

Abstract
Thyroid hormones (THs) play a pivotal role in regulating cardiovascular homeostasis. To provide a better understanding of the coordinated processes that govern cardiac TH bioavailability, this study investigated the influence of serum and cardiac TH status on the expression of TH transporters and cytosolic binding proteins in the myocardium. In addition, we sought to determine whether the administration of T3 (instead of T4) improves the relationship between THs in serum and cardiac tissue and cardiac function over a short-term treatment period. Adult female Sprague Dawley rats were made hypothyroid by 7 weeks treatment with the antithyroid drug 6-n-propyl-2-thiouracil (PTU). After establishing hypothyroidism, rats were assigned to 1 of 5 graded T3 dosages plus PTU for a 2-week dose-response experiment. Untreated, age-matched rats served as euthyroid controls. PTU was associated with depressed serum and cardiac tissue T3 and T4 levels, arteriolar atrophy, altered TH transporter and cytosolic TH binding protein expression, fetal gene reexpression, and cardiac dysfunction. Short-term administration of T3 led to a mismatch between serum and cardiac tissue TH levels. Normalization of serum T3 levels was not associated with restoration of cardiac tissue T3 levels or cardiac function. In fact, a 3-fold higher T3 dosage was necessary to normalize cardiac tissue T3 levels and cardiac function. Importantly, this study provides the first comprehensive data on the relationship between altered TH status (serum and cardiac tissue), cardiac function, and the coordinated in vivo changes in cardiac TH membrane transporters and cytosolic TH binding proteins in altered TH states.
Thyroid hormones (THs) play a pivotal role in regulating cardiovascular homeostasis and the peripheral vasculature (1). THs regulate cardiac function both directly (heart rate and myocardial contractility) and indirectly [systemic vascular resistance, blood volume, erythropoietin secretion], which ultimately influences cardiac output (1, 2). Both overt and subclinical hypothyroidism can lead to cardiac dysfunction characterized by impaired contractility, diastolic dysfunction, left ventricular (LV) chamber remodeling, impaired blood flow, and increased peripheral vascular resistance and eventually may lead to, or exacerbate, heart failure (1, 3). Recent clinical studies suggest that overt and subclinical hypothyroidism are associated with an increased all-cause and cardiovascular mortality (4–7).
There is considerable debate regarding the most appropriate approach to treat and restore serum and tissue TH homeostasis in settings of overt and subclinical hypothyroidism (8–10). Clinically most patients receiving TH supplementation are treated with oral T4. Typically T4 is preferred over biologically active T3 because it mimics the main secretory product of the thyroid gland. However, data from our laboratory and others have shown that T4 treatment in thyroidectomized rodents can lead to a mismatch between serum and cardiac tissue TH levels and cardiac function (10, 11). Presently the relationship between serum and cardiac TH status, TH transporter, and cytosolic TH binding protein expression has not been well characterized, and their interrelationship is virtually unknown within intact cardiac tissue. A better understanding of the coordinated processes that regulate TH bioavailability and function in the heart may improve the clinical efficacy of treating low thyroid conditions and provide a basis for identifying pathological derangements in its regulation.
The purpose of this study was to determine the influence of serum and cardiac TH status on the expression of key myocardial genes, including TH transporters and cytosolic TH binding proteins. In addition, we sought to determine whether the administration of biologically active T3 (instead of T4) improves the relationship between THs in serum and cardiac tissue and cardiac function in the short-term. We hypothesized that administering biologically active T3, thereby bypassing the need for peripheral T4 to T3 conversion, would clarify the relationship between serum and cardiac tissue TH levels and cardiac function. Results from the current investigation demonstrate that administration of biologically active T3 was unable to correct the mismatch between serum and cardiac tissue TH levels. Elevated physiological T3 dosing was required to restore both cardiac tissue T3 levels and cardiac function during short-term treatment of primary hypothyroidism. To our knowledge, this is the first study to comprehensively characterize the relationship between altered TH status (serum and cardiac tissue), cardiac function, and the coordinated in vivo changes in cardiac TH membrane transporters and cytosolic TH binding proteins in altered TH states.
Materials and Methods
For a detailed description of the methods, please see Supplemental Methods and Materials, published on The Endocrine Society's Journals Online web site at http://endo.endojournals.org.
Animal model and experimental design
Adult female Sprague Dawley rats (Harlan, Indianapolis, Indiana) were randomly assigned to either hypothyroid or euthyroid groups. Hypothyroidism was established by 7 weeks of treatment with the antithyroid drug 0.025% 6-n-propyl-2-thiouracil (PTU; Sigma, St Louis, Missouri) dissolved in drinking water as previously described (3). After 7 weeks of PTU treatment, hypothyroid rats were further assigned to one of the following T3 (Sigma) dosages to the drinking water for a 2-week period (n = 12–14/group): 0 μg/mL T3 (PTU), 0.03 μg/mL T3 (PTU + 0.03 T3), 0.05 μg/mL (PTU + 0.05 T3), 0.10 μg/mL (PTU + 0.10 T3), or 0.30 μg/mL (PTU + 0.30 T3). Assuming no impairment in T3 intestinal absorption, these graded oral T3 concentrations correspond to approximately 3, 5, 10, and 26 μg/kg·d T3, respectively (Supplemental Table 1). Non-PTU-treated, age-matched rats served as euthyroid controls (control). PTU was administered for the entire duration of the experiment to avoid endogenous TH elevations associated with PTU discontinuation (12). T3 was administered as solution dissolved in drinking water, as previously described (13). Water consumption was monitored twice per week, and the T3 concentration was adjusted based on individual body weight and water consumption. After 2 weeks of T3 treatment (9 weeks of PTU), terminal cardiac function was assessed by echocardiography and LV hemodynamics. All animals were kept on a 12-hour light, 12-hour dark cycle, and food and water were provided ad libitum. All experiments and protocols were performed in accordance with the Guide for the Care and Use of Laboratory Animals (US Department of Health, Education, and Welfare, Department of Health and Human Services, National Institutes of Health Publication 85-23) and were approved by the University of South Dakota Animal Care and Use Committee.
Measurement of serum and cardiac TH levels
Serum TH levels were measured using commercial ELISA kits (TSH: ALPCO, Salem, New Hampshire; T3, free T3 (FT3), T4, and free T4: Monobind, Lake Forest, California) according to manufacturer specifications. Cardiac TH extracts from pooled LV tissues homogenates were measured using tandem HPLC-mass spectrometry as previously described (14, 15).
Measurement of cardiac function
LV echocardiography and hemodynamics were performed in each animal before the animals were killed as previously described (16–18).
Real-time PCR
Gene expression was evaluated by a custom-designed primer plate (Supplemental Table 2; catalog number CAPR10931G; SABiosciences, QIAGEN INC, Valencia, California) for fetal genes, deiodinase (D) 1–3, TH transporters, and cytosolic TH binding proteins as previously described (19). Gene expression was normalized using the housekeeping genes cyclophilin A and Rplp1.
Western blot
Western blotting was performed as previously described (3).
Quantification of arteriolar remodeling
Formalin-fixed, paraffin-embedded LV tissue sections (5–7 μm) were used to evaluate changes in arteriolar resistance vessels as reported previously (19). Sections were deparaffinized with xylene and rehydrated with graded ethanol incubation. LV tissue sections were then placed in IHC select citrate buffer (pH 6.0; EMD Millipore, Billerica, Massachusetts) and boiled in a rice cooker for 20 minutes for antigen retrieval. Fluorescein isothiocyanate-conjugated isolectin B4 (IB4; Vector Laboratories, Burlingame, California) and Cy3-conjugated α-smooth muscle actin (α-SMA; Sigma, Saint Louis, Missouri) were used in combination to label endothelial cells and vascular smooth muscle cells, respectively. Arteriolar length density (LD) was calculated from approximately 30–35 random fields per heart based on the following formula: LD (millimeters per cubic millimeter) = Σ(A/B)/M, where A and B are the maximum and minimum external arteriolar diameters, and M is the total tissue area (20). Arterioles with B dimensions between 5 and 30 μm were used to calculate arteriolar LD. Arteriolar LD was converted to total arteriolar length by multiplying LD (millimeters per cubic millimeter) by LV weight expressed in cubic millimeters (∼ 1 mg of cardiac tissue is equivalent to 1 mm3).
Data analysis
All data are expressed as means ± SD. Statistical analysis was performed using a paired Student's t test [pre- vs posttreatment body weights (BW)], 1-way ANOVA followed by Holm-Sidak post hoc analysis, or nonparametric 1-way ANOVA (Kruskal-Wallis) ranked measurements with Dunn's post hoc analysis. Statistical analysis was performed using SigmaPlot 11 (Systat Software, San Jose, California). Gene expression was analyzed by the ΔΔcycle threshold (Ct) method using online software from SABiosciences. Values of P < .05 were considered statistically significant.
Results
Physical data
Physical data are summarized in Table 1. Significant BW reductions were observed in all groups receiving PTU. In addition, heart weight (HW; P < .01), LV weight (LVW; P < .01), HW to BW ratio (P < .01), and LVW to BW ratio (P < .01) were significantly reduced with PTU treatment (29%, 32%, 10%, and 16% reduction vs control, respectively). T3 treatment was associated with dose-dependent increases in all the above-mentioned parameters except for body weight.
Table 1.
Physical Data
| Control | PTU | PTU + 0.03 T3 | PTU + 0.05 T3 | PTU + 0.10 T3 | PTU + 0.30 T3 | |
|---|---|---|---|---|---|---|
| BW-pre, g | 238 (11) | 202 (9)a | 202 (8)a | 209 (9)a | 205 (9)a | 203 (9)a |
| BW-post, g | 247 (8) | 194 (13)a | 209 (11)a,b | 209 (9)a,b | 206 (10)a | 200 (9)a |
| HW, mg | 850 (48) | 601 (53)a | 667 (53)a,b | 723 (33)a,b | 779 (73)a,b,c,d | 863 (79)b,c,d,e |
| LVW, mg | 586 (45) | 400 (30)a | 451 (38)a,b | 487 (29)a,b | 532 (51)a,b,c,d | 583 (52)b,c,d,e |
| HW/BW, mg/g | 3.44 (0.2) | 3.09 (0.2)a | 3.18 (0.2) | 3.46 (0.2)b | 3.79 (0.3)b,c | 4.31 (0.3)a,b,c,d |
| LVW/BW, mg/g | 2.38 (0.2) | 2.06 (0.1)a | 2.17 (0.2)a,b | 2.33 (0.2)b | 2.59 (0.2)a,b,c,d | 2.91 (0.2)f |
Abbreviations: BW pre, body weight prior to T3 treatment/after 7 weeks PTU; BW post, body weight at terminal experiments. Values are means (SD) (n = 10–14/group).
P < .05 vs control.
P < .05 vs PTU.
P < .05 vs PTU + 0.03 T3.
P < .05 vs PTU + 0.05 T3.
P < .05 vs PTU + 0.10 T3.
P < .05 vs all other groups.
Restoration of serum T3 levels is inadequate to ensure cardiac tissue euthyroidism during short-term T3 supplementation
TH concentrations in serum and cardiac tissue are shown in Figure 1 and Supplemental Table 3. The effectiveness of PTU treatment was confirmed by a significant reduction in serum THs (T3, FT3, and T4: 53%, 39%, and 92% reduction vs control, respectively) and a 46-fold elevation in serum TSH levels in the non-T3-treated PTU group (Figure 1B). Moreover, serum T4 suppression was observed in all groups receiving PTU, further establishing the efficacy of the PTU treatment (Supplemental Table 3). T3 treatment reduced serum TSH levels in a dose-dependent manner, whereas normalization occurred at 0.10 μg/mL T3 (PTU + 0.10 T3; Figure 1B). The lowest administered T3 dosage (0.03 μg/mL T3) normalized serum T3 levels, despite serum TSH remaining 32-fold above euthyroid control. Significant elevations in serum T3 above normal levels did not occur until T3 dosages of 0.10 μg/mL (PTU + 0.10 T3) and higher (Figure 1A).
Figure 1.

Restoration of normal serum T3 levels is inadequate to ensure cardiac tissue euthyroidism during short-term T3 supplementation. Values represent means (SD). A–D, Serum T3 in serum (A); serum TSH, thyroid-stimulating hormone in serum (B); cardiac T3 in cardiac tissue (C); cardiac T4 in cardiac tissue (D) n = 3–12/group; n = 2–3 pooled samples/group for panels C and D. Each pooled LV sample within an experimental group consisted of pooled LV tissue from 2–4 hearts (6–8 hearts/group). ND, not detectible by assay. *, P < .05 vs control; †, P < .05 vs PTU; €, P < .05 vs PTU + 0.03 T3; ‡, P < .05 vs PTU + 0.05 T3; Σ, P < .05 vs all other groups. Mean differences in pooled samples (panels C and D) were not statistically tested.
As expected, PTU treatment was associated with a substantial reduction in cardiac tissue T3 levels (60% reduction vs control) and T3 supplementation restored cardiac tissue T3 levels in a dose-dependent manner (PTU + 0.03 T3, PTU + 0.05 T3: 52% and 31% reduction vs control, respectively; PTU + 0.10 T3, PTU + 0.30 T3: 5% and 7.54-fold increase vs control, respectively; Figure 1C). In line with serum observations, cardiac tissue T4 levels were suppressed in all groups receiving PTU treatment (Figure 1D).
LV echocardiography
LV echocardiography is summarized in Table 2. PTU was associated with significant reductions in LV posterior wall thickness both in systole and diastole (LVPWs and LVPWd; 21% and 26% reduction vs control, respectively; P < .01) and a reduction in anterior wall thickness in diastole (AWd; 20% reduction vs control; P < .01). The lowest T3 dosage, 0.03 μg/mL T3 (PTU + 0.03 T3), partially restored AWd and LVPWs thickness, whereas 0.05 μg/mL (PTU + 0.05 T3) was necessary for normalization of LVPWd.
Table 2.
LV Echocardiography
| Control | PTU | PTU + 0.03 T3 | PTU + 0.05 T3 | PTU + 0.10 T3 | PTU + 0.30 T3 | |
|---|---|---|---|---|---|---|
| n | 10 | 12 | 12 | 12 | 12 | 12 |
| AWd, mm | 1.28 (0.3) | 1.02 (0.1)a | 1.16 (0.1) | 1.12 (0.1) | 1.25 (0.1)b | 1.34 (0.3)b |
| LVIDd, mm | 7.13 (0.6) | 7.00 (0.4) | 7.04 (0.5) | 7.07 (0.4) | 7.05 (0.6) | 6.78 (0.4) |
| LVPWd, mm | 1.56 (0.3) | 1.16 (0.2)a | 1.23 (0.3)a | 1.30 (0.2) | 1.36 (0.2) | 1.56 (0.2)b,c |
| AWs, mm | 2.18 (0.4) | 1.73 (0.3) | 1.98 (0.2) | 2.03 (0.3) | 2.22 (0.3)b | 2.57 (0.5)b,c |
| LVIDs, mm | 4.24 (0.5) | 4.59 (0.4) | 4.52 (0.6) | 4.36 (0.4) | 4.14 (0.6) | 3.71 (0.5)b,c,d |
| LVPWs, mm | 2.33 (0.5) | 1.83 (0.2)a | 1.97 (0.4) | 2.12 (0.2) | 2.23 (0.3) | 2.58 (0.5)b,c |
Abbreviations: AWs, LV anterior wall in systole; LVIDd, LV internal diameter in diastole; LVIDs, LV internal diameter in systole. Values are means (SD).
P < .05 vs control.
P < .05 vs PTU.
P < .05 vs PTU + 0.03 T3.
P < .05 vs PTU + 0.05 T3.
Restoration of serum T3 levels is not associated with reestablishment of LV hemodynamic function in experimental hypothyroidism
LV hemodynamic data are shown in Figure 2. PTU led to cardiac dysfunction as evidenced by significant reductions in heart rate (HR; 42% reduction vs control; P < .01), LV systolic pressure (LVSP; 25% reduction vs control, P < .01), maximal rate of pressure development (dP/dT max; 61% reduction vs control, P < .01), and maximal rate of pressure decline (dP/dT min; 56% reduction vs control, P < .01), with a significantly increased time constant of isovolumic relaxation (Tau; 110% increase vs control, P < .01). Although T3 treatment restored parameters of cardiac function in a dose-dependent manner, normalization of cardiac function did not occur until treatment with 0.10 μg/mL T3 (PTU + 0.10 T3). It is important to note that full restoration of LV function was not achieved until a T3 dosage, which normalized cardiac tissue T3 levels.
Figure 2.

Reestablishment of normal serum T3 levels is not associated with restoration of LV hemodynamic function in experimental hypothyroidism. Values are means (SD). A–F, HR (A); LVSP (B); LVEDP, LV end-diastolic pressure (C); dP/dT max (D); dP/dT min (E); Tau (F) (n = 11–14/group). *, P < .05 vs control; †, P < .05 vs PTU; €, P < .05 vs PTU + 0.03 T3; ‡, P < .05 vs PTU + 0.05 T3; ¥, P < .05 vs PTU + 0.10 T3; Σ, P < .05 vs all other groups.
Cardiac gene expression
To evaluate the influence of serum and cardiac TH status on myocardial gene expression, RT-PCR was used to profile gene expression in the following groups: 1) control (euthyroid); 2) PTU (hypothyroid); 3) PTU + 0.03 T3 (T3 dosage, which restored serum T3 levels); and 4) PTU + 0.10 T3 (T3 dosage, which restored cardiac tissue T3 levels and cardiac function).
Fetal gene reexpression
Analyses of fetal genes are shown in Figure 3 and Supplemental Figure 1. Hypothyroid hearts showed the reexpression of the fetal gene phenotype typically observed during cardiac hypertrophy, heart failure, and tissue hypothyroidism (1, 21). PTU treatment led to a significant reduction in expression of α-myosin heavy chain (MHC; P < .01) and increased β-MHC (P < .01) gene and protein expression (Figure 3, A and B, and Supplemental Figure 1). This altered expression profile was associated with a major shift in the MHC isoform distribution when compared with control hearts (Figure 3C). In addition, hypothyroid rats also had a tendency for a nonsignificant reduction in sarco(endo)plasmic reticulum Ca2+-ATPase (Figure 3D). No changes were observed in thyroid receptor-α, thyroid receptor-β, or ryanodine receptor expression with the PTU treatment (data not shown).
Figure 3.

Reexpression of fetal genes. Values represented as means (SD) or percentage. A–D, α-MHC (A); β-MHC (B); relative MHC isoform distribution as a % of total MHC expression (C). The percentage was calculated as the ratio of either α- or β-MHC divided by the total MHC expression (α- + β-MHC) using their 2[−Avg.(Delta(Ct)] values; sarco(endo)plasmic reticulum Ca2+-ATPase expression (D) (n = 5–6/group). *, P < .05 vs control; †, P < .05 vs PTU; €, P < .05 vs PTU + 0.03 T3.
Despite normalizing serum T3 levels, β-MHC gene expression (increased vs control; P < .01) but not protein expression and the distribution of MHC isoforms were still significantly altered in rats treated with 0.03 μg/mL T3 (PTU + 0.03 T3). T3 treatment, which restored cardiac tissue T3 levels (PTU + 0.10 T3), was associated with normalization of α- and β-MHC gene and protein expression and reversed the shift in MHC isoform distribution observed in the hypothyroid rats.
Expression of cardiac TH transporters
To determine the influence of serum and cardiac tissue TH status on TH transporter expression in the myocardium, we analyzed the expression of known TH transporters [monocarboxylate transporter (MCT); solute carrier transporter family; solute carrier organic anion transporter family (SLCO); fatty acid translocase; large neutral amino acid transporter (LAT); Figure 4]. Hypothyroidism led to a significant elevation in the expression of MCT-10 (P < .01) and a trend for increased MCT-8 expression (P = .16) when compared with euthyroid control. Hypothyroidism was not associated with significant changes in any of the other measured TH transporters. Restoration of serum T3 (PTU + 0.03 T3) normalized the expression of MCT-10; however, there was still a tendency for this parameter to remain elevated (P = .09 vs control). Of note, 0.03 μg/mL T3 further augmented the gene expression of MCT-8 (P = .07 vs control).
Figure 4.
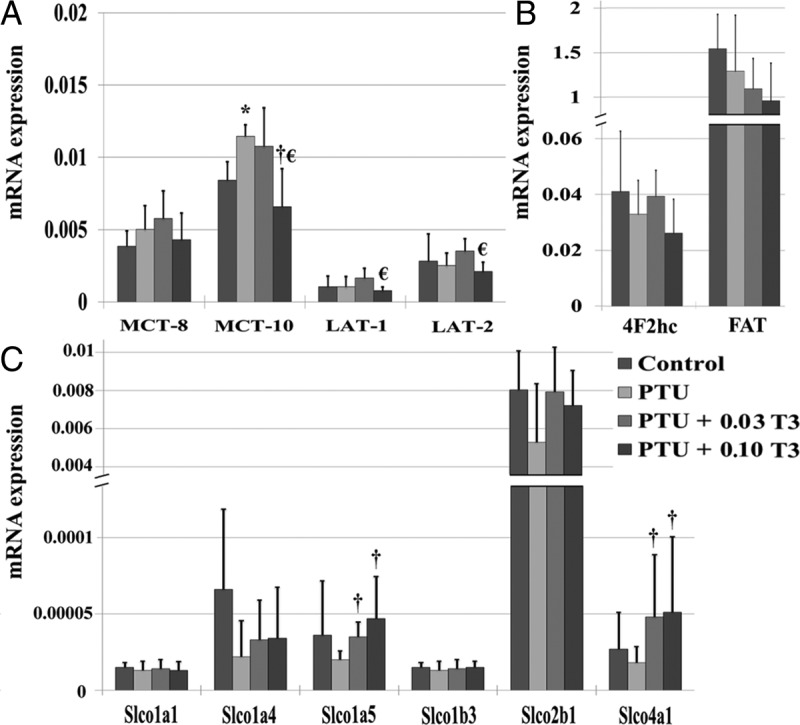
Relationship between TH status and expression of TH transporters in the myocardium. Values are means (SD). A–C, MCTs (A); large neutral amino acid transporters (LAT; A);, 4F2 cell surface antigen heavy chain (LAT escort protein; 4F2hc) (B); fatty acid translocase (FAT; B); SLCO (C). All transporters from the SLCO transporter family had low cardiac gene expression levels (Ct values > 30) (n = 5–6/group). *, P < .05 vs control; †, P < .05 vs PTU; €, P < .05 vs PTU + 0.03 T3.
Among TH transporters with normal relative expression (Ct values < 30), no significant differences in gene expression were observed between rats receiving 0.03 μg/mL T3 (PTU + 0.03 T3) and hypothyroid (PTU) rats. On the other hand, significant differences were observed in the expression of MCT-10, LAT-1, and LAT-2 when comparing rodent hearts receiving T3 dosages, which restored serum (PTU + 0.03 T3) and cardiac tissue T3 (PTU + 0.10 T3) levels. Interestingly, little to no gene expression (Ct values > 30) was detectible for members of the solute carrier organic anion transporter family. Despite low levels of expression, significant differences were observed in the expression of SLCO1a5 and SLCO4a1 when comparing PTU treatment to T3 dosages, which restored serum (PTU + 0.03 T3) and cardiac tissue T3 (PTU + 0.10 T3) levels (Figure 4C).
Expression of cytosolic TH binding proteins
Expression of the cytosolic TH binding proteins in the myocardium is shown in Figure 5. PTU led to a significant reduction in μu-crystallin (Crym; P < .05) and nonsignificant reductions in aldehyde dehydrogenase (Aldh)-1 isoforms. PTU also led to a significant increase in glutathione-s-transferase (GST)(m1) (P < .01 vs control). No changes were observed in the expression of any other cytosolic TH binding protein isoforms with PTU treatment. Restoration of serum T3 levels (PTU + 0.03 T3) normalized the expression of Crym and GST(m1). Restoration of cardiac tissue T3 (PTU + 0.10 T3) led to significant reductions in GST(m1 and a4) isoforms when compared with PTU treatment (Figure 5D). Although alterations in GST isoforms appear to be influenced by thyroid status, the interpretation of the present findings must be made with caution. Previous reports have shown that PTU can augment GST expression and inhibit proper enzymatic activity (22, 23).
Figure 5.

Relationship between TH status and expression of cytosolic TH binding proteins in the myocardium. Values are means (SD). A–D, Crym (A); PK(m2), pyruvate kinase, m2 isoform (B); Aldh isoforms (C); Gst, GST isoforms (D) (n = 5–6/group). *, P < .05 vs control; †, P < .05 vs PTU.
Expression of cardiac deiodinases
We measured the gene expression of D1-D3 deiodinases to evaluate their potential influence on cardiac tissue TH levels (Supplemental Figure 2 and Supplemental Table 4). No significant differences were observed in D1 or D3 expression among treatment groups. In fact, relatively low (Ct values 30–35) or nondetectable (Ct values > 35) expression levels were observed in most tested samples (Supplemental Table 4). Interestingly, high D3 expression (Ct value < 20) was observed in 1 sample from each of the PTU and PTU + 0.03 T3 groups, respectively (Supplemental Table 4).
In contrast, expression of intracardiac D2 was more robust in all groups. Cardiac samples from the PTU and PTU + 0.03 T3 groups had a strong tendency for elevated D2 expression (P = .07 and P = .099 vs control, respectively), whereas PTU + 0.10 T3 was associated with reduced D2 expression when compared with control hearts. In fact, D2 expression was significantly reduced in the PTU + 0.10 T3 group when compared with the PTU and PTU + 0.03 T3 groups (Supplemental Figure 2).
Arteriolar morphometry
To evaluate the influence of serum and cardiac TH status on arteriolar remodeling, morphometric analysis was conducted on the following groups: 1) control; 2) PTU (hypothyroid); 3) PTU + 0.03 T3 (T3 dosage, which restored serum T3 levels); and 4) PTU + 0.10 T3 (T3 dosage, which restored cardiac tissue T3 levels and cardiac function; Figure 6). Contrary to our previous reports, which used single antibody labeling only (3, 11), PTU treatment in the current study was not associated with a reduction in arteriolar LD. However, hypothyroid (PTU) rats had an approximately 25% reduction in total myocardial arteriolar length (LD × LVW; P < .05) due to reduced LV mass. T3 treatment restored the arteriolar vasculature in proportion to LV growth.
Figure 6.

Arteriolar resistance vessel remodeling in the myocardium. Values are means (SD). A and B, Arteriolar LD (A); arteriolar length, total arteriolar length (B). Arteriolar LD and arteriolar length were calculated using previously described methods (19). Arteriolar LD (millimeters per cubic millimeter) = Σ(a/b)/M, where a and b are the maximum and minimum external arteriolar diameters, and M is the tissue area. Arteriolar length = LD (millimeters per cubic millimeter) × LV weight per cubic millimeter; ∼1 mg of cardiac tissue is equivalent to 1 mm3) (n = 6/group). *, P < .05 vs control.
Discussion
We evaluated the relationship between serum and tissue TH restoration and cardiac function during short-term, graded T3 administration in experimental hypothyroidism in rodents. In a previous report, we noted a mismatch between the T4 supplementation required for restoration of serum and cardiac tissue TH levels and cardiac function in thyroidectomized rats (11). It has previously been shown that in certain regions, such as the blood-brain barrier, T3 uptake can occur more rapidly than T4 (24). Therefore, we tested the hypothesis that short-term TH supplementation with biologically active T3 would more closely match serum and tissue T3 levels and cardiac function. In addition, we sought to investigate the relationship between serum and cardiac tissue TH status on the integrated gene expression of cardiac TH transporters and cytosolic TH binding proteins in the intact myocardium.
PTU treatment led to significant serum and cardiac tissue TH suppression (T3 and T4), cardiac atrophy, contractile dysfunction, relaxation/diastolic abnormalities, LV remodeling, and fetal gene reexpression. As expected, administration of biologically active T3 reestablished the above-mentioned parameters in a dose-dependent manner. However, it is important to note that we observed a substantial mismatch between the T3 supplementation required for restoration of serum and cardiac tissue T3 levels. In the present investigation, T3 dosing, which restored serum T3 levels (PTU + 0.03 T3), failed to normalize cardiac tissue T3 levels (52% reduction vs control). In fact, a 3-fold larger T3 dosage (PTU + 0.10 T3) was necessary to fully normalize cardiac tissue T3 levels (Figure 1).
Restoration of cardiac tissue T3 corresponded with the T3 dosage that was necessary for the normalization of serum TSH levels. However, this same dosage was also associated with a significant elevation in serum T3 levels (37% elevation vs control). Previous investigations in rodent models have demonstrated that both circulating T3 (thyroidal secretion and T4 to T3 conversion via D1 deiodinase) and T4 via D2 deiodinase-mediated conversion to T3 contribute to negative feedback inhibition of hypothalamic TRH synthesis and pituitary TSH secretion (25, 26). Our results are in agreement with the investigation by Escobar-Morreale et al (10), who reported that the amount of T4 supplementation required to normalize serum TH levels was inadequate to normalize serum TSH or tissue T3 levels in thyroidectomized rats over a 13-day treatment period. Thus, it appears that to fully restore cardiac tissue T3 levels during short-term supplementation, an elevated physiological T3 dosage, capable of producing significant serum T3 elevations, is required.
Despite restoring serum T3 levels, 0.03 μg/mL T3 (PTU + 0.03 T3) was insufficient to reverse the severe cardiac impairment associated with PTU treatment (HR, LVSP, dP/dT max, and dP/dT min: 33%, 24%, 49%, and 48% reduction vs control, respectively; Tau, 63% increase vs control; Figure 2). Not surprisingly, the normalization of cardiac function did not occur until the same T3 replacement dosage that fully restored cardiac tissue T3 levels (PTU + 0.10 T3). This confirms previous findings, which suggest that restoring cardiac tissue TH levels is essential to reestablishing proper cardiac function in settings of hypothyroidism (11). As noted previously, it is important to draw attention to the finding that full restoration of both cardiac tissue T3 levels and LV hemodynamic function, including heart rate, was not achieved until a T3 dosage that produced a significant elevation in serum T3 levels.
We recognize that there will be considerable trepidation among the medical community to treat patients with T3 or T4 dosages that significantly elevate serum TH levels to acutely restore cardiac tissue T3. Moreover, we also acknowledge the adherent risk of overtreatment leading to overt or subclinical hyperthyroidism and in no way advocate extended treatment with elevated physiological dosages of THs. However, it is important for clinicians to be cognizant of a potential serum and cardiac tissue mismatch in the treatment of primary hypothyroidism. Results of the present study are in line with our previous investigation examining graded T4 supplementation after thyroidectomy, in which cardiac tissue TH levels were not fully restored until a T4 dosage was administered that significantly elevated serum TH levels (11).
It appears that short-term restoration of serum THs is inadequate to fully restore cardiac tissue TH levels, cardiac function, or suppress fetal gene expression caused by hypothyroidism. Whether the 0.03 μg/mL T3 replacement dose can adequately restore cardiac tissue TH levels when administered over longer treatment durations has yet to be determined. Exciting preliminary work from our laboratory suggests that the 0.03 μg/mL T3 serum-restoring dosage used here can adequately restore cardiac tissue TH levels in 2 rat models of heart disease without elevating serum THs or inducing signs of hyperthyroidism (eg, tachycardia; Weltman, N., and A. Gerdes, unpublished observation, 2012). This encouraging observation suggests there may not be a serum-tissue mismatch when low cardiac TH function occurs secondarily to heart disease as opposed to our observations for primary hypothyroidism in the current study.
To further investigate this discrepancy during acute settings, mathematical modeling was performed to identify the serum TH parameter or parameters (T3, FT3, T4, TSH) which most reliably predict cardiac function and presumably cardiac tissue TH status (Supplemental Table 5). Our results suggest that serum TSH levels may provide the most accurate predictor of cardiac function and presumably cardiac tissue TH levels in settings of acute TH restoration after chronic hypothyroidism. Not surprisingly, no single serum TH marker appears able to adequately correlate all parameters of cardiac function. This observation, in conjunction with previous investigations from our group and others, suggest that measures of cardiac function are likely more reliable markers of cardiac tissue TH levels than measured serum THs when cardiac tissue hypothyroidism is suspected. In this respect, monitoring of resting heart rate may be a useful clinical indicator of cardiac hypothyroidism.
After establishing the presence of a serum and cardiac tissue T3 mismatch, we sought to investigate the potential mechanisms responsible for this mismatch. TH bioavailability and transport into the myocardium is crucial for normal TH homeostasis. Growing evidence suggests that TH transport across the plasma membrane is rate limiting in cellular TH metabolism and directly influences binding of T3 to its nuclear receptors (27–29). TH transport is thought to be a highly regulated, often energy and/or Na+ dependent, and saturable process (30–33), which may contribute to the observed serum and cardiac tissue TH mismatch. Altered expression of TH transporters has been observed during critical illness and likely contributes to altered TH bioavailability and metabolism in certain disease states (30). At present, TH transporter expression has not been explored in great depth in the heart, and there is very limited information regarding the relationship between TH status (serum and cardiac) and compensatory changes in expression of cardiac TH transporters. Therefore, we sought to characterize the relationship between TH status (serum and cardiac tissue) and changes in key TH transporters. Two unique members of the monocarboxylate transporter family, MCT-8 and MCT-10, appear to be responsible for most plasma membrane TH transport (31–33). In the present study, hypothyroidism led to an apparent compensatory overexpression of MCT-10 and a strong trend for increased MCT-8 expression (Figure 4A). Restoration of serum T3 (PTU + 0.03 T3) normalized the expression of MCT-10 but further increased the expression of MCT-8. We recently observed a similar MCT-8 gene expression pattern in diabetic rat hearts treated with a comparable replacement dosage of T3 (0.03 μg/mL T3; Weltman, N., and A. Gerdes, unpublished observation, 2012). This observation, along with recent evidence showing that MCT-10 is arguably a more active TH transporter than MCT-8, suggests that MCT-10 might play a larger role than MCT-8 in plasma membrane TH transport during low thyroid conditions (33).
In addition to TH transporter expression, we investigated the relationship between TH status (serum and tissue) and the expression of cytosolic TH binding proteins that appear responsible for intracellular TH retention. Cytosolic TH binding proteins are thought to serve as intracellular reservoirs for THs and potentially facilitate intracellular transfer and/or metabolism of THs. To our knowledge, 4 cytosolic T3 proteins have been identified in the literature: GST, Aldh, Crym, and pyruvate kinase (22, 34–36). Crym is a cytosolic binding protein with 8–34 times greater T3 binding affinity than the other above-mentioned intracellular TH binding proteins (34). Crym has been shown to increase the uptake and decrease the efflux of T3 by binding intracellular T3 in the presence of reduced nicotinamide adenine dinucleotide phosphate (NADPH), thereby serving as a cytoplasmic T3 reservoir (33, 37). In addition, the nicotinamide adenine dinucleotide phosphate activated form of Crym is thought to facilitate import and retention of T3 within the nucleus (37, 38). Cotransfection studies have demonstrated that the cellular uptake of T3, mediated by the TH transporters MCT-8 and MCT-10, is considerably augmented in the presence of Crym (33). Therefore, our initial observation of Crym gene suppression in the PTU-treated rat hearts appears counterintuitive (Figure 5A). However, further examination suggests that despite intracellular T3 retention, Crym might actually suppress T3-mediated gene expression through the thyroid receptor-thyroid responsive element interaction (37). Using a thyroid responsive element-luciferase reporter assay, Mori et al (37) reported that T3-mediated gene expression was inversely related to the level of intracellular Crym expression. It is plausible that decreased Crym gene expression may help liberate intracellular T3, thereby maximizing T3-mediated gene expression from all available T3 sources.
In addition to alterations in plasma membrane transport and intracellular retention of THs, it is plausible that exogenous TH administration may augment intracardiac T3 degradation by induction of D1 and D3 deiodinases. This, in turn, may contribute to the observed serum and cardiac tissue T3 mismatch. In the present study, no significant differences were observed in the cardiac expression of D1 or D3 among the varying groups. Based on these findings, it is highly unlikely that excess intracardiac T3 degradation by the induction of D1 and D3 deiodinases is responsible for the serum and tissue mismatch observed in the present experimental setting. This view is further strengthened by the well-established inhibitory influence of PTU on peripheral D1 activity (26). In contrast, cardiac D2 expression was elevated in hearts with confirmed cardiac tissue hypothyroidism (PTU and PTU + 0.03 T3), whereas D2 expression was suppressed with higher T3 dosing (PTU + 0.10 T3). Our findings are in agreement with Wagner et al (39), who reported elevated D2 expression and activity in hypothyroid rodent hearts and subsequent D2 suppression after a short duration T3 treatment.
Finally, we examined the influence of hypothyroidism and T3 replacement on the arteriolar resistance vessel network in the myocardium. THs are known regulators of the cardiac vasculature and directly influence vasoreactivity and coronary blood flow (1, 3, 19, 40). We have previously shown that hypothyroidism caused by PTU treatment or surgical thyroidectomy leads to significant reductions in both resting and maximal coronary blood flow (3, 11). This reduction in blood flow was attributed to a seemingly robust loss of small arteriolar resistance vessels when measured using α-SMA single labeling (3, 11). However, recent studies using IB4 and α-SMA colabeling suggest that single labeling of arterioles may underestimate the contribution of the smallest resistance vessels (19). In the present study, hypothyroidism was not associated with a reduction in length density when arterioles were colabeled with IB4 and α-SMA. However, after correcting for cardiac atrophy/LV mass reduction, PTU-treated rats had a significant reduction in total arteriolar length (Figure 6B). Restoration of LV mass with T3 treatment led to proportional and physiological arteriolar growth as evidenced by the dose-dependent changes in total arteriolar length. These findings are in agreement with our recent report, which used IB4 and α-SMA colabeling to examine arteriolar remodeling in thyroidectomized animals (19).
Limitations
Our study has several limitations. First, due to the high tissue requirement for our current tissue TH analysis method (∼350–500 mg), LV samples from animals within the same treatment groups were pooled together. Unfortunately, this does not allow for individual correlation between TH status (serum and tissue) and cardiac function, thus limiting statistical power. Second, rates of T3 uptake and cellular retention by the TH transporters and cytosolic TH binding proteins were not measured in the present study. Although other investigations have examined these transport properties and binding characteristics in culture or isolated myocyte systems, the aim of the present study was much different. Our goal was to characterize the influence of both systemic and local cardiac tissue TH status on the integrated expression profile of TH transporters and cytosolic binding proteins in a global organ setting. Finally, despite devoting considerable time and resources, we were unable to find reliable commercial antibodies needed to confirm protein expression changes for the reported TH transporters and cytosolic binding proteins. Future studies are needed to examine the influence of TH status on cardiac TH transporter and binding characteristics and to confirm changes in the protein expression of TH transporters and cytosolic TH binding proteins in intact cardiac models.
Conclusions
Our results demonstrate that the short-term administration of biologically active T3, designed to restore serum THs, led to a mismatch between the serum and cardiac tissue TH levels. Moreover, elevated physiological T3 dosing was required to fully normalize cardiac function. Additionally, this study provides the first in vivo characterization of coordinated changes in cardiac TH membrane transporters and cytosolic TH binding proteins during altered TH states.
Acknowledgments
We thank Dr Jie Li (University of South Dakota) for her assistance with LV echocardiography and Dr Yi-Fan Li (University of South Dakota) for his generosity in providing laboratory space for this experiment. The monoclonal antibody mAB s58 for MHC-β was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the Eunice Kennedy Shriver National Institute of Child Health and Human Development[b] and maintained by Department of Biology, The University of Iowa, Iowa City, Iowa. Author contributions included the following: N.Y.W., O.V.S., Y.-F.C., E.H.S., R.Z., A.S., D.C., and C.J.P. performed the experiments; N.Y.W. and A.M.G. analyzed the data and interpreted the results; N.Y.W. wrote the manuscript; N.Y.W., K.O., O.V.S., E.H.S., and A.M.G. edited and revised the manuscript; and N.Y.W., O.V.S., and A.M.G. developed the concept and the design of the research.
This study was supported by the National Institutes of Health Grants RO1 HL093160-01A1 (to A.M.G.) and RO1 HL103671-01A1 (to A.M.G.) and an American Diabetes Association Clinical Scientist Training Award 7-10-CST-01 (to N.Y.W. and A.M.G.). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Aldh
- aldehyde dehydrogenase
- AWd
- anterior wall thickness in diastole
- BW
- body weight
- Crym
- μu-crystallin
- Ct
- cycle threshold
- D
- deiodinase
- dP/dT max
- maximal rate of pressure development
- dP/dT min
- maximal rate of pressure decline
- FT3
- free T3
- GST
- glutathione-s-transferase
- HR
- heart rate
- HW
- heart weight
- IB4
- isolectin B4
- LAT
- large neutral amino acid transporter
- LD
- length density
- LV
- left ventricular
- LVSP
- LV systolic pressure
- LVW
- LV weight
- LVPWd
- LV posterior wall thickness in diastole
- LVPWs
- LV posterior wall thickness in systole
- MCT
- monocarboxylate transporter
- MHC
- myosin heavy chain
- PTU
- 6-n-propyl-2-thiouracil
- SLCO
- solute carrier organic anion transporter family
- α-SMA
- α-smooth muscle actin
- Tau
- time constant of isovolumic relaxation
- TH
- thyroid hormone.
References
- 1. Klein I, Ojamaa K. Mechanisms of disease: thyroid hormone and the cardiovascular system. N Engl J Med. 2001;344:501–509 [DOI] [PubMed] [Google Scholar]
- 2. Kahaly G, Dillmann W. Thyroid hormone action in the heart. Endocr Rev. 2005;26:704–728 [DOI] [PubMed] [Google Scholar]
- 3. Tang Y, Kuzman J, Said S, Anderson B, Wang X, Gerdes A. Low thyroid function leads to cardiac atrophy with chamber dilatation, impaired myocardial blood flow, loss of arterioles, and severe systolic dysfunction. Circulation. 2005;112:3122–3130 [DOI] [PubMed] [Google Scholar]
- 4. Tseng F, Lin W, Lin C, et al. Subclinical hyopthyroidism is associated with increased risk for all-cause and cardiovascular mortality in adults. J Am Coll Cardiol. 2012;60:730–737 [DOI] [PubMed] [Google Scholar]
- 5. McQuade C, Skugor M, Brennan D, Hoar B, Stevenson C, Hoogwerf B. Hypothyroidism and moderate subclinical hypothyroidism are associated with increased all-cause mortality independent of coronary heart disease risk factors: a PreCIS database study. Thyroid. 2011;21:837–843 [DOI] [PubMed] [Google Scholar]
- 6. Iervasi G, Molinaro S, Landi P, et al. Association between increased mortality and mild thyroid dysfunction in cardiac patients. Arch Intern Med. 2007;167:1526–1532 [DOI] [PubMed] [Google Scholar]
- 7. Gerdes A, Iervasi G. Thyroid replacement therapy and heart failure. Circulation. 2010;122:385–393 [DOI] [PubMed] [Google Scholar]
- 8. Wiersinga W. Do we need still more trials on T4 and T3 combination therapy in hypothyroidism? Eur J Endocrinol. 2009;161:955–959 [DOI] [PubMed] [Google Scholar]
- 9. Grozinsky-Glasberg S, Fraser A, Nahshoni E, Weizman A, Leibovici L. Thyroxine-triiodothyronine combination therapy versus thyroxine monotherapy for clinical hypothyroidism: meta-analysis of randomized controlled trials. J Clin Endocrinol Metab. 2006;91:2592–2599 [DOI] [PubMed] [Google Scholar]
- 10. Escobar-Morreale H, Obregon M, del Rey F, de Escobar G. Replacement therapy for hypothyroidism with thyroxine alone does not ensure euthyroidism in all tissues, as studied in thyroidectomized rats. J Clin Invest. 1995;96:2828–2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu Y, Redetzke R, Said S, Pottala J, de Escobar G, Gerdes A. Serum thyroid hormone levels may not accurately reflect thyroid tissue levels and cardiac function in mild hypothyroidism. Am J Physiol Heart Circ Physiol. 2008;294:H2137–H2143 [DOI] [PubMed] [Google Scholar]
- 12. Cooper D, Kieffer D, Halpern R, et al. Propylthiouracil (PTU) pharmacology in the rat, II. Effects of PTU on thyroid function. Endocrinology. 1983;113:921–928 [DOI] [PubMed] [Google Scholar]
- 13. East-Palmer J, Szkudlinski MW, Lee J, Totakura NR, Weintraub BD. A novel, nonradioactive in vivo bioassay of thyrotrophin (TSH). Thyroid. 1995;5:55–59 [DOI] [PubMed] [Google Scholar]
- 14. Saba A, Chiellini G, Frascarelli S, et al. Tissue distribution and cardiac metabolism of 3-iodothyronamine. Endocrinology. 2010;151:5063–5073 [DOI] [PubMed] [Google Scholar]
- 15. Pol C, Muller A, Zuidwijk M, et al. Left-ventricular remodeling after myocardial infarction is associated with a cardiomyocyte-specific hypothyroid condition. Endocrinology. 2011;152:669–679 [DOI] [PubMed] [Google Scholar]
- 16. Tamura T, Said S, Harris J, Lu W, Gerdes A. Reverse remodeling of cardiac myocyte hypertrophy in hypertension and failure by targeting of the renin-angiotensin system. Circulation. 2000;102:253–259 [DOI] [PubMed] [Google Scholar]
- 17. Weltman N, Wang D, Redetzke R, Gerdes A. Longstanding hyperthyroidism is associated with normal or enhanced intrinsic cardiomyocyte function despite decline in global cardiac function. Plos One. 2012;7:e46655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zimmer H, Gerdes A, Lortet S, Mall G. Changes in heart function and cardiac cell size in rats with chronic myocardial infarction. J Mol Cell Cardiol. 1990;22:1231–1243 [DOI] [PubMed] [Google Scholar]
- 19. Savinova O, Liu YH, Aasen G, et al. Thyroid hormone promotes remodeling of coronary resistance vessels. Plos One. 2011;6:e25054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Adair T, Wells M, Hang J, Montani J. A stereological method for estimating length density of the arterial vascular system. Am J Physiol. 1994;266:H1434–H1438 [DOI] [PubMed] [Google Scholar]
- 21. Kinugawa K, Minobe W, Wood W, et al. Signaling pathways responsible for fetal gene induction in the failing human heart—evidence for altered thyroid hormone receptor gene expression. Circulation. 2001;103:1089–1094 [DOI] [PubMed] [Google Scholar]
- 22. Ishigaki S, Abramovitz M, Listowsky I. Glutathione-S-transferases are major cytosolic thyroid hormone binding proteins. Arch Biochem Biophys. 1989;273:265–272 [DOI] [PubMed] [Google Scholar]
- 23. Lee E, Okuno S, Kariya K. Propylthiouracil inducible glutathione transferases. Selective induction of ligandin (glutathione transferase 1-1). Biochem Pharmacol. 1989;35:1835–1839 [DOI] [PubMed] [Google Scholar]
- 24. Chernow B, Burman K, Johnson D, et al. T3 may be a better agent than T4 in the critically ill hypothyroid patient: evaluation of transport across the blood-brain barrier in a primate model. Crit Care Med. 1983;11:99–104 [DOI] [PubMed] [Google Scholar]
- 25. Schneider M, Fiering S, Pallud S, Parlow A, St Germain D, Galton V. Targeted disruption of the type 2 selenodeiodinase gene (D102) results in a phenotype of pituitary resistance to T4. Mol Endocrinol. 2001;15:2137–2148 [DOI] [PubMed] [Google Scholar]
- 26. Bianco AC, Kim BS. Deiodinases: implications of the local control of thyroid hormone action. J Clin Invest. 2006;116:2571–2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hennemann G, Everts M, de Jong M, Lim C, Krenning E, Docter R. The significance of plasma membrane transport in the bioavailability of thyroid hormone. Clin Endocrinol (Oxf). 1998;48:1–8 [DOI] [PubMed] [Google Scholar]
- 28. Hennemann G, Docter R, Friesema E, De Jong M, Krenning E, Visser T. Plasma membrane transport of thyroid hormones and its role in thyroid hormone metabolism and bioavailability. Endocr Rev. 2001;22:451–476 [DOI] [PubMed] [Google Scholar]
- 29. Friesema E, Kuiper G, Jansen J, Visser T, Kester M. Thyroid hormone transport by the human monocarboxylate transporter 8 and its rate-limiting role in intracellular metabolism. Mol Endocrinol. 2006;20:2761–2772 [DOI] [PubMed] [Google Scholar]
- 30. Mebis L, Paletta D, Debaveye Y, et al. Expression of thyroid hormone transporters during critical illness. Eur J Endocrinol. 2009;161:243–250 [DOI] [PubMed] [Google Scholar]
- 31. Friesema E, Ganguly S, Abdalla A, Fox J, Halestrap A, Visser T. Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem. 2003;278:40128–40135 [DOI] [PubMed] [Google Scholar]
- 32. Visser W, Friesema E, Visser T. Minireview: thyroid hormone transporters: the knowns and the unknowns. Mol Endocrinol. 2011;25:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Friesema E, Jansen J, Jachtenberg JW, Visser W, Kester M, Visser T. Effective cellular uptake and efflux of thyroid hormone by human monocarboxylate transporter 10. Mol Endocrinol. 2008;22:1357–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Suzuki S, Suzuki N, Mori J, Oshima A, Usami S, Hashizume K. mu-Crystallin as an intracellular 3,5,3′-triiodothyronine holder in vivo. Mol Endocrinol. 2007;21:885–894 [DOI] [PubMed] [Google Scholar]
- 35. Ashizawa K, Cheng S. Regulation of thyroid-hormone receptor-mediated transcription by a cytosol protein. Proc Natl Acad Sci USA. 1992;89:9277–9281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yamauchi K, Nakajima J, Hayashi H, Horiuchi R, Tata J. Xenopus cytosolic thyroid hormone-binding protein (xCTBP) is aldehyde dehydrogenase catalyzing the formation of retinoic acid. J Biol Chem. 1999;274:8460–8469 [DOI] [PubMed] [Google Scholar]
- 37. Mori J, Suzuki S, Kobayashi M, et al. Nicotinamide adenine dinucleotide phosphate-dependent cytosolic T3 binding protein as a regulator for T3-mediated transactivation. Endocrinology. 2002;143:1538–1544 [DOI] [PubMed] [Google Scholar]
- 38. Hashizume K, Miyamoto T, Ichikawa K, et al. Counterregulation of nuclear 3,5,3′-triiodo-L-thyronine (T3) binding by oxidized and reduced-nicotinamide adenine dinucleotide phosphates in the presence of cytosolic T3-binding protein in vitro. Endocrinology. 1989;124:1678–1683 [DOI] [PubMed] [Google Scholar]
- 39. Wagner M, Morimoto R, Dora J, Benneman A, Pavan R, Maia A. Hypothyroidism induces type 2 iodothyronine deiodinase expression in mouse heart and testis. J Mol Endocrinol. 2003;31:541–550 [DOI] [PubMed] [Google Scholar]
- 40. Luidens M, Mousa S, Davis F, Lin H, Davis P. Thyroid hormone and angiogenesis. Vasc Pharmacol. 2010;52:142–145 [DOI] [PubMed] [Google Scholar]