

Abstract
Objectives
Adult subjects with untreated, lifetime, isolated GH deficiency (IGHD) due to a homozygous GHRH receptor gene mutation (MUT/MUT) residing in Itabaianinha, Brazil, present with lower BMI, higher prevalence of impaired glucose tolerance (IGT), increased insulin sensitivity (IS), and reduced β-cell function (βCF) when compared with non-BMI-matched homozygous normal controls. However, the prevalence of diabetes mellitus (DM) in this cohort is unknown. Comparing their IS and βCF with BMI-matched individuals heterozygous for the same mutation (MUT/N) may be useful to elucidate the role of the GH–IGF1 axis in IS and βCF. The purposes of this work were to verify the prevalence of IGT and DM in adult MUT/MUT subjects from this kindred and to compare IS and βCF in MUT/MUT and MUT/N.
Design
Cross-sectional study.
Methods
We studied most (51) of the living IGHD adults of this kindred who are GH naive. The oral glucose tolerance test (OGTT) could be performed in 34 subjects, fasting glucose was measured in 15, while two had a previous diagnosis of DM. The OGTT results of 24 MUT/MUT subjects were compared with those of 25 BMI-matched MUT/N subjects. IS was assessed by homeostatic model assessment of insulin resistance (HOMA–IR), quantitative IS check index, and oral glucose IS index for 2 and 3 h. βCF was assayed by HOMA-β, insulinogenic index, and the area under the curve of insulin:glucose ratio.
Results
The prevalence of DM and IGT in IGHD was 15.68 and 38.23% respectively. IS was increased and βCF was reduced in MUT/MUT in comparison with MUT/N.
Conclusions
Lifetime, untreated IGHD increases IS, impairs βCF, and does not provide protection from diabetes.
Keywords: insulin sensitivity, β-cell function, diabetes, GH deficiency
Introduction
GH regulates β-cell function (βCF) and insulin sensitivity (IS) both directly and via complex interactions with its principal mediator, insulin-like growth factor 1 (IGF1) (1). While GH reduces IS, IGF1 increases it. The hyperglycemic action of GH occurs mainly in the liver, and the hypoglycemic action of IGF1 occurs mainly in muscle (2). At least in animal models, IGF1 also has an important role in the development of β-cell mass (3, 4, 5). Genetic forms of GH resistance or GH deficiency (GHD) may be useful to clarify the role of the GH–IGF1 axis in regulating insulin secretion and action in humans.
In contrast to obese individuals, who present with reduced IS, GH-resistant (Laron syndrome) dwarfs present with normal IS – as measured by homeostatic model assessment of insulin resistance (HOMA–IR) – despite increased fat mass percentage (6). In these subjects, βCF – assessed by the insulin response during an oral glucose tolerance test (OGTT) – also seems to be impaired, especially in subjects older than 23 years, with a case of diabetes mellitus (DM) diagnosed in a 38-year-old patient due to βCF exhaustion (7). However, recently, in a large Ecuadorian study of kindred with Laron syndrome, none of the GH-resistant individuals self-reported to have DM (8). This has attracted the attention of scientific and lay media, as has been interpreted as showing that in the absence of GH action, DM does not occur.
We have previously described a large extended kindred with congenital isolated GHD (IGHD) due to the c.57+1G>A mutation in the GHRH receptor (GHRHR) gene (GHRHR), who reside in Itabaianinha county in northeast Brazil (9). Individuals homozygous for this mutation (MUT/MUT) have very low serum GH and IGF1 levels (10), resulting in severe proportionate dwarfism. Despite several cardiovascular risk factors (11), they exhibit normal longevity (12) and no evidence of premature atherosclerosis (13). We have recently studied 24 IGHD subjects and found that, despite an increase in percentage fat mass they do not have insulin resistance, but have impaired βCF and a higher prevalence of impaired glucose tolerance (IGT) when compared with homozygous (N/N) normal controls (14). One important limitation of our previous report was that the MUT/MUT and N/N groups were not matched for BMI. It is difficult to match MUT/MUT with N/N individuals, as reduced BMI is a hallmark of congenital GHD. However, subjects who are heterozygous for the mutation (MUT/N) have a partial phenotype. Despite normal stature and serum IGF1 levels, they have reduced lean body mass and a similar BMI compared with MUT/MUT subjects (15). This renders the MUT/N group an ideal one to compare with the MUT/MUT group.
The purposes of this work were i) to extend the study of prevalence of impaired fasting glucose (IFG) and DM to all the adult IGHD subjects from Itabaianinha and ii) to compare the prevalence of IGT and the degree of IS and βCF in MUT/MUT and MUT/N subjects.
Materials and methods
Design
This was a cross-sectional study performed in Itabaianinha county in the northeastern Brazilian state of Sergipe. Subjects were recruited by an advertisement, placed in the local dwarfs' association building, and by word of mouth. Two groups were enrolled: MUT/MUT (IGHD homozygous individuals for the GHRHR c.57+1G>A mutation) and MUT/N (heterozygous for the same mutation living in the same community). Both the Federal University of Sergipe and the Johns Hopkins University Institutional Boards approved this study. All subjects signed a written informed consent. A two-step protocol was performed: the first epidemiological and the second physiological.
Experiment 1
We have identified 52 living untreated adult MUT/MUT individuals, 26 females, age 45.08 (20.03) years, range 18–97 years. Two of them had a previous diagnosis of DM (one on metformin and the other on metformin and glimepiride) and did not undergo OGTT. Depending on logistics and subjects' availability, some had only fasting glucose, some partial, and some complete OGTT. One individual declined to participate. We measured fasting glucose in 15 individuals, performed OGTT with glucose measurements at 0 and 120 min in 10 individuals, and complete OGTT (with glucose and insulin measurements at 0, 30, 60, 90, 120, and 180 min) in 24 additional individuals, for a total of 51 subjects. Data of this experiment are descriptive and were compared with Brazilian epidemiological studies.
Experiment 2
The results of previously reported OGTT performed in 24 MUT/MUT subjects (14) were compared with those of 25 age- and sex-matched MUT/N individuals. Glucose and insulin were measured at 0, 30, 60, 90, 120, and 180 min. Exclusion criteria were as follows: age <25 or more than 60 years, known history of DM, excessive alcohol use, chronic and systemic diseases, previous GH use, use of drugs that could impact IS and βCF, and fasting glucose ≥126 mg/dl.
Laboratory methods and calculations
Glucose was measured by the enzymatic Trinder colorimetric test on the day of each test. Sera for insulin were frozen at −40 °C and assayed together. Insulin was measured by an immunofluorometric assay having a sensitivity of 0.5 μU/ml (PerkinElmer Life and Analytical Sciences, Turku, Finland), and intra-assay and interassay variabilities were 2.5 and 3% respectively. IS was assessed by three methods: i) HOMA–IR, measured by the formula: fasting serum insulin (μU/ml)×fasting plasma glucose (mmol/l)/22; ii) quantitative IS check index (QUICK), measured by the formula: 1/(log insulin (μU/ml)+log glucose (mg/dl)); and iii) oral glucose IS index for 2 h (OGIS2) and 3 h (OGIS3). βCF was estimated using the basal glucose and insulin values by HOMA-β (20×fasting insulin)/(fasting glucose (mmol/l)−3.5); the first rapid phase of insulin secretion, using the insulinogenic index (IGI) (insulin time 30−insulin time 0 (μU/ml))/(glucose time 30−glucose time 0 (mg/dl)); and the total glucose-adjusted insulin response during the OGTT, by the area under the curve of insulin:glucose ratio (AUC I (μU/ml):G (mg/dl)) (14).
Anthropometric measurements
Height, body weight, waist circumference (W) at half the distance between the last rib and the superior margin of iliac crest, and hip circumference (H) at the level of femoral trochanters were measured. The W:H ratio was calculated, and BMI was obtained by the formula: weight (kg)/height (m2).
Prediabetes and diabetes diagnosis
Current American Diabetes Association criteria were followed. Prediabetes was defined by the presence of either IFG (fasting glucose levels between 100 and 125 mg/dl) and/or IGT (2-h glucose level during OGTT between 140 and 199 mg/dl). DM was defined by a fasting glucose level ≥126 mg/dl or a 2-h glucose level during OGTT ≥200 mg/dl.
Statistical analysis
Variables with normal and not-normal distribution were compared by t-test and Mann–Whitney U test respectively. Data of normal distribution are reported as mean (s.d.). Data with not-normal distribution (HOMA–IR, HOMA-β, IGI, QUICK-I, and BMI) are reported as median (interquartile range). Statistical analysis was performed using the Software SPSS/PC 15.0 (SPSS, Inc.). P values ≤0.05 were considered statistically significant.
Results
Experiment 1
The prevalence of IFG was 9.8% (five cases). The prevalence of DM was 15.7% (eight cases: two cases were previously diagnosed, two were diagnosed by fasting glucose ≥126 mg/dl, and four by 2-h post-glucose value ≥200 mg/dl). The prevalence of IGT in the MUT/MUT subject who underwent OGTT was 13/34 (38.2%). However, four of these subjects had IFG. Therefore, the prevalence of IGT in subjects with normal fasting glucose is 9/34 (26.4%).
Experiment 2
As expected, height and weight were lower in MUT/MUT subjects than in MUT/N subjects. BMI was similar, while W:H ratio was higher in MUT/MUT subjects. HOMA–IR was lower and QUICKI was higher in MUT/MUT subjects than in MUT/N subjects (P=0.004 and P=0.005 respectively). OGIS2 had a trend toward being higher in MUT/MUT subjects than in MUT/N subjects (P=0.054), while OGIS3 was similar (Table 1). HOMA-β, IGI, and AUC I:G were lower in MUT/MUT subjects than in MUT/N subjects (P=0.005, P=0.001, and P=0.002 respectively; Table 1 and Fig. 1).
Table 1.
Anthropometric and metabolic data in 24 MUT/MUT subjects and 25 MUT/N subjects involved in experiment 2. Data are expressed as mean (s.d.), except for HOMA–IR, HOMA-β, and IGI, which are expressed as median (interquartile range).
| MUT/MUT | MUT/N | P | |
|---|---|---|---|
| Age (years) | 39.25 (11.73) | 40.08 (10.87) | 0.798 |
| Sex (males/females) | 12/12 | 12/13 | 1.000 |
| Height (cm) | 128.21 (9.42) | 160.66 (9.27) | <0.001 |
| Weight (kg) | 37.2 (5.95) | 59.35 (10.26) | <0.001 |
| BMI (kg/m2) | 22.9 (4.68) | 22.88 (2.79) | 0.983 |
| Waist (cm) | 76.5 (9.99) | 80.46 (7.72) | 0.139 |
| Hip (cm) | 81.94 (8.01) | 91.95 (7.9) | <0.001 |
| W:H ratio | 0.93 (0.09) | 0.88 (0.09) | 0.033 |
| HOMA–IR | 0.55 (0.49) | 0.96 (0.57) | 0.004 |
| QUICKI | 0.43 (0.04) | 0.39 (0.04) | 0.005 |
| OGIS2 | 444.3 (49.6) | 413.18 (59.74) | 0.054 |
| OGIS3 | 453.6 (62.14) | 430.14 (72.2) | 0.230 |
| HOMA-β | 28.1 (14.52) | 52.24 (42.4) | 0.005 |
| IGI | 0.4 (0.36) | 0.99 (0.98) | 0.001 |
| AUC I (μU/ml):G (mg/dl) | 0.22 (0.08) | 0.36 (0.17) | 0.001 |
Figure 1.
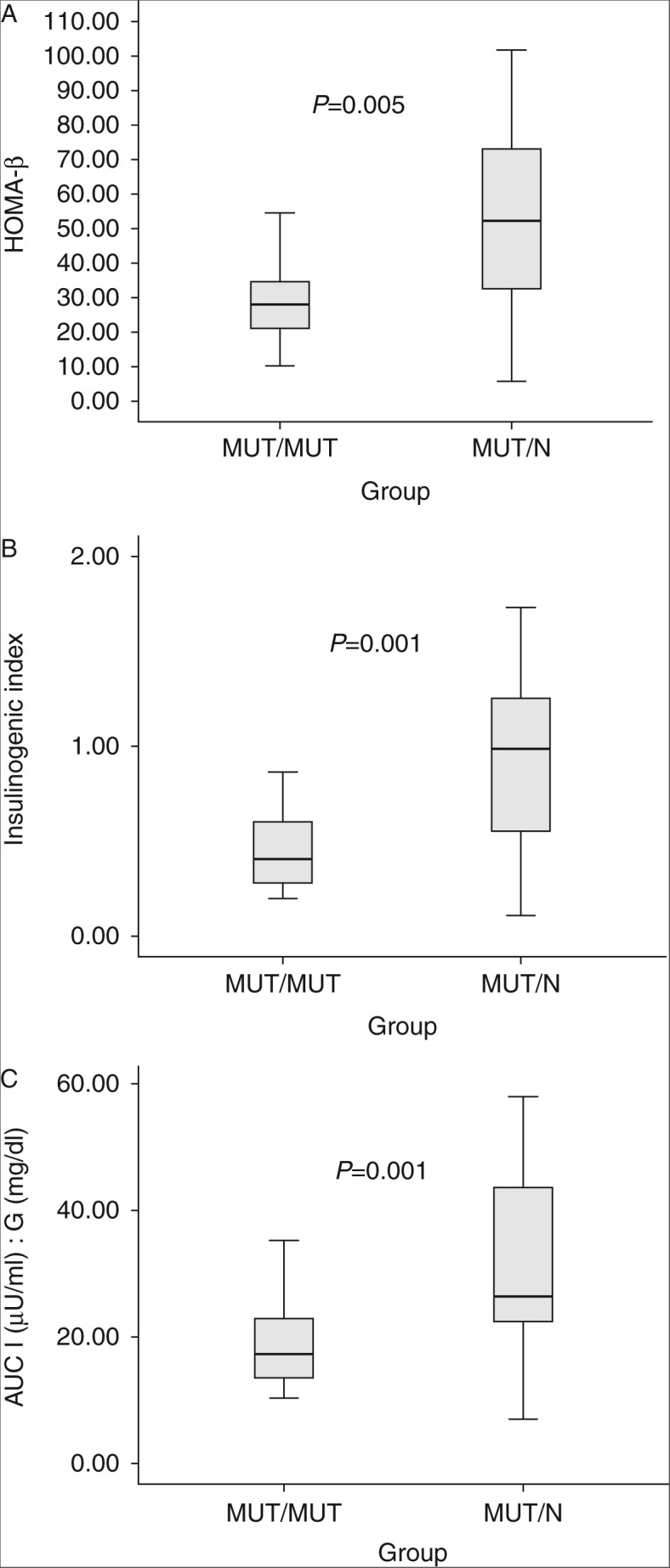
Boxplot of (A) HOMA-β, (B) IGI, and (C) AUC I (μU/ml):G (mg/dl) in 24 MUT/MUT subjects and 25 MUT/N subjects. The horizontal line in the middle of the rectangle is the median, the low margin of the rectangle is the 25th percentile, and the high margin of the rectangle is the 75th percentile.
Discussion
Recently, there has been great interest – both in the medical literature and in the lay press – about the report that adult dwarf individuals with GH resistance from a large Ecuadorian kindred do not have diabetes or cancer (8). Conversely, the prevalence of DM and IGT in our IGHD cohort was 15.7 and 38.2% respectively. Although the prevalence of DM we have observed was similar to that of DM in two population studies in Brazil (12.1 and 13.5%), the prevalence of IGT is even higher than that reported in these studies (5 and 7.7% respectively) (16, 17). The DM prevalence in our cohort may have been even higher if we had been able to perform an OGTT in all the 51 subjects. These findings prove that lifetime IGHD due to a GHRHR mutation – contrary to what has been reported in a large GH-resistant cohort – does not protect against development of DM.
It is possible that the metabolic consequences of GH resistance and GHD are different, particularly because patients with GHRHR mutations secrete a small but detectable amount of GH (18), which may contribute to a lower IS than the Laron model (where the GH effect is likely to be completely absent). It is conceivable that the lack of a GHRH effect per se may have additional consequences on glucose metabolism independent of GHD. It is also possible that the self-reporting approach may have underestimated the real prevalence of DM in the Ecuadorian subjects. Indeed, diabetes and its complications have been described in patients with Laron dwarfism (19, 20, 21). Accordingly, if we had relied only on self-reporting, we would have concluded that only two subjects (3.92%) have diabetes.
Interestingly, despite reduced glucose tolerance, our IGHD individuals present increased IS in comparison with a BMI-matched MUT/N group. These data are similar to the previous finding of increased IS in MUT/MUT in comparison to N/N. The limitation of the previous paper was that the MUT/MUT and N/N groups were not perfectly matched for BMI (14). Reduced BMI is a hallmark of congenital GHD (22) and GH resistance (6), due to small muscles and bones, and it is very difficult to match MUT/MUT with N/N individuals by BMI. In this study, we were fortunate to have a MUT/N group with a BMI similar to that of MUT/MUT group. Therefore, the present data confirm our previous conclusion that IGHD per se, despite increase in percentage fat mass, and contrary to what has been reported in adult-onset GHD (AOGHD), does not cause insulin resistance. We hypothesize that the difference in IS (increased in our IGHD individuals and reduced in AOGHD individuals) is probably caused by different degrees of GHD, and possibly influenced by co-morbidities, such as a lack of other pituitary hormones and associated therapies in AOGHD (23), and is not due to methodological issues. Indeed, the OGTT with simultaneous glucose and insulin measurement has an excellent correlation with the euglycemic–hyperinsulinemic clamp (24), the ‘gold standard’ method to assess IS, used in acquired AOGHD (25).
The increase of IS in the IGHD group may be caused by the marked reduction in GH levels and thereby lack of insulin antagonism. Additionally, increased muscle IS may be due to an increase in the molar ratio of total IGF to IGF binding protein type 3 (10), similar to what seen in healthy centenarians (26).
Such an increase in IS does not result in the prevention of diabetes. We speculate that this is due to the important effect of the GH–IGF1 axis on βCF, whose measures were reduced in MUT/MUT subjects. This reduction in βCF may be the decisive factor for the high prevalence of IGT in IGHD subjects. Conversely, as MUT/N subjects have normal IGF1 levels, they may have normal βCF.
Similar to our IGHD subjects, little mice (also carrying a homozygous GHRHR mutation like our subjects) (27) and mice with GH receptor gene ablation (GHRKO) (3) are hypersensitive to insulin, but have IGT due to a reduced number of pancreatic β cells and reduced insulin secretion, probably due to reduced pancreatic β-cell mass. Accordingly, Igf1 gene overexpression in GHRKO animals restores β-cell mass, normalizing insulin production, and glucose tolerance (4).
In conclusion, severe congenital lifetime IGHD increases IS but impairs βCF and does not provide protection from DM, whose prevalence is similar to that of the general Brazilian population. Therefore, the dream of a ‘diabetes-free life’ is not present even in severe congenital IGHD.
Acknowledgements
The authors thank the ‘Associação do Crescimento Físico e Humano de Itabaianinha’ for their assistance.
Footnotes
(These authors contributed equally to this work)
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
References
- 1.Vijayakumar A, Yakar S, LeRoith D. The intricate role of growth hormone in metabolism. Frontiers in Endocrinology. 2011;2:32. doi: 10.3389/fendo.2011.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clemmons DR. The relative roles of growth hormone and IGF1 in controlling insulin sensitivity. Journal of Clinical Investigation. 2004;113:25–27. doi: 10.1172/JCI200420660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu JL, Coschigano KT, Robertson K, Lipsett M, Guo Y, Kopchick JJ, Kumar U, Liu YL. Disruption of growth hormone receptor gene causes diminished pancreatic islet size and increased insulin sensitivity in mice. American Journal of Physiology. Endocrinology and Metabolism. 2004;287:405–413. doi: 10.1152/ajpendo.00423.2003. [DOI] [PubMed] [Google Scholar]
- 4.Guo Y, Lu Y, Houle D, Robertson K, Tang Z, Kopchick JJ, Liu YL, Liu JL. Pancreatic islet-specific expression of an insulin-like growth factor-I transgene compensates islet cell growth in growth hormone receptor gene deficient mice. Endocrinology. 2005;146:2602–2609. doi: 10.1210/en.2004-1203. [DOI] [PubMed] [Google Scholar]
- 5.Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, LeRoith D. Normal growth and development in the absence of hepatic insulin-like growth factor I. PNAS. 1999;96:7324–7329. doi: 10.1073/pnas.96.13.7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanety H, Hemi R, Ginsberg S, Pariente C, Yissachar E, Barhod E, Funahashi T, Laron Z. Total and high molecular weight adiponectin are elevated in patients with Laron syndrome despite marked obesity. European Journal of Endocrinology. 2009;161:837–844. doi: 10.1530/EJE-09-0419. [DOI] [PubMed] [Google Scholar]
- 7.Laron Z, Avitzur Y, Klinger B. Carbohydrate metabolism in primary growth hormone resistance (Laron syndrome) before and during insulin-like growth factor-I treatment. Metabolism. 1995;44(Suppl 4):113–118. doi: 10.1016/0026-0495(95)90231-7. [DOI] [PubMed] [Google Scholar]
- 8.Guevara-Aguirre P, Balasubramanian VD, Guevara-Aguirre M, Wei M, Madia F, Cheng C-W, Hwang D, Martin-Montalvo A, Saavedra J, Ingles S, et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Science Translational Medicine. 2011;3:70ra13. doi: 10.1126/scitranslmed.3001845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salvatori R, Hayashida CY, Aguiar-Oliveira MH, Phillips JA, III, Souza AH, Gondo RG, Toledo SP, Conceicão MM, Prince M, Maheshwari HG, et al. Familial dwarfism due to a novel mutation of the growth hormone-releasing hormone receptor gene. Journal of Clinical Endocrinology and Metabolism. 1999;84:917–923. doi: 10.1210/jc.84.3.917. [DOI] [PubMed] [Google Scholar]
- 10.Aguiar-Oliveira MH, Gill MS, de A Barretto ES, Alcântara MR, Miraki-Moud F, Menezes CA, Souza AH, Martinelli CE, Pereira FA, Salvatori R, et al. Effect of severe growth hormone (GH) deficiency due to a mutation in the GH-releasing hormone receptor on insulin-like growth factors (IGFs), IGF-binding proteins, and ternary complex formation throughout life. Journal of Clinical Endocrinology and Metabolism. 1999;84:4118–4126. doi: 10.1210/jc.84.11.4118. [DOI] [PubMed] [Google Scholar]
- 11.Barreto-Filho JA, Alcântara MR, Salvatori R, Barreto MA, Sousa AC, Bastos V, Souza AH, Pereira RM, Clayton PE, Gill MS, et al. Familial isolated growth hormone deficiency is associated with increased systolic blood pressure, central obesity, and dyslipidemia. Journal of Clinical Endocrinology and Metabolism. 2002;87:2018–2023. doi: 10.1210/jc.87.5.2018. [DOI] [PubMed] [Google Scholar]
- 12.Aguiar-Oliveira MH, Oliveira FT, Pereira RM, Oliveira CR, Blackford A, Valenca EH, Santos EG, Gois MB, Jr, Meneguz-Moreno RA, Araujo VP, et al. Longevity in untreated congenital growth hormone deficiency due to a homozygous mutation in the GHRH receptor gene. Journal of Clinical Endocrinology and Metabolism. 2010;95:714–721. doi: 10.1210/jc.2009-1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menezes Oliveira JL, Marques-Santos C, Barreto-Filho JA, Ximenes Filho R, de Oliveira Britto AV, Oliveira Souza AH, Prado CM, Pereira Oliveira CR, Pereira RM, Ribeiro Vicente Tde A, et al. Lack of evidence of premature atherosclerosis in untreated severe isolated growth hormone (GH) deficiency due to a GH releasing hormone receptor mutation. Journal of Clinical Endocrinology and Metabolism. 2006;91:2093–2099. doi: 10.1210/jc.2005-2571. [DOI] [PubMed] [Google Scholar]
- 14.Oliveira CR, Salvatori R, Barreto-Filho J, Rocha IES, Mari A, Pereira RM, Campos VC, Menezes M, Gomes E, Meneguz-Moreno RA, et al. Insulin sensitivity and beta-cell function in adults with lifetime, untreated isolated growth hormone deficiency. Journal of Clinical Endocrinology and Metabolism. 2012;97:1013–1019. doi: 10.1210/jc.2011-2590. [DOI] [PubMed] [Google Scholar]
- 15.Pereira RM, Aguiar-Oliveira MH, Sagazio A, Oliveira CR, Oliveira FT, Campos VC, Farias CT, Vicente TA, Gois MB, Jr, Oliveira JL, et al. Heterozygosity for a mutation in the growth hormone releasing hormone receptor gene does not influence adult stature, but affects body composition. Journal of Clinical Endocrinology and Metabolism. 2007;92:2353–2357. doi: 10.1210/jc.2007-0092. [DOI] [PubMed] [Google Scholar]
- 16.Torquato MT, Montenegro Júnior RM, Viana LA, de Souza RA, Lanna CM, Lucas JC, Bidurin C, Foss MC. Prevalence of diabetes mellitus and impaired glucose tolerance in the urban population aged 30–69 years in Ribeirão Preto (São Paulo), Brazil. Arquivos Brasileiros de Endocrinologia e Metabologia. 2003;47:69–74. doi: 10.1590/S1516-31802003000600002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bosi PL, Carvalho AM, Contrera D, Casale G, Pereira MA, Gronner MF, Diogo TM, Torquarto MT, Oishi J, Leal AM. Prevalence of diabetes and impaired glucose tolerance in the urban population of 30 to 79 years of the city of São Carlos, São Paulo. Arquivos Brasileiros de Endocrinologia e Metabologia. 2009;53:726–732. doi: 10.1590/S0004-27302009000600006. [DOI] [PubMed] [Google Scholar]
- 18.Salvatori R, Serpa MG, Parmigiani G, Britto AV, Oliveira JL, Oliveira CR, Prado CM, Farias CT, Almeida JC, Vicente TA, et al. Growth hormone (GH) response to hypoglycemia and clonidine in the GH-releasing hormone resistance syndrome. Journal of Endocrinological Investigation. 2006;29:805–808. doi: 10.1007/BF03347374. [DOI] [PubMed] [Google Scholar]
- 19.Laron Z. Insulin secretion and carbohydrate metabolism in patients with Laron syndrome: from hypoglycemia to diabetes mellitus. In: Laron Z, Kopchick JJ, editors. Laron Syndrome from Man to Mouse. Springer-Verlag; Berlin–New York: 2011. pp. 259–272. [Google Scholar]
- 20.Laron Z, Weinberger D. Diabetic retinopathy in two patients with congenital IGFI deficiency (Laron syndrome) European Journal of Endocrinology. 2004;151:103–106. doi: 10.1530/eje.0.1510103. [DOI] [PubMed] [Google Scholar]
- 21.Laron Z, Weinberger D. Diabetic retinopathy, nephropathy and cardiovascular disease in a patient with GH gene deletion. Clinical Endocrinology. 2005;63:699–700. doi: 10.1111/j.1365-2265.2005.02402.x. [DOI] [PubMed] [Google Scholar]
- 22.Aguiar-Oliveira MH, Salvatori R. Lifetime growth hormone (GH) deficiency: impact on growth, metabolism, body composition and survival capacity. In: Preedy VR, editor. Handbook of Growth and Growth Monitoring in Health and Disease. Springer Science+Business Media, LLC; New York: 2012. pp. 2699–2710. [Google Scholar]
- 23.Rosén T, Bengtsson BA. Premature mortality due to cardiovascular disease in hypopituitarism. Lancet. 1990;336:285–288. doi: 10.1016/0140-6736(90)91812-O. [DOI] [PubMed] [Google Scholar]
- 24.Mari A, Pacini G, Murphy E, Ludvik B, Nolan JJ. A model based method for assessing insulin sensitivity from the oral glucose tolerance test. Diabetes Care. 2001;24:539–548. doi: 10.2337/diacare.24.3.539. [DOI] [PubMed] [Google Scholar]
- 25.Hew FL, Koschmann M, Christopher M, Rantzau C, Vaag A, Ward G, Beck-Nielsen H, Alford F. Insulin resistance in growth hormone-deficient adults: defects in glucose utilization and glycogen synthetase activity. Journal of Clinical Endocrinology and Metabolism. 1996;81:555–564. doi: 10.1210/jc.81.2.555. [DOI] [PubMed] [Google Scholar]
- 26.Paolisso C, Ammendola M, Del Buono A, Gambardella A, Riondino M, Tagliamonte MR, Rizzo MR, Carella C, Varricchio M. Serum levels of insulin-like growth factor-I (IGF1) and IGF-binding protein-3 in healthy centenarians: relationship with plasma leptin and lipid concentrations, insulin action, and cognitive function. Journal of Clinical Endocrinology and Metabolism. 1997;82:2204–2209. doi: 10.1210/jc.82.7.2204. [DOI] [PubMed] [Google Scholar]
- 27.Fleenor D, Oden J, Kelly P, Mohan S, Alliouachene S, Pende M, Wentz S, Kerr J, Freemark M. Roles of the lactogens and somatogens in perinatal and postnatal metabolism and growth: studies of a novel mouse model combining lactogen resistance and growth hormone deficiency. Endocrinology. 2005;146:103–112. doi: 10.1210/en.2004-0744. [DOI] [PubMed] [Google Scholar]