

Key Points
Profiling of the Wnt/β-catenin pathway reveals overexpression of Wnt5a, LEF-1 and TCF-1 in ATL patient cells.
ATL cells overexpress Wnt5a, which enhances osteoclastogenesis and may contribute to the osteolytic bone lesions and hypercalcemia.
Abstract
Adult T-cell leukemia/lymphoma (ATL) is etiologically linked to infection with the human T-cell leukemia/lymphoma virus type 1 (HTLV-I). ATL is classified into 4 distinct clinical diseases: acute, lymphoma, chronic, and smoldering. Acute ATL is the most aggressive form, representing 60% of cases and has a 4-year survival of <5%. A frequent complication and cause of death in acute ATL patients is the presence of lytic bone lesions and hypercalcemia. We analyzed the Wnt/β-catenin pathway because of its common role in cancer and bone remodeling. Our study demonstrated that ATL cells do not express high levels of β-catenin but displayed high levels of LEF-1/TCF genes along with elevated levels of β-catenin (LEF-1/TCF target genes) responsive genes. By profiling Wnt gene expression, we discovered that ATL patient leukemia cells shifted expression toward the noncanonical Wnt pathway. Interestingly, ATL cells overexpressed the osteolytic-associated genes—Wnt5a, PTHLH, and RANKL. We further show that Wnt5a secreted by ATL cells favors osteoclast differentiation and expression of RANK. Our results suggest that Wnt5a is a major contributing factor to the increase in osteolytic bone lesions and hypercalcemia found in ATL patients. Anti-Wnt5a therapy may prevent or reduce osteolytic lesions found in ATL patients and improve therapy outcome.
Introduction
Wnt signaling has a role in embryonic development, adult homeostasis, and disease and acts through the canonical and noncanonical β-catenin pathways. Wnt signaling is required for the self-renewal of normal and neoplastic stem cells in the hematopoietic system, and activation of β-catenin may contribute to acquisition of the self-renewal capacity of leukemia stem cells.1,2 The noncanonical Wnt pathway results in changes in cell polarity, motility, migration, and axon guidance. The noncanonical Wnt pathway also antagonizes the canonical Wnt pathway, which is β-catenin–dependent and plays an important role in cellular proliferation, fate, and differentiation. Activation of the β-catenin pathway has been demonstrated in several cancer types and is involved in the pathogenesis of leukemia/lymphomas, such as acute myelogenous leukemia (AML), chronic lymphocytic leukemia, mantle cell lymphoma, and a subset of T-cell non-Hodgkin lymphomas.3-5 In addition, primary patient samples from acute lymphoblastic leukemia (ALL), AML, and multiple myeloma (MM) have abnormal methylation of Wnt antagonists.6-8 In the absence of Wnt signaling, β-catenin is phosphorylated and degraded by a complex consisting of glycogen synthase kinase 3 (GSK3-β), adenomatous polyposis coli, casein kinase 1, and axin.9 Wnt signaling can relieve β-catenin degradation through activation of Dishevelled, which blocks phosphorylation by GSK3-β.
Wnt signaling is also recognized as a key developmental pathway involved in osteoblast differentiation. Deregulation of, or mutations in, low-density lipoprotein receptor–related protein-5 (LRP5), LRP6, Frizzled-9, Wnt10b, and Wnt5a have been shown to disrupt bone regulation.10 Activation of the noncanonical Wnt pathway by Wnt5a also leads to enhanced osteoclastogenesis by increasing the expression of RANK, which is impaired in mouse knock-outs of Wnt5a or its receptor, Ror2.11
We have previously shown that overexpression of the human T-cell leukemia/lymphoma virus type 1 (HTLV-1) posttranscriptional regulator p30 leads to an increase in phosphorylated GSK3-β on Ser9.12 Phosphorylation of Ser9 on GSK3-β by AKT results in GSK3-β inhibition and subsequent β-catenin activation.13 However, the effects of p30 and Tax, another viral gene involved in initiating tumorigenesis, have not been studied in Wnt/β-catenin signaling. Furthermore, no study has been performed to examine the role of Wnt ligands or the downstream pathways they alter in HTLV-I–infected cells or adult T-cell leukemia/lymphoma (ATL) patient samples. One of the most serious and frequent complications arising in ATL patients is hypercalcemia as a result of increased osteolytic bone lesions. Bone resorption releases growth factors that drive proliferation of tumorigenic cells. Studies have shown that overexpression of bone resorption factors, RANKL, PTHrP, interleukin (IL)-1, and MIP-1α, frequently occur in acute ATL patient samples and are linked to a worsening of symptoms in ATL.14,15
Given the link between Wnt signaling and osteoclastogenesis, we examined the Wnt/β-catenin pathway in HTLV-I–infected cells and ATL patients. Here, we report that in vitro established HTLV-I cell lines activate the Wnt canonical pathway through Tax and p30-mediated increases in β-catenin transcriptional activity. In contrast, primary ATL patient samples do not express detectable β-catenin and instead overexpress LEF-1/TCF factors.
We also found that leukemic cells from ATL patients express high levels of the pro-osteoclastic genes, Wnt5a, RANKL, and PTHLH. Importantly, Wnt5a secreted by ATL cells was able to induce osteoclast differentiation in vitro, which could be prevented by the addition of Wnt5a antibodies to the culture media. Overall, our data support a model in which osteolytic lesions in ATL patients are byproducts of an altered Wnt/β-catenin pathway and overexpression of Wnt5a.
Methods
Cells culture and patient samples
293T and the pre-osteoblastic mouse cell line, C2C12, were obtained from the ATCC (Manassas, VA) and cultured in DMEM with 10% fetal bovine serum. HTLV-I cell lines and the human T-cell lymphoblast cell line, Jurkat, were maintained in RPMI supplemented with 10% fetal bovine serum. The ATL-like cell lines, MT1, TL-Om1, and ED-40515(−), were kindly provided by Dr Michiyuki Maeda (Kyoto University). IL-2 (50 U/mL) was added to the HTLV-I immortalized cell lines, 1185 and LAF. Cells were treated with 2.5 µM MG132 (Sigma-Aldrich, St. Louis, MO) and 10 mM LiCl (Invitrogen, Carlsbad, CA) (see figure legends). Patient samples were obtained after informed consent was provided and in agreement with regulations for the protection of human subjects according to the National Institutes of Health (NIH) guidelines.
RNA extraction, PCR, and real-time quantitative PCR
RNA was extracted using TRIzol (Invitrogen) and treated with DNaseI (Roche Applied Science, Indianapolis, IN). Total RNA was reverse-transcribed using the RNA-to-cDNA synthesis kit (Applied Biosystems, Carlsbad, CA). cDNA was used in polymerase chain reaction (PCR) tests, with GAPDH amplification in nonsaturating conditions used as a control. PCR products were visualized by electrophoresis using 1.8% agarose gels stained with ethidium bromide. Quantitative real-time PCR was performed using RT2 Real-time SYBR Green PCR master mix (SABiosciences, Valencia, CA) on the StepOnePlus Real-time PCR System (Applied Biosystems), with GAPDH expression serving as an internal control. Fold change is calculated as the ratio of normalized expression of the target gene divided by the normalized expression of the control sample. Primers are provided as supplemental data (supplemental Table 1).
Luciferase assays
293T cells were transfected using Polyfect Transfection reagent (Qiagen, Valencia, CA). PMH-p30, pc-Tax, and TOPflash luciferase (which contains 3 copies of the β-catenin–responsive, TCF binding site) were used in transfections. Cell extracts were lysed in 1× passive lysis buffer (Promega, Madison, WI) and analyzed using the Luciferase Reporter Assay System (Promega), according to the manufacturer’s instructions. Luciferase assays were performed at least twice from independent experiments, and results were normalized to Renilla luciferase or protein concentration, as described in the figure legends.
Western blot analysis
Total protein extracts were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and probed with the following antibodies: HA/3F10 (Roche Applied Science), β-catenin (BD Transduction Laboratories, San Jose, CA), pGSK3-β Ser 9 (Cell Signaling, Danvers, MA), GSK3-β(H-76) (Santa-Cruz, Santa Cruz, CA), and actin (C11) (Santa-Cruz). Anti-mouse–Tax antibody (NIH AIDS Reagent Program) was kindly provided by Dr John Brady (NCI).
Cell cycle and flow cytometry
1185 cells were grown with 20% serum and 50 U/mL IL-2 or removed of serum, IL-2, and treated with 0.1µM hydroxyurea (Sigma-Aldrich) for 48 hours. RNA was extracted for real-time PCR or lysates were stained with propidium iodine (Invitrogen) for cell-cycle analysis using the LSRII flow cytometer.
Coculture system and alkaline phosphatase assay
C2C12 cells were seeded onto 0.4-µM Transwell plates one day before induction. MT1-pTRIPZ or MT1-DKK-1 Tet-inducible cell lines were added to the Transwells inserts. When indicated, Wnt5a antibody (R&D Systems, Minneapolis, MN) or Rabbit IgG control antibody (Santa-Cruz), 50 ng/mL rWnt3a (R&D Systems), 30 ng/mL recombinant RANKL (rRANKL) (PeproTech, Rocky Hill, NJ), or 100 ng/mL rBMP-2 (R&D Biosystems) was added to the Transwells. After 48 hours, RNA was extracted from the C2C12 cells and used for real-time quantitative PCR analysis. For alkaline phosphatase (ALP) activity, C2C12 cells were cultured for 5 days, as stated before. Cells were lysed in assay buffer, and enzyme activity was determined after incubation at 30 C using the Alkaline Phosphatase Activity Colorimetric Assay Kit (BioVision, Milpitas, CA). Measurements were taken at 405 nm using an Anthos 2010 microplate reader (Biochrom, Holliston, MA). Enzyme activity was normalized to total protein concentration (Bio-Rad, Hercules, CA). The MT1-DKK-1 cell line was established by cloning DKK-1 into the pTRIPZ inducible, lentiviral vector (Thermo Scientific, Waltham, MA). High titer virus was produced in 293T cells by cotransfection with the VSV-G and pDLN packaging system and used to infect MT1 cells. Stable cell lines were obtained by puromycin selection. DKK-1 expression was induced with 2 µg/mL doxycycline.
Results
The Wnt/β-catenin pathway is constitutively activated in HTLV-I–transformed cells
We first performed Western blot analysis on total cellular extracts derived from HTLV-I cell lines and examined the level of β-catenin. As a reference, we compared expression to the non–HTLV-I-infected t-cell line, Jurkat, because it expresses high levels of β-catenin and has an active Wnt-canonical pathway. Peripheral blood mononuclear cells (PBMCs) were used as a negative control. Results showed significant levels of β-catenin protein expression in the HTLV-I cell lines, MT2, MT4, 1185, and LAF, and lower levels of β-catenin in 2 of the 3 ATL cell lines (Tl-Om1 ED-40515(–)) (Figure 1A and supplemental Figure 1). We then determined whether the HTLV-I cells were responsive to Wnt signaling by examining the expression of nuclear and cytoplasmic β-catenin. β-catenin localization was highly variable among the HTLV-I cell lines, with the majority of cell lines containing both nuclear and cytoplasmic β-catenin (data not shown).
Figure 1.
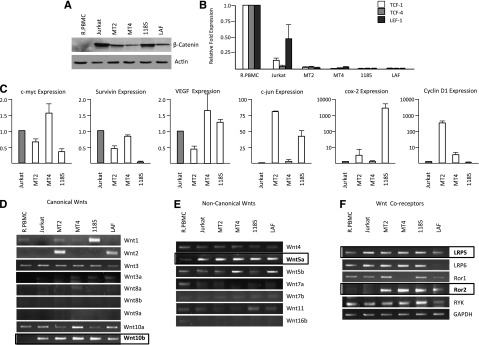
HTLV-I cell lines express varying levels of β-catenin but remain responsive to Wnt stimulation. (A) Western blot analysis of β-catenin expression from total cellular extracts of PBMCs, Jurkat, and HTLV-I cell lines. Extracts were normalized to actin expression. (B) Real-time PCR was performed on β-catenin cotranscription factors, TCF-1, TCF-4, and LEF-1 from cDNA derived from HTLV-I cell lines. Real-time PCR was performed in duplicate and samples were normalized to GAPDH expression. Fold change was calculated by comparing values with resting PBMCs (R.PBMCs). (C) Real-time PCR expression of Wnt/β-catenin downstream target genes, c-myc, survivin, VEGF, c-jun, COX-2, and cyclin D1 from total cDNA from HTLV-I cell lines. Real-time PCR was performed in duplicate. Fold change was calculated by comparing values with Jurkat normalized gene expression. (D-F) PCR amplification of typical canonical Wnts (D), noncanonical Wnts (E), and their coreceptors (F) from cDNA derived from HTLV-I–transformed (MT2 and MT4) and immortalized (1185 and LAF) cell lines. Resting PBMCs (R.PBMCs) and the non–HTLV-I T-cell line, Jurkat, were used as controls. GAPDH amplification in nonsaturating conditions served as a control for the quality and quantity of the samples.
β-catenin interacts with transcriptional coactivators, such as LEF-1, TCF-1, and TCF-4, to activate TCF/LEF-1-target genes. In general, expression of these coactivators in HTLV-I–infected cells were readily detectable (as determined by low Ct values). However, LEF-1/TCF genes were expressed at significantly lower levels when compared with Jurkat T cells or primary PBMCs (Figure 1B). Because of the expression of β-catenin in HTLV-I cell lines, we then examined downstream targets that are representative of an active Wnt/β-catenin pathway. Consistent with the elevated levels of β-catenin, we found high expression of the target genes, c-myc, survivin, vascular endothelial growth factor (VEGF), c-jun, COX-2, and cyclin D1 in HTLV-I–transformed cells (Figure 1C). Jurkat was chosen as a reference because these genes are all expressed at high levels in this cell line, and Jurkat cells have an active canonical Wnt pathway. The fact that β-catenin/TCF target genes are differentially expressed among different cell lines suggests that additional cellular pathways or viral proteins may regulate the expression of these genes.
We next sought to investigate whether defects in canonical or noncanonical Wnt signaling might explain the constitutive activation of the β-catenin pathway in HTLV-I–transformed cells. Canonical Wnt signaling is typically associated with an active β-catenin signal. Gene expression for canonical Wnts (Wnt1, Wnt2, Wnt3, Wnt3a, Wnt8a, Wnt8b, Wnt9a, Wnt10a, and Wnt10b) was monitored by PCR analysis on cDNA from HTLV-I cells (MT2, MT4, 1185, and LAF) and ATL cell lines (MT1, ED-40515(–), and TL-Om1) and was compared with the HTLV-I–negative controls Jurkat and PBMCs. We also monitored the expression of noncanonical Wnts (Wnt4, Wnt5a, Wnt5b, Wnt7a, Wnt7b, Wnt11, and Wnt16b), which are typically independent of β-catenin and regulate diverse pathways such as planar cell polarity or calcium signaling, as well as various Wnt coreceptors (LRP5, LRP6, Ror1, Ror2, and RYK). Our results showed increased expression of Wnt1 and/or Wnt2 in some HTLV-I cell lines (Figure 1D). Wnt3 and Wnt10a were expressed in all HTLV-I cell lines, including controls, whereas Wnt3a, Wnt8a, Wnt8b, and Wnt9a were not detected in any of the HTLV-I cell lines, ATL cell lines (supplemental Figure 1) or the controls PBMCs and Jurkat. The only canonical Wnt gene that was expressed in HTLV cells compared with PBMCs was Wnt10b. Previous studies showed that Wnt10b promotes osteoblastogenesis and acts as a regulator of bone formation.16 Analyses of the noncanonical Wnts revealed no change in Wnt4 and absence of Wnt7a, Wnt7b, Wnt11, and Wnt16b expression (Figure 1E and supplemental Figure 1). However, Wnt5a was strongly expressed in all HTLV-I cell lines (Figure 1E) and ATL lines (supplemental Figure 1). Wnt5a is a key noncanonical Wnt ligand and has roles in β-catenin–independent pathways, including proliferation, differentiation, migration, adhesion, and polarity.17 The analyses of Wnt coreceptors demonstrated similar expression patterns to PBMCs, with the exception of LRP5 and tyrosine kinase–like orphan receptor 2 (Ror2). The data suggested that Ror2 is highly expressed in HTLV-I cell lines (Figure 1F).
Viral proteins Tax and p30 increase β-catenin transcriptional activity in HTLV-I–infected cells
Previous studies showed that the assembly of a signalosome-based Wnt/β-catenin complex inhibits GSK3-β activation, a negative regulator of β-catenin. This process involves Wnt5a, Frizzled-7, Ror2, and Dishevelled, triggering phosphorylation of low-density LRP, which then acts as a competitive inhibitor of GSK-3β. Accordingly, the elevated levels of Wnt5a and Ror2 found in HTLV-I–transformed cells would predict decreased phosphorylation of GSK3-β at Ser9. Our results, however, clearly indicated that GSK3-β is inactivated by increased Ser9 phosphorylation in HTLV-I–transformed cells (Figure 2A) and some ATL lines (supplemental Figure 1). This also suggests that although the combination of high Wnt5a and Ror2 expression may induce Wnt5a-mediated inhibition of canonical signaling, it may not be the case in HTLV-I cell lines. In fact, recent studies with Ror1 and Ror2 double knockouts demonstrate that these receptors are not required for Wnt5a-mediated effects on canonical Wnt signaling.18 Instead Wnt5a may be competing with other Wnts in binding to LRP5/6 or other Frizzled receptors, and may have effects other than targeting GSK3-β.
Figure 2.
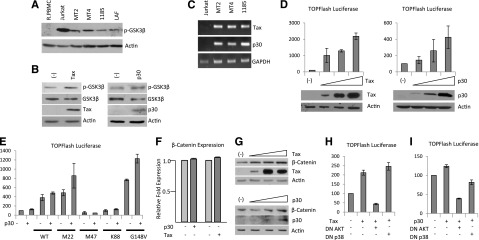
Tax and p30 increase β-catenin transcriptional activity through AKT and/or p38 pathways. (A) Western blot analysis of phosphorylated GSK3-β (Ser9) expression from total cellular extracts of R.PBMCs, Jurkat, and HTLV-I cell lines. Extracts were normalized to actin expression. (B) Western blot analysis of total cellular extract for phosphorylated GSK3-β (Ser9) from 293T cells transfected with pc-Tax (0.5 µg) or PMH-p30 (2.0 µg). Extracts were analyzed 48 hours after transfection and normalized to actin expression. Western blots were stripped and reprobed for GSK3-β expression. Tax and HA-p30 expression were demonstrated in the transfected lysates. (C) PCR analysis of Tax and p30 expression in HTLV-I cell lines. Jurkat cDNA was used as a negative control. (D) pc-Tax (0.1, 0.2, and 0.5 µg) and PMH-p30 (0.5, 1.0, and 2.0 µg) were transfected into 293T cells along with the TOPflash luciferase reporter plasmid. Cellular extracts were used in luciferase assays to measure the level of β-catenin activity. All experiments were performed in duplicate, and values represent the average reading normalized to Renilla luciferase or protein concentration. Error bars represent the population standard deviation for each sample. (E) 293T cells were transfected with or without 1.0 µg PMH-p30, along with 0.1 µg of wild-type or mutant Tax and the TOPflash reporter plasmid, and analyzed as in (D). (F) 293T cells were transfected as in (B) and analyzed for β-catenin expression by real-time PCR. Real-time PCR was performed in duplicate, and samples were normalized to GAPDH expression. Fold change was calculated by comparing values with PMH-transfected cell–normalized gene expression. (G) 293T cells were transfected as in (D). Total extracts were analyzed 48 hours after transfection and normalized to actin expression. Tax and HA-p30 expression were demonstrated in the transfected lysates. (H-I) 293T cells were transfected in duplicate with 0.1 µg pc-Tax (H) or 1.0 µg PMH-p30 (I), along with 1.0 µg of dominant-negative AKT (DN AKT) or dominant-negative p38 (DN p38), and the TOPflash reporter plasmid. Values represent the average reading normalized to protein concentration. Error bars represent the population standard deviation for each sample.
Several pathways are known to target GSK3-β for degradation, including growth factors that stimulate the mitogen-activated protein kinase, phosphoinositide-3-kinase (PI3K)/AKT, or Wnt pathways. Given the fact that the PI3K/AKT pathway is active in HTLV cells, we hypothesized that PI3K/AKT may be inhibiting GSK3-β.19 We found that inhibition of PI3K kinase activity with LY294002 led to a loss of p-GSK3β, resulting in decreased β-catenin expression in all cell lines (with the exception of MT2, for which LY294002 did not inhibit GSK3-β) (data not shown). Together these data suggest that in HTLV-I cell lines, β-catenin expression is in part regulated through the inhibition of GSK3-β activity. Previous studies have demonstrated that MT2, MT4, 1185, LAF, and MT1– express detectable levels of Tax, whereas ED-404515(–) and Tl-Om1 have no detectable Tax expression (and low levels of β-catenin; supplemental Figure 1), suggesting that Tax may play a role in inactivation of GSK3-β. In addition, we have previously published that another viral factor, p30, increases phosphorylation and inactivates GSK3-β in human monocytes.12 Therefore, we hypothesized that both Tax and p30 might be responsible for the constitutive activation of the β-catenin pathway in HTLV-I–transformed cells. Transient transfection of Tax or p30 independently was sufficient to increase the levels of inactive p-GSK3-β at Ser9 (Figure 2B). Consistent with these results and the data presented in Figure 1C, HTLV-I cell lines express Tax and p30 (Figure 2C).
To confirm increased β-catenin transcriptional activity by Tax and p30, we transfected 293T cells with a TCF-reporter plasmid and measured the level of β-catenin activity. Tax expression led to a dose-dependent increase in β-catenin transcriptional activity (Figure 2D). Similar to Tax, we found p30 could also induce β-catenin transcriptional activity, albeit at a lower level (Figure 2D). In addition, we found that the combination of Tax and p30 viral genes could stimulate expression of β-catenin target genes (data not shown).
To gain insights into the mechanism(s) used by Tax, we used Tax and Tax mutants, which are impaired in the activation of either the NF-κB (M22 and G148V) or cAMP-responsive element binding protein/activating transcription factor (CREB/ATF) (M47 and K88) pathways. Tax mutants M22 and G148V enhanced β-catenin transcriptional activity in the presence or the absence of added p30 (Figure 2E). However, M47 and K88 were not able to significantly activate β-catenin transcriptional activity above basal levels (Figure 2E). These results demonstrate that Tax-mediated activation of β-catenin occurs in a CREB/ATF-dependent manner, and independently of NF-κB activation. Interestingly, coexpression of p30 enhanced Tax-induced β-catenin transcriptional activity (Figure 2E). Our data also suggest that p30 increases Tax’s effect on β-catenin transcriptional activity, only when a Tax mutant retaining CREB activity is used. These results further imply that signaling pathways used by p30 and Tax are distinct. Although neither Tax nor p30 altered β-catenin gene expression (Figure 2F), we found that Tax and p30 expression led to an increase in the total amount of β-catenin protein (Figure 2G).
Because phosphorylation on serine 9 of GSK3-β is mediated by the MAP kinase-activated protein kinase-1 or AKT pathways to suppress GSK3-β–mediated degradation of β-catenin, we transfected 293T cells with Tax and a dominant-negative form of AKT or p38. Dominant-negative AKT inhibited Tax-mediated transactivation of the TOPflash reporter plasmid, whereas a dominant-negative form of p38 did not (Figure 2H). This suggests that Tax-mediated activation of β-catenin transcriptional activity occurs in an AKT-dependent, p38-independent manner. In contrast, both AKT and p38 are involved in p30-mediated activation of β-catenin activity, because activation of β-catenin transcriptional activity by p30 was inhibited by both AKT and p38 dominant-negative plasmids (Figure 2I).
Absence of active β-catenin is compensated by elevated levels of LEF-1/TCF transcriptional coactivators in ATL patient samples
Surprisingly, we found no detectable full-length β-catenin protein in freshly isolated ATL patient samples (Figure 3A). However, high levels of cleaved β-catenin were detected. This observation was not the result of nonspecific protein degradation in our protein samples, as shown by detection of STAT3, a protein with similar size to full-length β-catenin. In contrast to HTLV-I cell lines transformed in vitro, only a few ATL patients displayed inactivated phosphorylated GSK3-β (Figure 3A). Because the low levels of full-length β-catenin observed in ATL patients correlated with increased active GSK3-β, and not with β-catenin RNA expression levels (Figure 3B), we hypothesized that GSK3-β may target β-catenin to the proteasome. Treatment with MG132, to inhibit proteasome-mediated processing, led to a significant increase in the levels of full-length and mono-ubiquitinated β-catenin in ATL cell lines, ED-40515(–) and TL-Om1 (Figure 3C). These results suggest that the absence of full-length β-catenin in ATL cells was related to intracellular processing through the proteasome. We further hypothesized that if β-catenin is targeted for degradation by nonphosphorylated, active GSK3-β, treatment of cells with lithium chloride (LiCl), an inhibitor of GSK3-β, should restore β-catenin expression. In fact when ATL cells ED-40515(–) or Tl-Om1 were treated with LiCl, the levels of full-length β-catenin significantly increased (Figure 3D). Despite the low levels of full-length β-catenin expression in ATL cells, the β-catenin target genes, c-myc, survivin, VEGF, c-jun, COX-2, and cyclin D1, were highly expressed in ATL cells with little Tax or p30 expression (Figure 3E). We also found that in general, LEF-1/TCF-1 were overexpressed and higher than TCF-4 gene expression, except in the cases of 2 different patients (ATL5 and ATL7) (Figure 3F). In 2 additional ATL patients, the pattern was the opposite, and TCF-4 was expressed at higher levels than LEF-1/TCF-1 (ATL6 and ATL15).
Figure 3.
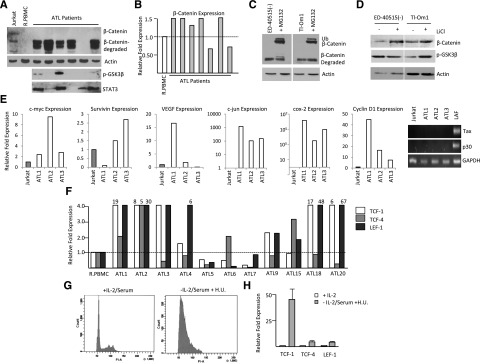
ATL patient samples express high levels of β-catenin cotranscriptional genes, TCF-1 and LEF-1. (A) Western blot analysis of β-catenin, phosphorylated GSK3-β (Ser9), and STAT3 expression from total cellular extracts of 6 ATL patient samples. Extracts were normalized to actin expression. (B) Real-time PCR analysis of β-catenin expression from cDNA from ATL patients. Samples were normalized to GAPDH expression, and the fold change was calculated by comparing values with R.PBMCs’ normalized gene expression. (C) ATL cell lines were treated with MG132 (2.5 µM) or dimethyl sulfoxide control for 8 hours, followed by Western blot analysis on total cellular extract with β-catenin antibody. The upper band corresponds to mono-ubiquitinated β-catenin, the middle band corresponds to wild-type β-catenin, and the lower band corresponds to degraded β-catenin after MG132 treatment. (D) ATL cell lines were treated with 10 mM LiCl for 24 hours. Total cell lysates were normalized to actin expression before Western blot analysis with β-catenin or p-GSK3β (Ser9). (E) Real-time PCR expression of Wnt/β-catenin downstream target genes, c-myc, survivin, VEGF, c-jun, COX-2, and cyclin D1from total cDNA from ATL patients. Fold change was calculated by comparing values with Jurkat-normalized gene expression. The ATL patient cDNAs were additionally analyzed for Tax and p30 expression by qualitative PCR. Jurkat and LAF cDNA were used as negative and positive controls, respectively. (F) Real-time PCR for TCF-1, TCF-4, and LEF-1 from cDNA derived from ATL patients. Real-time PCR was performed in duplicate and samples were normalized to GAPDH expression. Fold change was calculated by comparing values with R.PBMC-normalized gene expression. (G) Cell cycle analysis of 1185 cells treated with or without IL-2, serum, and hydroxyurea after 48 hours. (H) Cells were treated as in (G) and analyzed by real-time PCR analysis. Real-time PCR was performed in duplicate and samples were normalized to GAPDH expression. Fold change was calculated by comparing values with nontreated 1185 cells’ normalized gene expression.
We then analyzed the underlying mechanism by which LEF-1 and TCF expression was increased in ATL patient samples compared with HTLV-I cell lines. In vitro, HTLV-I cell lines are highly proliferative, whereas ATL patient samples exhibit moderate growth and do not exhibit excessive proliferation ex vivo. To test whether LEF-1 and TCF expression was dependent on the level of proliferation, we removed IL-2 and serum from the HTLV-I immortalized cell line, 1185, which is dependent on exogenous IL-2 for growth.20 The cells were treated with hydroxyurea to arrest cells in G0/G1 (Figure 3G) to determine the effect on LEF-1, TCF-1, and TCF-4 expression. After cell-cycle arrest, we found a significant increase in LEF-1 (5-fold), TCF-4 (5-fold), and TCF-1 (50-fold) expression, as seen by real-time PCR analysis (Figure 3H). This suggests that in slow or nonproliferating cells, such as ATL patient samples, LEF-1/TCF gene expression is enhanced and may compensate for the lack of a constitutively active Wnt/β-catenin pathway.
Profiling of the Wnt pathway in ATL patient samples demonstrates activation of noncanonical Wnts and high levels of Wnt5a
We next analyzed ATL patient samples for expression of the Wnt pathway. Gene expression for canonical Wnts (Wnt1, Wnt2, Wnt3, Wnt3a, Wnt8a, Wnt8b, Wnt9a, Wnt10a, and Wnt10b) was monitored by PCR analysis on cDNA from freshly isolated ATL PBMC samples and compared with HTLV-I–negative PBMCs as a control. We also monitored the expression of noncanonical Wnts (Wnt4, Wnt5a, Wnt5b, Wnt7a, Wnt7b, Wnt11, and Wnt16b) and Wnt coreceptors (LRP5, LRP6, Ror1, Ror2, and RYK). Overall, some differences were observed between ATL samples and HTLV-I–transformed cell lines in vitro. Wnt10b and Ror2 were highly expressed in cell lines but were absent in ATL samples (Figure 4A,C). A common feature to both HTLV-I cell lines and ATL samples was the high levels of Wnt5a expression (Figure 4B). To further confirm this data and get more accurate results, we performed real-time PCR for Wnt5a. Our data showed overexpression of Wnt5a in approximately 75% of acute ATL samples (Figure 4D). In addition to Wnt5a’s role in Wnt signaling, it has also been shown to enhance osteoclastogenesis.11 This is significant because hypercalcemia and osteolytic bone lesions are frequently reported in patients with acute ATL.21 This suggests that Wnt5a may play a role in bone resorption and the subsequent hypercalcemia in acute ATL patients. Thus, we compared the levels of PTHLH (PTHrP, parathyroid hormone–like hormone) and RANKL (TNFSF11, tumor necrosis factor [ligand] superfamily, member 11), which have previously been reported to be elevated in ATL patients and are involved in hypercalcemia.15,22 We found that these genes were also overexpressed in about 85% of ATL patients (Figure 4E-F). These results suggest that ATL cells display constitutive activation of the noncanonical Wnt pathway and that elevated Wnt5a may be implicated in bone resorption.
Figure 4.
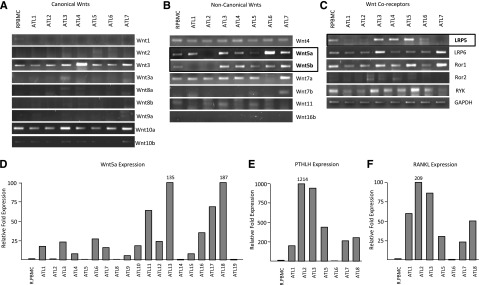
Wnt gene profiling demonstrates elevated expression of Wnt5a in ATL patient samples. (A-C) PCR amplification of typical canonical Wnts (A), noncanonical Wnts (B), and their coreceptors (C) from cDNA derived from ATL patient samples. R.PBMCs were used as controls. GAPDH amplification in nonsaturating conditions served as a control for the quality and quantity of the samples. Open boxes highlight genes differentially expressed between R.PBMCs and ATL patients. (D-F) Real-time PCR analysis of Wnt5a, PTHLH, and RANKL expression from cDNA derived from ATL patients. Samples were normalized to GAPDH expression and the fold change was calculated by comparing values to R.PBMCs normalized gene expression.
Wnt5a secreted by ATL cells induces osteoclast differentiation
In addition to ATL patient samples, we also found that Wnt5a was overexpressed in ATL cell lines (Figure 5A and data not shown). The levels were significantly elevated compared with PBMCs and Jurkat cells, which are reported to have low levels of Wnt5a because of promoter methylation. To test the role of the canonical and noncanonical Wnt pathways in osteoclastogenesis in ATL patients, we used the myoblast cell line, C2C12, which can differentiate into osteoblast or osteoclasts. Because C2C12 cells are very sensitive to culture conditions and spontaneously differentiate if left to reach confluency, we first demonstrated the C2C12 pre-osteoblastic state with the potential to differentiate into osteoclasts in our experimental conditions. We seeded cells from the Wnt5a-expressing ATL cell line, MT1, into a Transwell insert placed over C2C12 cells. As has been previously reported, an increase in the activity of the osteoblastic marker, ALP, was detected in C2C12 cells 5 days after stimulation with recombinant bone morphogenic protein (rBMP-2) (Figure 5B).23 To demonstrate the specificity of these data, we established a DKK-1, Tet-inducible MT1 cell line, in which DKK-1 expression could be induced by doxycycline. In agreement with previously published studies, we found that DKK-1 secreted by MT1 cells was able to inhibit osteoblastic differentiation of C2C12 after rBMP-2 incubation, as shown by reduced levels of ALP activity (Figure 5B). Real-time PCR confirmed that DKK-1 was induced in the MT1 cells (Figure 5C).
Figure 5.
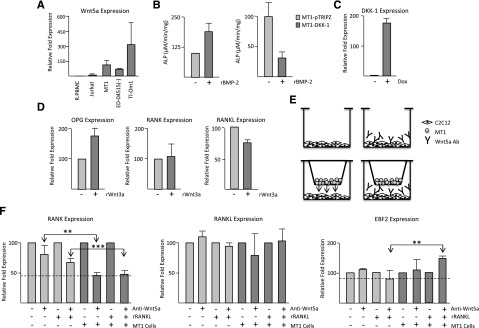
Wnt5a is involved in osteoblast differentiation in ATL cells. (A) Real-time PCR analysis of Wnt5a expression from cDNA derived from ATL cell lines. Samples were normalized to GAPDH expression and the fold change was calculated by comparing values with R.PBMCs’ normalized gene expression. Jurkat expression served as a negative control. (B) C2C12 and doxycycline-treated MT1-DKK1 or MT1-pTRIPZ cells were cultured in 0.4 µM Transwell plates with or without 100 ng/mL rBMP-2. After 5 days, C2C12 cells were lysed and used in alkaline phosphatase assays. ALP activity was measured as the amount of pNP generated in µmol per volume of the sample per reaction time and normalized to protein concentrations. Results are representative of 2 independent experiments. (C) MT1 cells stably expressing DKK-1 were induced with or without 2 µg/mL doxycycline (Dox) for 72 hours. Real-time PCR was performed on cDNA for DKK-1 expression and normalized to GAPDH expression. Results are representative of 2 independent inductions. (D) C2C12 cells were cocultured with MT1-pTRIPZ cells on 0.4 µM Transwell plates. Three hours after culturing, 50 ng/mL rWnt3a was added and cells were grown at subconfluence for an additional 48 hours. RNA was extracted from C2C12 cells and used for real-time PCR analysis. Results are representative of 2 independent experiments. Samples were normalized to GAPDH expression and the fold change was calculated by comparing values with C2C12 cells without rWnt3a-normalized gene expression. (E) Model demonstrating the C2C12 Transwell system used in the study. MT1 cells were seeded in a Transwell insert and C2C12 cells were seeded at subconfluency on the bottom of the plate. Secreted Wnt5a was blocked by the addition of anti-Wnt5a antibody. (F) C2C12 cells were cultured in a Transwell plate with and without MT1-TRIPZ cells and with or without Wnt5a antibody. Three hours after the addition of MT1-pTRIPZ cells and Wnt5a antibody, 30 ng/mL of rRANKL or control was added. Cells were cultured for a further 48 hours, after which RNA was extracted and used for real-time PCR. Results are representative of 2 independent experiments. Samples were normalized to GAPDH expression and the fold change was calculated to control normalized gene expression.
Because ATL patients have decreased expression of the majority of canonical Wnts, we tested whether Wnt3a, the key inducer of Wnt/canonical signaling, could disrupt the OPG/RANKL/RANK system in myoblasts cultured with ATL cells, because this system is the final mediator of osteoclastogenesis.24 Addition of recombinant Wnt3a (rWnt3a) to C2C12 cells cocultured with MT1 cells increased the expression of the osteoblastic marker, OPG, but had no effect on RANK expression (Figure 5D). Consistent with the role of canonical signaling on osteoblastogenesis, rWnt3a mediated a slight downregulation of RANKL, a key activator of osteoclasts (Figure 5D).
We next aimed to demonstrate the role of noncanonical Wnt signaling, as mediated by the secretion of Wnt5a, in osteoclastogenesis in ATL cells. We took advantage of the fact that MT1 cells expressed high levels of Wnt5a (Figure 5A) and set up a Transwell system with myoblasts in the presence or absence of Wnt5a antibodies (Figure 5E). As we expected, blocking Wnt5a signaling with Wnt5a antibodies caused a reduction in RANK expression in the myoblast cells. Our results demonstrate a further, significant reduction in RANK receptor expression when Wnt5a secretion is blocked from MT1 cells in the presence of Wnt5a-specific antibodies (Figure 5F). This is in accordance with published results, demonstrating that Wnt5a mediates bone resorption by increasing RANK expression on pre-osteoblasts.11 Because MT1 cells cannot produce RANKL, we also performed these experiments in the presence of recombinant rRANKL and similar results were obtained. This demonstrates that the Wnt5a produced by ATL cells is sufficient to modulate the expression of RANK in the absence of RANKL. The decrease in RANK expression observed in the presence of Wnt5a antibodies was not linked to nonspecific toxicity, as was shown by unchanged levels of RANKL expression (Figure 5F). Finally, we tested early B-cell factor−2 (EBF2) gene expression, a key regulator of RANK-RANKL signaling and osteoblast-dependent differentiation of osteoclasts.25 We found that inhibition of Wnt5a secretion from ATL cells was sufficient to significantly elevate EBF2 expression in myoblasts (Figure 5F). Wnt5a mediation of EBF2 expression occurred only in the presence of rRANKL. Together our results suggest that in ATL cells, inhibition of canonical signaling and increases in noncanonical signaling through secretion of Wnt5a play an important role in bone lytic lesions and may contribute to hypercalcemia.
Discussion
Hypercalcemia is a severe complication arising in >80% of acute ATL patients and serves as a major prognostic factor for acute ATL disease outcome.26 Osteolytic bone lesions have been reported in acute ATL patients, along with elevated levels of RANKL, MIP-1α, PTHrP, and IL-6, and are believed to contribute to hypercalcemia.14,21,27-29 Our results described herein reveal differential regulation of Wnt/β-catenin signaling between leukemic cells from ATL patients and HTLV-I cell lines. We found readily detectable levels of β-catenin expression in HTLV-I cell lines, with no detectable expression of active β-catenin in ATL patient samples. The large variation in β-catenin expression between HTLV-I cell lines is in agreement with previous reports, which found variable levels of β-catenin protein expression between different leukemia cell lines and even within HTLV-I, Tax-expressing cells.30 Reports demonstrate that the maintenance of chronic myelogenous leukemia stem cells is dependent on the Wnt/β-catenin pathway, whereas other reports demonstrate that inhibition of β-catenin can have different effects on proliferation depending on the initial β-catenin levels and that β-catenin can even induce apoptosis.30,31 Because of the variable levels of β-catenin in HTLV-I cells and ATL patient samples, we believe that cellular dependency on β-catenin will be cell type–specific in HTLV-I–infected cells.
In general, activated β-catenin accumulates in the nucleus, where it acts as a cotranscription factor with LEF-1 and TCF to stimulate transcription of target genes. LEF-1 is expressed in various leukemias and can increase c-myc, cyclin D1, and COX-2 expression.32 Therefore, increased expression of LEF-1 and TCF-1 in ATL cells may sustain activation of target genes, even in the presence of low levels of active β-catenin. How the expression of LEF-1 and TCF-1 is regulated in ATL patients is under consideration but may reflect the undifferentiated state of these cells compared with established, differentiated cell lines. Hematopoietic stem cells also express high levels of LEF-1, whereas LEF-1 and TCF-1 expression are diminished in mature T cells.33 TCF-4 has also been shown to bind and activate the c-myc promoter.34 Furthermore, studies have demonstrated overexpression of TCF-4 in ATL patient samples and its role in activating the anti-apoptotic protein, BIRC5 promoter.35 Our analysis looked at the levels of LEF-1 but did not determine the expression of alternatively spliced isoforms of this gene. It is possible that alternative isoforms of LEF-1 may be expressed in HTLV-I–infected cells, but were not detected in this assay, and may serve an additional role in tumorigenesis.
We also found constitutive phosphorylation of GSK3-β (Ser9), in all HTLV-I–infected cell lines that demonstrated high levels of β-catenin, suggesting that inactivation of GSK3-β plays a vital role in elevating β-catenin protein levels in these cells. We found that the combination of p30 and Tax can act through the AKT and/or p38 pathways to increase β-catenin transcriptional activity. We found that Tax and p30 could increase the expression of genes activated by β-catenin, but whether this occurred indirectly or through β-catenin is still unknown. The loss of β-catenin in ATL patient samples is similar to what is observed in MM, where nuclear β-catenin is lost with progression of disease.36 However, leukemic cells from ATL patients still retained high expression levels of LEF-1 and TCF-1, along with downstream β-catenin targets (c-myc, survivin, VEGF, c-jun, COX-2, and cyclin D1), suggesting that although active β-catenin expression is absent or greatly reduced, leukemic cells from ATL patients have found alternative ways to elevate or mimic activation of β-catenin signaling. This supports the hypothesis that deregulation of at least 1 component of β-catenin signaling is characteristic of leukemic cells.30
The role of Wnt5a in cancer cells has been conflicting and seems to be cancer type–specific. Although Wnt5a is downregulated in AML, ALL, and breast and hepatocellular carcinomas, Wnt5a is overexpressed in melanoma, gastric, pancreatic, prostate, and non–small-cell lung cancers.37 Wnt5a cannot transform cells and instead is believed to advance, but not initiate, certain cancer types. In those cancers with increased Wnt5a expression, Wnt5a was shown to increase cell invasion and proliferation. Wnt5a can signal through Ror2 to induce migration and invasion; however, unlike in HTLV-I cell lines, we did not find expression of Ror2 in leukemic cells from ATL patients. In addition, the absence of Ror2 when LRP5 and Frizzled-4 are overexpressed, leads to Wnt5a-mediated increases in β-catenin transcriptional activities.38 Ror2, a transmembrane receptor for Wnt factors that activates noncanonical pathways, is frequently repressed by aberrant promoter hypermethylation in human colon cancer cell lines and primary tumors, and its epigenetic-dependent loss can be pro-tumorigenic. We also found that Wnt antagonists, DKK-1, -2, and -3, which bind to the LRP6 coreceptor, were downregulated by promoter hypermethylation in HTLV-I–transformed cells and ATL cells and could also be reexpressed after treatment with hypomethylating agents (data not shown). Epigenetic repression of other Wnt inhibitors such as WIF-1, DKK-1, sFRP-1, and sFRP-2 could directly promote tumorigenesis in colon cancer cells by promoting constitutive Wnt signaling.39
Given the role of canonical Wnt signaling in bone development, and the high percentage of ATL patients with bone lesions and hypercalcemia, we expected that expression of noncanonical, pro-osteolytic Wnts would be favored over canonical Wnts that are generally pro-osteoblastic. This is the first report to examine this pathway and the expression of Wnt ligands in HTLV-I–infected cells and leukemic cells from ATL patients. Our data demonstrate that the pro-osteolytic ligand, Wnt5a, is highly expressed in 75% of ATL patients. During the review of this manuscript, another group published a study demonstrating increased Wnt5a expression in HTLV-I cell lines and ATL patients associated with viral HBZ expression.40 Because Wnt5a has been shown to increase osteoclastogenesis by enhancing RANK expression in osteoclast precursors, we investigated whether secreted Wnt5a from ATL cells can have a similar effect. We also tested EBF2 because its expression has been linked to osteoblast differentiation. Our results demonstrated that ATL cells stimulate osteoclast differentiation and secreted Wnt5a is responsible for increases in RANK. In the presence of rRANKL expression, which is also overexpressed in ATL patients, Wnt5a could decrease EBF2 expression. The fact that Jurkat cells, an ALL cell line, do not have high expression of Wnt5a owing to methylation of the Wnt5a promoter further supports our study.41 ALL patients do not normally have lytic bone lesions, whereas ATL patients do, correlating elevated Wnt5a expression with osteoclastogenesis. Therefore, ATL patients may benefit from anti-Wnt5a therapy. Not only could anti-Wnt5a treatment suppress metastasis in ATL, as it has been shown to do in other cancers, it may also reduce osteolytic bone lesions and hypercalcemia levels in ATL patients.8,40,42
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (AI 058944, CA106258) (C.N.), the Intramural Research Program of the National Cancer Institute (T.A.W., J.B.T.), and the Biomedical Research Training Program (M.B.).
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: M.B. performed the experiments shown in Figures 1 through 5, designed and interpreted experiments, and wrote the manuscript; N.L.K. and Y.Y. performed the experiments shown in Figure 2; M.-J.L. and J.B.T. performed the experiments shown in Figure 2 and provided the reagents; T.A.W. provided patient samples; and C.N. designed and interpreted the results.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Christophe Nicot, Center for Viral Oncology, 3901 Rainbow Blvd, 3025 Wahl Hall West, Kansas City, KS 66160; e-mail: cnicot@kumc.edu.
References
- 1.Zhao C, Blum J, Chen A, et al. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell. 2007;12(6):528–541. doi: 10.1016/j.ccr.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guo W, Lasky JL, Chang CJ, et al. Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature. 2008;453(7194):529–533. doi: 10.1038/nature06933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simon M, Grandage VL, Linch DC, Khwaja A. Constitutive activation of the Wnt/beta-catenin signalling pathway in acute myeloid leukaemia. Oncogene. 2005;24(14):2410–2420. doi: 10.1038/sj.onc.1208431. [DOI] [PubMed] [Google Scholar]
- 4.Lu D, Zhao Y, Tawatao R, et al. Activation of the Wnt signaling pathway in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2004;101(9):3118–3123. doi: 10.1073/pnas.0308648100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gelebart P, Anand M, Armanious H, et al. Constitutive activation of the Wnt canonical pathway in mantle cell lymphoma. Blood. 2008;112(13):5171–5179. doi: 10.1182/blood-2008-02-139212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valencia A, Román-Gómez J, Cervera J, et al. Wnt signaling pathway is epigenetically regulated by methylation of Wnt antagonists in acute myeloid leukemia. Leukemia. 2009;23(9):1658–1666. doi: 10.1038/leu.2009.86. [DOI] [PubMed] [Google Scholar]
- 7.Chim CS, Pang R, Fung TK, Choi CL, Liang R. Epigenetic dysregulation of Wnt signaling pathway in multiple myeloma. Leukemia. 2007;21(12):2527–2536. doi: 10.1038/sj.leu.2404939. [DOI] [PubMed] [Google Scholar]
- 8.Roman-Gomez J, Jimenez-Velasco A, Cordeu L, et al. WNT5A, a putative tumour suppressor of lymphoid malignancies, is inactivated by aberrant methylation in acute lymphoblastic leukaemia. Eur J Cancer. 2007;43(18):2736–2746. doi: 10.1016/j.ejca.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 9.Camilli TC, Weeraratna AT. Striking the target in Wnt-y conditions: intervening in Wnt signaling during cancer progression. Biochem Pharmacol. 2010;80(5):702–711. doi: 10.1016/j.bcp.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Long F. Building strong bones: molecular regulation of the osteoblast lineage. Nat Rev Mol Cell Biol. 2012;13(1):27–38. doi: 10.1038/nrm3254. [DOI] [PubMed] [Google Scholar]
- 11.Maeda K, Kobayashi Y, Udagawa N, et al. Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat Med. 2012;18(3):405–412. doi: 10.1038/nm.2653. [DOI] [PubMed] [Google Scholar]
- 12.Datta A, Sinha-Datta U, Dhillon NK, Buch S, Nicot C. The HTLV-I p30 interferes with TLR4 signaling and modulates the release of pro- and anti-inflammatory cytokines from human macrophages. J Biol Chem. 2006;281(33):23414–23424. doi: 10.1074/jbc.M600684200. [DOI] [PubMed] [Google Scholar]
- 13.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378(6559):785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 14.Okada Y, Tsukada J, Nakano K, Tonai S, Mine S, Tanaka Y. Macrophage inflammatory protein-1alpha induces hypercalcemia in adult T-cell leukemia. J Bone Miner Res. 2004;19(7):1105–1111. doi: 10.1359/JBMR.040314. [DOI] [PubMed] [Google Scholar]
- 15.Nosaka K, Miyamoto T, Sakai T, Mitsuya H, Suda T, Matsuoka M. Mechanism of hypercalcemia in adult T-cell leukemia: overexpression of receptor activator of nuclear factor kappaB ligand on adult T-cell leukemia cells. Blood. 2002;99(2):634–640. doi: 10.1182/blood.v99.2.634. [DOI] [PubMed] [Google Scholar]
- 16.Cawthorn WP, Bree AJ, Yao Y, et al. Wnt6, Wnt10a and Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a β-catenin-dependent mechanism. Bone. 2012;50(2):477–489. doi: 10.1016/j.bone.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kikuchi A, Yamamoto H, Sato A, Matsumoto S. Wnt5a: its signalling, functions and implication in diseases. Acta Physiol (Oxf) 2012;204(1):17–33. doi: 10.1111/j.1748-1716.2011.02294.x. [DOI] [PubMed] [Google Scholar]
- 18.Ho HY, Susman MW, Bikoff JB, et al. Wnt5a-Ror-Dishevelled signaling constitutes a core developmental pathway that controls tissue morphogenesis. Proc Natl Acad Sci USA. 2012;109(11):4044–4051. doi: 10.1073/pnas.1200421109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeong SJ, Pise-Masison CA, Radonovich MF, Park HU, Brady JN. Activated AKT regulates NF-kappaB activation, p53 inhibition and cell survival in HTLV-1-transformed cells. Oncogene. 2005;24(44):6719–6728. doi: 10.1038/sj.onc.1208825. [DOI] [PubMed] [Google Scholar]
- 20.Bellon M, Nicot C. Central role of PI3K in transcriptional activation of hTERT in HTLV-I-infected cells. Blood. 2008;112(7):2946–2955. doi: 10.1182/blood-2008-01-134692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiyokawa T, Yamaguchi K, Takeya M, et al. Hypercalcemia and osteoclast proliferation in adult T-cell leukemia. Cancer. 1987;59(6):1187–1191. doi: 10.1002/1097-0142(19870315)59:6<1187::aid-cncr2820590626>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 22.Richard V, Lairmore MD, Green PL, et al. Humoral hypercalcemia of malignancy: severe combined immunodeficient/beige mouse model of adult T-cell lymphoma independent of human T-cell lymphotropic virus type-1 tax expression. Am J Pathol. 2001;158(6):2219–2228. doi: 10.1016/S0002-9440(10)64694-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katagiri T, Akiyama S, Namiki M, et al. Bone morphogenetic protein-2 inhibits terminal differentiation of myogenic cells by suppressing the transcriptional activity of MyoD and myogenin. Exp Cell Res. 1997;230(2):342–351. doi: 10.1006/excr.1996.3432. [DOI] [PubMed] [Google Scholar]
- 24.Khosla S. Minireview: the OPG/RANKL/RANK system. Endocrinology. 2001;142(12):5050–5055. doi: 10.1210/endo.142.12.8536. [DOI] [PubMed] [Google Scholar]
- 25.Kieslinger M, Folberth S, Dobreva G, et al. EBF2 regulates osteoblast-dependent differentiation of osteoclasts. Dev Cell. 2005;9(6):757–767. doi: 10.1016/j.devcel.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 26.Tsukasaki K, Hermine O, Bazarbachi A, et al. Definition, prognostic factors, treatment, and response criteria of adult T-cell leukemia-lymphoma: a proposal from an international consensus meeting. J Clin Oncol. 2009;27(3):453–459. doi: 10.1200/JCO.2008.18.2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujihira T, Eto S, Sato K, et al. Evidence of bone resorption-stimulating factor in adult T-cell leukemia. Jpn J Clin Oncol. 1985;15(2):385–391. [PubMed] [Google Scholar]
- 28.Takaori-Kondo A, Imada K, Yamamoto I, et al. Parathyroid hormone-related protein-induced hypercalcemia in SCID mice engrafted with adult T-cell leukemia cells. Blood. 1998;91(12):4747–4751. [PubMed] [Google Scholar]
- 29.Hino M, Yamane T, Ohta K, et al. Bone resorption associated with uncoupling of osteoclastic and osteoblastic activities in adult T cell leukemia with hypercalcemia: case report. Ann Hematol. 2001;80(7):426–429. doi: 10.1007/s002770100305. [DOI] [PubMed] [Google Scholar]
- 30.Chung EJ, Hwang SG, Nguyen P, et al. Regulation of leukemic cell adhesion, proliferation, and survival by beta-catenin. Blood. 2002;100(3):982–990. doi: 10.1182/blood.v100.3.982. [DOI] [PubMed] [Google Scholar]
- 31.Kim K, Pang KM, Evans M, Hay ED. Overexpression of beta-catenin induces apoptosis independent of its transactivation function with LEF-1 or the involvement of major G1 cell cycle regulators. Mol Biol Cell. 2000;11(10):3509–3523. doi: 10.1091/mbc.11.10.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jesse S, Koenig A, Ellenrieder V, Menke A. Lef-1 isoforms regulate different target genes and reduce cellular adhesion. Int J Cancer. 2010;126(5):1109–1120. doi: 10.1002/ijc.24802. [DOI] [PubMed] [Google Scholar]
- 33.Skokowa J, Cario G, Uenalan M, et al. LEF-1 is crucial for neutrophil granulocytopoiesis and its expression is severely reduced in congenital neutropenia. Nat Med. 2006;12(10):1191–1197. doi: 10.1038/nm1474. [DOI] [PubMed] [Google Scholar]
- 34.He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281(5382):1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 35.Pise-Masison CA, Radonovich M, Dohoney K, et al. Gene expression profiling of ATL patients: compilation of disease-related genes and evidence for TCF4 involvement in BIRC5 gene expression and cell viability. Blood. 2009;113(17):4016–4026. doi: 10.1182/blood-2008-08-175901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chien AJ, Moore EC, Lonsdorf AS, et al. Activated Wnt/beta-catenin signaling in melanoma is associated with decreased proliferation in patient tumors and a murine melanoma model. Proc Natl Acad Sci USA. 2009;106(4):1193–1198. doi: 10.1073/pnas.0811902106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDonald SL, Silver A. The opposing roles of Wnt-5a in cancer. Br J Cancer. 2009;101(2):209–214. doi: 10.1038/sj.bjc.6605174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mikels AJ, Nusse R. Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol. 2006;4(4):e115. doi: 10.1371/journal.pbio.0040115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Veeck J, Dahl E. Targeting the Wnt pathway in cancer: the emerging role of Dickkopf-3. Biochim Biophys Acta. 2012;1825(1):18–28. doi: 10.1016/j.bbcan.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 40.Ma G, Yasunaga J, Fan J, Yanagawa S, Matsuoka M. HTLV-1 bZIP factor dysregulates the Wnt pathways to support proliferation and migration of adult T-cell leukemia cells. Oncogene. 2012 doi: 10.1038/onc.2012.450. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 41.Martín V, Valencia A, Agirre X, et al. Epigenetic regulation of the non-canonical Wnt pathway in acute myeloid leukemia. Cancer Sci. 2010;101(2):425–432. doi: 10.1111/j.1349-7006.2009.01413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hanaki H, Yamamoto H, Sakane H, et al. An anti-Wnt5a antibody suppresses metastasis of gastric cancer cells in vivo by inhibiting receptor-mediated endocytosis. Mol Cancer Ther. 2012;11(2):298–307. doi: 10.1158/1535-7163.MCT-11-0682. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.