer Res. 2010;12:R80. doi: 10.1186/bcr2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Azare J, Doane A, Leslie K, Chang Q, Berishaj M, Nnoli J, Mark K, Al-Ahmadie H, Gerald W, Hassimi M, et al. Stat3 mediates expression of autotaxin in breast cancer. PLoS One. 2011;6:e27851. doi: 10.1371/journal.pone.0027851. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.