

Abstract
Ribonuclease HI (RNase H) is a member of the nucleotidyl-transferase superfamily and endo-nucleolytically cleaves the RNA portion in RNA/DNA hybrids and removes RNA primers from Okazaki fragments. The enzyme also binds RNA and DNA duplexes but is unable to cleave either. Three-dimensional structures of bacterial and human RNase H catalytic domains bound to RNA/DNA hybrids have revealed the basis for substrate recognition and the mechanism of cleavage. In order to visualize the enzyme’s interactions with duplex DNA and to establish the structural differences that afford tighter binding to RNA/DNA hybrids relative to dsDNA, we have determined the crystal structure of Bacillus halodurans RNase H in complex with the B-form DNA duplex [d(CGCGAATTCGCG)]2. The structure demonstrates that the inability of the enzyme to cleave DNA is due to the deviating curvature of the DNA strand relative to the substrate RNA strand and the absence of Mg2+ at the active site. A subset of amino acids engaged in contacts to RNA 2′-hydroxyl groups in the substrate complex instead bind to bridging or non-bridging phosphodiester oxygens in the complex with dsDNA. Qualitative comparison of the enzyme’s interactions with the substrate and inhibitor duplexes is consistent with the reduced binding affinity for the latter and sheds light on determinants of RNase H binding and cleavage specificity.
Keywords: A-form, B-form, DNA sliding, minor groove, metal ions, ribose, X-ray crystallography
Introduction
Ribonuclease H (RNase H) hydrolyzes the RNA portion of RNA/DNA hybrids and of heteroduplexes between chimeric RNA-DNA strands and DNA non-sequence specifically.1 The RNase H family of enzymes comprises members of various molecular masses, but endonuclease activity and substrate requirements are similar for all of them and include the dependence on metal ions (Mg2+, Mn2+) and cleavage products with 5′-phosphate and 3′-hydroxyl termini.2 RNases H can be classified into type 1 and type 2 enzymes, whereby bacterial RNase HI, yeast RNase H1, mammalian RNase H2 and reverse transcriptase RNase H domains are of type 1, and bacterial RNase HII and HIII, archaeal RNase HII, yeast RNase H2 and mammalian RNases H1 constitute type 2 enzymes.3 In eukaryotes, type 2 enzymes are active during replication and are necessary for removal of RNA primers during lagging strand synthesis (ref. 4 and cited refs.). Among the type 1 bacterial enzymes, RNase HI from E. coli has been studied in particular detail.5 It is noteworthy that the enzyme not only binds RNA/DNA hybrids, but in addition displays affinity for both RNA and DNA duplexes (dsRNA and dsDNA, respectively) as well as single-stranded oligonucleotides.6 RNA/DNA and dsRNA are bound with similar affinities and the KD for the complex with a 17mer dsRNA is ca. 1 µM. This binding strength exceeds that to dsDNA ca. 60-fold and single strands are bound with even lower affinity (ca. 300-reduction in KD relative to the natural substrate). Importantly, non-cleavable dsRNA, dsDNA, and heteroduplexes between RNA and chemically modified antisense oligonucleotides (AONs) all function as competitive inhibitors.6 The activities of E. coli6,7 and human RNase H8 with the latter duplexes were assayed extensively because the enzyme is considered a crucial factor for potent antisense activity by artificial oligonucleotides.9,10 However, with a few exceptions, i.e. phosphorothioate-DNA (PS-DNA)11 and 2′-deoxy-2′-fluoroarabinonucleic acid (2′-FANA)12, chemically modified AONs do not elicit RNase H action. Thus, RNase H does not cleave RNAs bound to complementary AONs with 2′-modified carbohydrate moieties.13,14
The crystal structure of E. coli RNase H (Ec-RNase H) alone was determined many years ago.15,16 However, structures of complexes remained elusive until recently, despite many attempts that were undoubtedly made to grow diffraction-quality crystals of the enzyme bound to either substrate (i.e. refs. 17, 18) or inhibitor duplexes (dsRNA).19 Instead, recent crystal structures for the catalytic domains of B. halodurans RNase H (Bh-RNase HC)18 and for human RNase H1 (Hs-RNase HC)20 in complex with RNA/DNA hybrids have shed light on the conformation of the substrate duplex at the active site of the enzyme. Despite similar affinities for RNA/DNA and dsRNA by RNase H, the geometry of the former duplex clearly deviates from the canonical A-form and the majority of 2′-deoxyriboses adopt puckers more typical of a B-form backbone. The specificity for the RNA strand is established by contacts to five consecutive 2′-hydroxyl groups in the complex of Bh-RNase HC. Hs-RNase H and Ec-RNase H both feature a basic protrusion that is absent in Bh-RNase H. The structure of the Hs-RNase H-substrate complex revealed extensive interactions between enzyme and DNA strand and variations in the conformation of the DNA between the Band A-forms in the vicinity of the above protrusion that are missing in the complex with Bh-RNase H.20 These structures together with those of product complexes with different mutant proteins21 have also helped settle the role of metal ions in RNase H catalysis and clearly support a two metal-ion mechanism.
It was pointed out that the specificity of RNase H depends both on the binding interaction (duplex shape) and the catalytic process.6 Differential binding strengths to dsRNA, RNA/DNA and dsDNA allow the enzyme to selectively bind RNA-containing duplexes despite an excess of double-stranded DNA. A relatively weak interaction between RNase H and nucleic acid duplexes, particularly with dsDNA, would allow the enzyme to more efficiently scan double strands in order to locate the hybrid duplex substrate and thus lead to improved turnover. The productive binding interaction in the minor groove must entail contacts to both the RNA and the DNA strands18,20,22 and cleavage is sensitive to steric factors and conformational variations in the geometry of the sugarphosphate backbone, particularly as they affect the minor groove width.8,18,20,23–27 The interactions with non-target DNA sequences, mainly by electrostatic interactions, of enzymes that scan dsDNA in order to locate the intended substrate have been studied in some detail with repressor proteins,28 endonucleases involved in DNA repair,29,30 and restriction enzymes.31–34 However, the interactions between the above proteins and dsDNA are sequence-nonspecific. By comparison, the difference between specific RNase H:RNA/DNA and non-speficic RNase H:dsDNA binding does not concern the presence or absence, respectively, of interactions with nucleobase atoms, but relates to the nature of the carbohydrate moiety in the backbone (2′-ribose vs. 2′-deoxyribose) and the duplex shape (A-form vs. B-form).
To shed light on the non-specific binding mode between RNase H and dsDNA (inhibitor) we have determined the crystal structure of the complex between Bh-RNase HC and the dodecamer duplex [d(CGCGAATTCGCG)]2. More crystallographic analyses of this DNA duplex, the so-called Dickerson-Drew Dodecamer (DDD), have been carried out than for any other DNA (i.e. refs. 35–39) during the last 25 years. The availability of detailed structures of the DNA alone facilitates the identification of putative conformational adaptations of the duplex as a result of the interaction with RNase H. Comparison of the structure of the DDD complex with that of the published complex between Bh-RNase HC and an RNA/DNA 12mer18 allows an amino acid-by-amino acid visualization of the changes at the protein-duplex interfaces. As pointed out above, the enzyme does not cleave RNA sequence-specifically and the fact that the sequences of the two duplexes are different is therefore irrelevant in terms of an interpretation of the altered binding modes. Our structure helps rationalize the lower binding affinity of the enzyme to dsDNA compared with an RNA/DNA hybrid. Moreover, it implicates not only minor groove width and presence of ribose 2′-hydroxyl groups but also strand curvature as a major determinant of RNase H substrate recognition and cleavage specificity.
Results
Overall Structure of the RNase H:dsDNA Complex
The crystal structure reveals two independent DDD duplexes that both interact with two protein molecules. One of these duplex : protein complexes with 1:2 stoichiometry is depicted in Fig. 1A. The duplexes are located on crystallographic dyads and the asymmetric unit is therefore composed of two dodecamer strands and two RNase H molecules. All 24 nucleotides and 133 (134 in the second subunit) of the 139 residues per Bh-RNase HC are resolved in the electron density map; an example of the quality of the final density is depicted in Fig. 1B. The space group, C2, of the RNase H:DDD complex is the same as that of the published crystal structure of the complex between the identical protein (Asp132→Asn mutant) and a dodecamer RNA/DNA hybrid.18 Crystal data and refinement statistics are listed in Table 1. Although the crystallographic unit cell constants of the two complex crystals are different, their unit cell volumes are very similar. Thus, the binding of two protein molecules per duplex is a shared feature of both structures. However, the RNA/DNA hybrid lies in a general location and the binding modes of the two independent RNase H molecules are slightly different in the enzyme:substrate complex. By contrast, they are identical in the DDD structure (Fig. 1A). Because the RNase H complexes with the two independent DDD duplexes are also very similar (Fig. 2), it is sufficient to analyze the interactions between a single Bh-RNase HC molecule and six base pairs of the dodecamer duplex (Fig. 3) in order to compile a unique set of protein-DNA contacts.
Figure 1.


Overall structure of the Bh-RNase HC:dsDNA complex and quality of the model. (A) The DDD complexed by two RNase H molecules (ribbon cartoons colored gray and blue) is viewed into the A-tract minor groove and nucleotides are colored green (G), yellow (C), red (A) and blue (T). Protein chain termini and terminal nucleotides are labeled and solid lines in black trace the helix axis. Asterisks indicate that the two strands of the DDD are symmetry-related in the crystal structure of the complex. (B) Final Fourier (2Fo-Fc) sum electron density contoured at the 1σ level in the region of Trp139 that interacts with the phosphate group of A5 (upper red residue).
Table 1.
Selected crystal data, data collection and refinement parameters*
| Data collection | |
|---|---|
| Space group | C2 |
| Cell dimensions a, b, c (Å), β (°) | 98.40, 66.66, 76.93, 122.3 |
| Wavelength (Å) | 0.9785 Å |
| Resolution (last shell; Å) | 42.0–1.80 (1.86-1.80) |
| Unique reflections | 37,768 (3,643) |
| Completeness (%) | 97.3 (94.7) |
| R-merge | 0.061 (0.248) |
| I/σ(I) | 42.7 (4.6) |
| Refinement | |
| Working set reflections | 35,877 |
| Test set reflections (5 %) | 1,886 |
| No. protein / DNA / H2O / Na+ | 2,169 / 486 / 167 / 1 |
| R-work / R-free (5%) | 0.215 / 0.241 |
| Average B-factors (Å2) | |
| Protein / DNA / solvent | 30. 9 / 29.4 / 37.3 |
| R.m.s. deviations | |
| bond lengths (Å) | 0.016 |
| bond angles (°) | 1.7 |
Values in parentheses refer to the last resolution shell.
Figure 2.
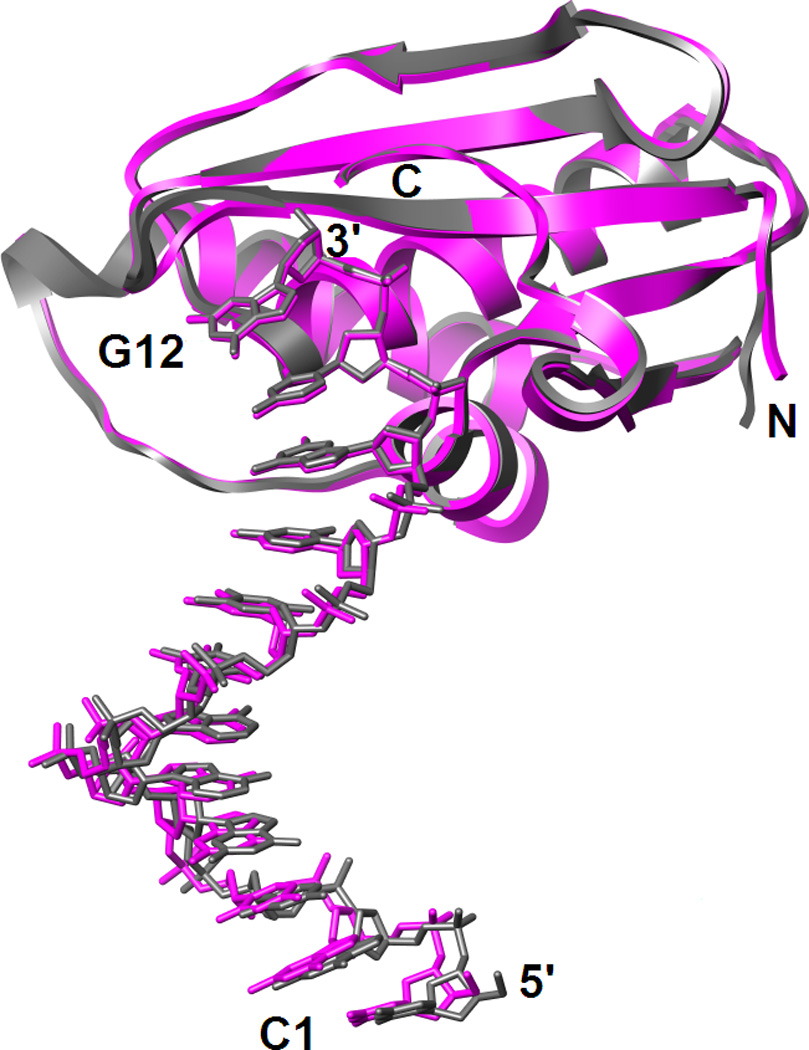
The two independent Bh-RNase HC:DDD complexes per crystallographic asymmetric unit adopt virtually identical conformations as revealed by the superimposition of complexes 1 (magenta) and 2 (gray). The two complexes are related by a translation of ca. 43 Å along the ac diagonal.
Figure 3.
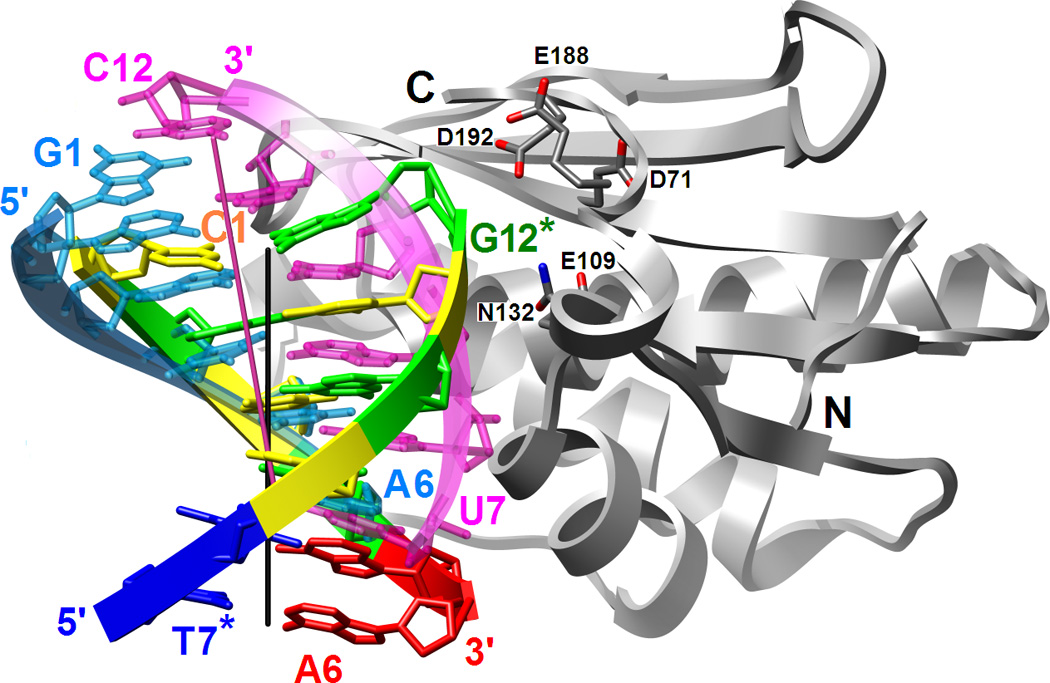

Binding mode of RNase H in the minor groove of the DDD G-tract and comparison between the relative orientations of the RNA/DNA and DNA duplexes bound to the enzyme. (A) Superimposition of the Bh-RNase HC molecules in the complexes with DDD and RNA/DNA hybrid (PDB ID code 1zbi18) reveals a two base-pair shift of the duplexes relative to one another. The enzyme adopts virtually identical conformations in the two structures and only one of the superimposed RNase H molecules is depicted. Only six base pairs per duplex are shown; the color code of residues in the DDD is the same as in Fig. 1 and DNA and RNA strands in the hybrid are colored cyan and pink, respectively. Chain termini and active site Asp and Glu(Gln) residues as well as 5′- and 3′-terminal nucleotides are labeled and solid lines in black and pink represent the overall helical axes for dsDNA and hybrid hexamers, respectively. The view is across the major and minor grooves. (B) The superimposed complexes rotated by 90° around the vertical relative to panel A and viewed into the major grooves of hexameric duplexes.
Bh-RNase HC binds to the G-C portion of the DDD from the minor groove side and kinks the dodecamer symmetrically into the major groove at GC/AT junctions (Fig. 1A). The roll-bend between A-tract and G-tracts amounts to 17°, thereby illustrating the rigid nature of the central AATT portion and confirming the junction regions as the source of A-tract bending (ref. 40 and cited refs.). The A-tract minor groove is very narrow and the central tetramer acts as a spacer between the G-tract protein binding sites that exhibit a wider minor groove (Fig. 1A). Consistent with the positioning of RNase H above the minor groove of the G-C portion of the duplex, the majority of interactions involve the sugar-phosphate backbone of G and C residues (Fig. 3). Additional contacts are established to the phosphates of nucleotides A5 and A6. An overview of direct and water-mediated protein-DDD interactions is provided in Table 2. Nucleotides in the DDD duplex are numbered C1 to G12 in the first strand and C1* to G12* in the symmetry-related second strand. The protein forms contacts to five residues of the first strand (G2 to A6), of which two involve hydrogen bonds to the 4′-oxygen of the deoxyribose moiety. Contacts to the opposite strand are made to bridging and non-bridging phosphate oxygens from residues C11* and the terminal G12*. The phosphate group of the latter is located in the immediate vicinity of the active site although no Mg2+ ions were observed in the structure of the Bh-RNase HC:DDD complex. In addition to these direct protein-DNA contacts 12 water-mediated interactions are found at the binding interface. Besides the asymmetry in the number of nucleotides from opposite strands probed by the enzyme, the absence of basic amino acids among those gripping the DDD backbone is most noteworthy. Thus, residues establishing hydrogen bonds to phosphate groups include Asn, Gln, Ser, Thr and Trp. We will discuss the potentially unfavorable interaction between the active site residue Asp192 and the phosphate of G12* below (Table 2). None of the direct interactions between amino acid side chains and DNA residues involve nucleobase atoms, consistent with the observation that the enzyme hydrolyzes the RNA substrate strand nonsequence specifically. The observation that the enzyme binds in the minor groove of both RNA/DNA substrate and DNA inhibitor duplexes indicates that the spacing of the sugar-phosphate backbones is important for RNase H recognition and binding.
Table 2.
Protein-nucleic acid interactions in the Bh-RNase HC complexes with the DDD and RNA/DNA hybrid duplexes.*
| DDD base- pairs |
DNA-protein interactions |
H2O-mediated DNA-protein interactions |
DNA-protein interactions |
H2O-mediated DNA-protein interactions |
DNA/RNA base- pairs |
DNA-protein interactions |
H2O-mediated DNA-protein interactions |
RNA-protein interactions |
|---|---|---|---|---|---|---|---|---|
| C1→A6 | C1→A6 | G12*←T7* | G12*←T7* | A2→G7 | A2→G7 | U11←C6 | ||
| A2-U11 | O2′ - 76 G-NH | |||||||
| C1-G12* | O2 - W1 - 77 N |
O3′ - 74 S (O1P -192 D) (O2P -192 D) |
O3′ - W116 - 193 Y O1P - W62 - 72 V/192 D/193 Y O1P - W25 - 72 V O2P - W72 - 192 D |
A3-U10 | N3 - W143 - 77 N |
O2′ - 74 S O4′ - 105 N O1P - Mg2+a/ 71 D O1P - 74 S |
||
| G2-C11* | O4′ - 77 N | O4′ - W1 - 77 N |
O3′ - 132 N O2P - 183 T |
T4-A9 | O2 - 77 N | O4′ - W143 - 77 N |
N3 - 105 N O2′ - 109 E O1P - 180 K |
|
| C3-G10* | O4′ - 77 N | O2P - W2 - 148 T | C5-G8 | O2 - 106 N O4′ - 77 N |
O2P - W135 - 148 T | O2′-134 Q-NH | ||
| G4-C9* | O4′ - 106 N O1P - 148 T O2P - 104 T O2P - 147 S |
N2 - W74 - 106 N/135 T N3 - W26 - 135 T |
A6-U7 | O4′ - 106 N O1P - 104 T O1P - 147 S O2P - 148 T |
N3 - W127 - 125 T | O2′ - 134 Q | ||
| A5-T8* | O4′ - 135 T O1P - 139 W |
G7-C6 | O1P - 139 W | O4′ - W127 - 134 Q/ 135 T O2P - W174 - 146 K |
||||
| A6-T7* | O1P - 134 Q | |||||||
Ribonucleotides/atoms in the hybrid18 and DNA nucleotides/atoms in the DDD strand that corresponds to the hybrid RNA strand are in bold font and symmetry-related DDD residues are marked by an asterisk. The upper distance cut-off for hydrogen bonds was 3.3 Å.
Comparison between the Protein-Nucleic Acid Binding Interfaces in RNase H Complexes with RNA/DNA and dsDNA
The Bh-RNase HC enzyme adopts very similar conformations in the structures of the complexes with the DDD and a 12mer RNA/DNA hybrid18 (Fig. 4). To determine the relative orientations of the hybrid and DDD duplexes we superimposed the protein molecules from the two complexes. As illustrated in Fig. 3, such a comparison reveals a shift as well as a slight rotation of the DDD relative to the RNA/DNA hybrid duplex bound to RNase H. Shift and rotation amount to ca. 5 Å (or two base pairs) and 10°, respectively, with the phosphate of the 3′-terminal G12 of the DDD being positioned closest to active site Asp and Glu residues (Fig. 3A). By comparison, in the structure of the complex with the RNA/DNA substrate r(GACACCUGAUUC)/d(GAATCAGGTGTC) (residues are numbered 1 through 12 in both strands), the phosphate of residue rU10 is lodged at the active site. These differences are accompanied by a marked reduction in the area of the binding interface in the DDD (ca. 600 Å2) compared to the RNA/DNA complex (ca. 900 Å2). The reduced binding interface with dsDNA is most likely also a consequence of the different curvature of the DDD and RNA/DNA duplexes (Fig. 3).
Figure 4.
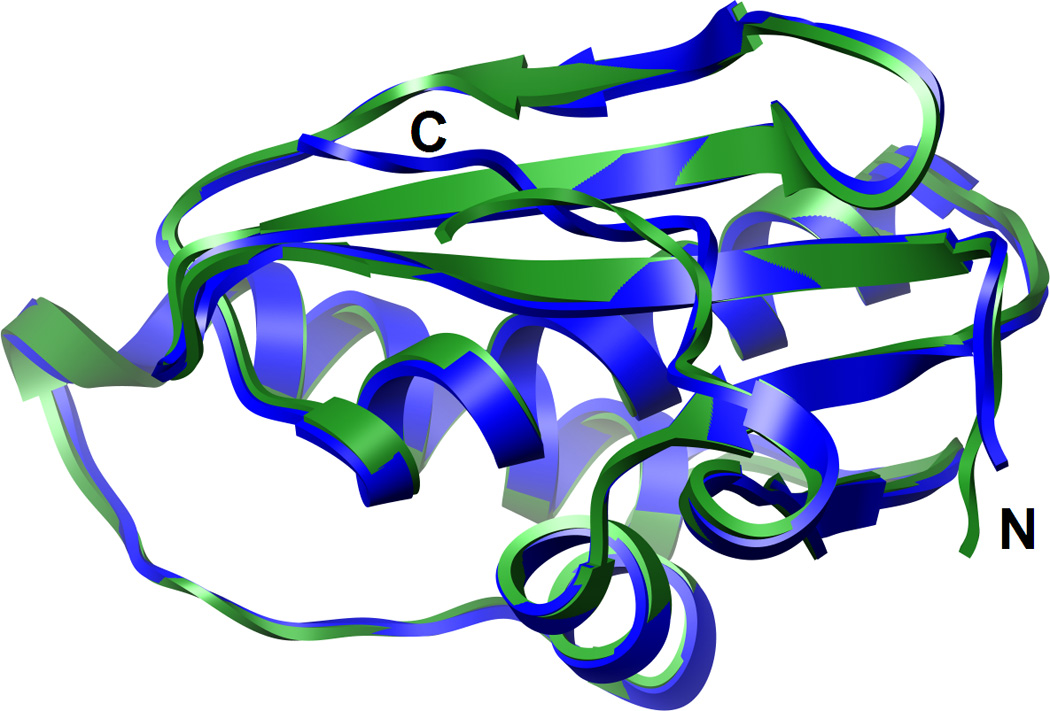
The superimposition of the Bh-RNase HC proteins in the complexes with the DDD (blue) and the RNA/DNA hybrid duplex (green) demonstrates that they adopt very similar conformations.
Of the 12 direct contacts between RNase H and duplex, only 3 are to the strand that corresponds to the RNA dodecamer in the complex with the hybrid (Table 2). A total of 17 direct protein-nucleic acid interactions are observed in the complex with the hybrid duplex of which 9 are formed to the RNA strand. Availability of RNase H structures bound to substrate and inhibitor duplexes provides an opportunity to directly compare the roles of individual amino acids at the binding interfaces. For example, in the complex with RNA/DNA the enzyme established hydrogen bonds to 2′-hydroxyl groups of five successive RNA residues, rU7, rG8, rA9, rU10 and rU11. The amino acids participating in these contacts are Gln134, Gln134-NH, Glu109, Ser74 and Gly76-NH (in the above order). Of these, Ser74 contacts a bridging 3′-oxygen atoms and Gln134 forms a hydrogen bond to a phosphate from the opposite strand in the DDD complex. This switching of strands is a consequence of the different orientation and topology of the DNA duplex relative to RNA/DNA. Glu109 is part of the active site (Fig. 5) and is coordinated to the Mg2+ ion occupying the B site in the substrate complex besides hydrogen bonding to the 2′-OH of A9 (18). In the DDD complex, the side chain of Glu109 is rotated away and does not lie close enough to the DNA backbone to form a direct interaction (Fig. 5B). Thus, whereas five residues in the RNA strand are contacted by amino acids in the complex with the RNA/DNA hybrid, only two residues of the corresponding strand in the DDD duplex are probed by the enzyme.
Figure 5.

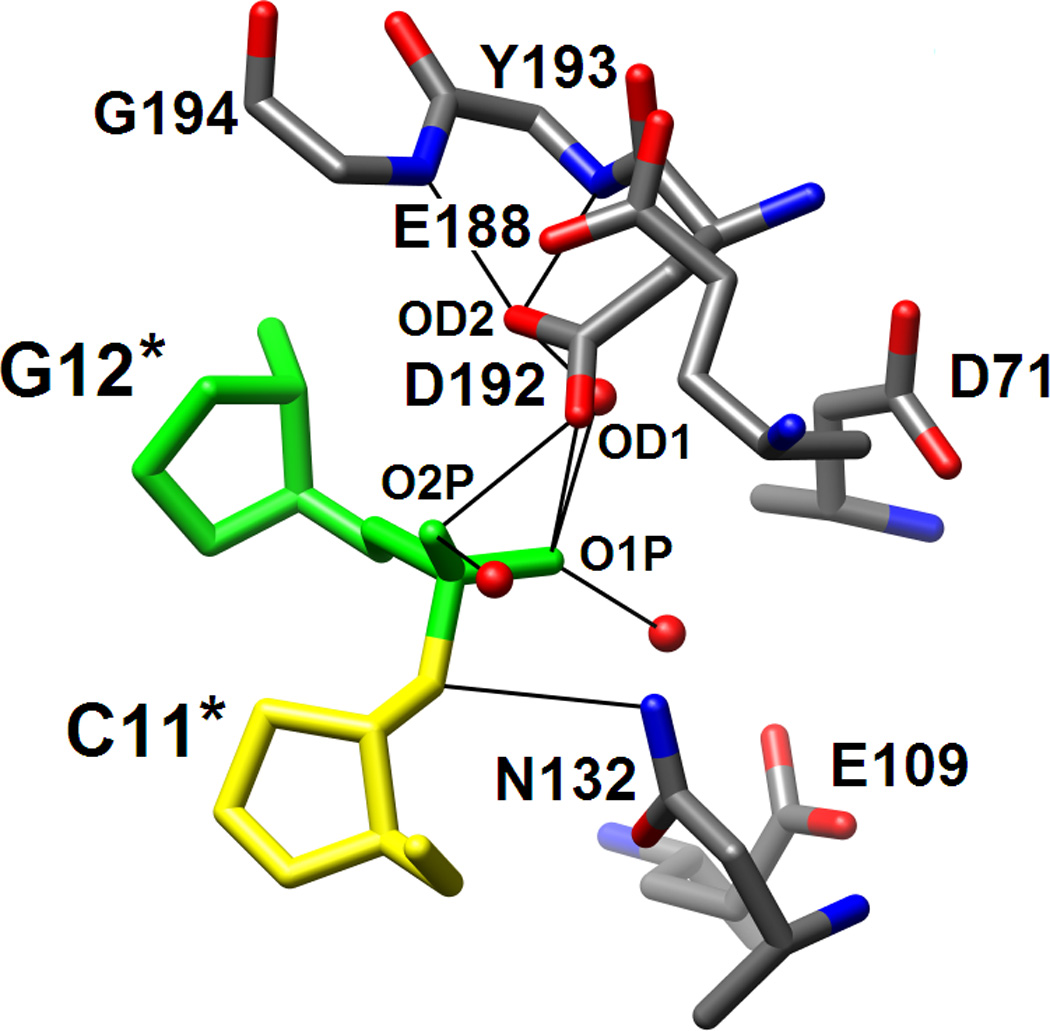
RNase H active site location and configurations in the complexes with DDD and RNA/DNA hybrid. (A) Electrostatic surface potential of Bh-RNase H; red and blue regions indicate negative and positive charge, respectively, and the potential scale ranges from −50 to +50. Only half the DDD duplex is shown and the color code of nucleotides is identical to that in Fig. 1. The locations of the active site (circled) and the phosphate binding site (boxed) can easily be recognized as clefts accommodating the phosphates of residues G12* and G4, respectively. (B) Active site configuration in the DDD complex. Sugar moieties and amino acids as well as phosphate and Asp192 carboxyl oxygen atoms are labeled. Hydrogen bonds and short contacts (Asp192…phosphate; see Results text) are indicated by thin lines. (C) Active site configuration in the RNA/DNA hybrid complex. Sugar moieties, amino acids and A- and B-site Mg2+ ions are labeled. Hydrogen bonds and Mg2+ coordination spheres are indicated by thin solid lines. The path of the water nucleophile to carry out the attack at the phosphate group of U10 is indicated by an arrow and the scissile bond is marked by an asterisk. The viewing direction in panels B and C is identical to that in Fig. 3A.
Most of the amino acids that contact the DNA strand of the RNA/DNA hybrid engage in similar interactions in the complex with the DDD, albeit with nucleotides that are closer to the 5′-end of the strand in the latter (Fig. 3, Table 2). Thus, Asn77 hydrogen bonds to 4′-oxygen atoms of G2 and C3 (O2[T4] and O2/O4′ [C5], RNA/DNA), Thr104 H-bonds to O2P of G4 (O1P[A6], R/D), Asn106 H-bonds to O4′ of G4 (O4′[A6] R/D), Ser147 H-bonds to O2P of G4 (O1P[A6], R/D), Thr148 H-bonds to O1P of G4 (O2P[A6], R/D), and Trp139 H-bonds to O1P of A5 (O1P[G7], R/D). Overall, ten amino acids form very similar interactions in the RNase H-DDD and -RNA/DNA complexes, whereby 2′-hydroxyl groups in the latter are traded for bridging and non-bridging phosphate oxygens in the complex with dsDNA. Among the features that are virtually conserved in the substrate and inhibitor complexes is the so-called phosphate-binding pocket18 In the DDD complex, the phosphate of G4 is surrounded by residues Thr104, Ser147 and Thr148 in a fashion very similar to the situation in the substrate complex. The two base-pair spacing between the DNA residue whose phosphate is trapped in this pocket and the ‘scissile’ phosphate on the RNA (DNA) strand is the same in the two complexes (Fig. 5A). Additional interactions involving G4 in the DDD complex include a contact to O4′ by Asn106 and to its minor groove edge by the same amino acid and Thr135 via water bridges (Table 2). The corresponding residue A6 in the DNA strand of the hybrid also features a water-mediated contact between the minor groove edge of its base and the protein, but the partner amino acid is Thr125.
No direct protein-DNA nucleobase interactions are observed in the DDD complex compared with 3 such interactions in the complex with RNA/DNA hybrid. This disparity is related to another key difference between the complexes with substrate RNA/DNA and inhibitor dsDNA, namely the number of observed water-mediated amino acid-backbone or -base interactions. No fewer than 12 water-mediated interactions between protein and DNA exist in the DDD complex compared with only 7 interactions of this type in the RNA/DNA complex (Table 2). The presence of a sheath of 12 water molecules at the protein-DNA interface in the inhibitor complex and a comparably much drier binding interface in the substrate complex (7 water molecules) is consistent with the tighter binding of RNA/DNA relative to dsDNA by RNase H and a more optimal fit between enzyme and hybrid duplex. Although the sum of direct and indirect protein-nucleic acid interactions is the same in the complexes with RNA/DNA and dsDNA (24), 70% of these are direct contacts in the complex with the hybrid compared to only 50% direct contacts in the DDD complex.
Configuration of the Active Site
The most obvious difference between the active sites in the DDD (Fig. 5B) and RNA/DNA (Fig. 5C) complexes is the presence of two Mg2+ ions in the latter and their absence in the former. In both complexes Asp132 is mutated to asparagine and the resulting change in local electrostatics is therefore unlikely to be the cause of this difference. The side chains of Asp71, Glu109 and Glu188 that coordinate to Mg2+ ions in the substrate complex point away from the phosphate group of G12 that is lodged adjacent to the active site in the DDD complex. In the case of Glu109 the conformational change is probably linked to the absence of a 2′-hydroxyl group in DNA; in the substrate complex Glu109 is hydrogen bonded to the 2′-OH of A9. The lack of the ribose hydroxyl groups prevents the precise alignment of the DNA strand with respect to active site residues and the optimal spacing of coordination sites that is needed for binding of two Mg2+ ions and subsequent phosphodiester cleavage. The DNA backbone geometry deviates from that adopted by the RNA strand, resulting in a significantly different orientation of the phosphodiester moiety linking C11 and G12 in the DDD complex compared with the A9-U10 dimer in the substrate complex (Figs. 3,5).
Unlike the side chains of the above amino acids that are farther removed from the DNA backbone relative to the situation in the complex with RNA/DNA hybrid, Asp192 and Asn132 lie closer to the G12 phosphate in the DDD complex compared with their position vis-à-vis the U10 phosphate in the substrate complex. In fact their carboxyl and carboxamide groups, respectively, practically occupy the positions of Mg2+ ion A (Asp192) and Mg2+ ion B (Asn132) (Fig. 5B,C). In the case of Asn132, the close association with the phosphate backbone is due to formation of a hydrogen bond between the amino group and O3′(C11). The close spacing of phosphate and Asp192 carboxyl group - the OD1…O1P and OD1…O2P distances are 3.27 Å and 2.69 Å, respectively - may indicate that the side chain is protonated (the crystallization pH was 4.6). The OD1 oxygen displays no other close contacts whereas OD2 is engaged in three hydrogen bonds, two to backbone amide NH groups (Tyr193 and Gly194) and an additional one to a water molecule (Fig. 5B). This environment suggests that OD1 is the carboxylic acid (C-)OH oxygen and OD2 is the carbonyl oxygen. Whatever the precise nature of the interaction, it is clear that the stable and cleavage-competent association of active site residues and RNA mediated by coordinated Mg2+ ions and 2′-hydroxyl groups in the substrate complex is replaced by interactions in the inhibitor complex that, although weakly stabilizing, are obviously not conducive to cleavage. If the side chain of Asp192 is not protonated, the close contact with the phosphate of G12 has to be considered repulsive, thus likely rendering the interaction between this DNA strand and RNase H destabilizing in the absence of Mg2+ coordination.
Perturbation of the DDD Structure upon RNase H Binding
Comparison between the crystal structures of the DDD in complex with RNase H and in the free state shows that the latter duplex is bent into the major groove only at one G-/A-tract junction (Fig. 6A). ‘Facultative’ bending (reviewed in ref. 40) at this site in the free DDD is induced by crystal packing forces and stabilized by a Mg2+ coordinating to G2 and G22 from opposite strand in the major groove.37–39 Due to its location on a dyad in the crystal of the complex, the DDD here is symmetrically bent into the major groove at both junctions. However, instead of crystal packing, it is the binding of RNase H in the minor groove of G-tracts that causes the bends into the major groove. The DDD A-tract minor groove is exceptionally narrow and changes little on binding by the enzyme (Fig. 6). The minor groove in GC-rich stretches is generally wider but opens up further as a result of bending into the major groove induced by RNase H (Fig. 6B). Perhaps unexpectedly, the minor groove in the DDD at the binding interface is slightly wider on average (9.1 Å) than in the RNA/DNA hybrid (8.5 Å). The average minor groove width of the G-tract at the bent end in the free DDD duplex amounts to 8.3 Å. Thus, RNase H pries open the minor groove of the DNA duplex by about 1 Å. This widening is accompanied by a reduction in minor groove depth (average 3.5 Å) relative to the free DDD (average 4.7 Å). By comparison, the minor groove of the RNA/DNA hybrid bound to RNase H is even shallower (average 2.3 Å). We conclude that the Bh-RNase HC perturbs the geometry of the DDD duplex despite its inability to cleave DNA. The observed structural changes are subtle but clearly significant given the resolution of the structural data. Unavailability of the structure of the RNA/DNA dodecamer duplex in its free form prevents us from analyzing the structural changes induced by enzyme binding.
Figure 6.
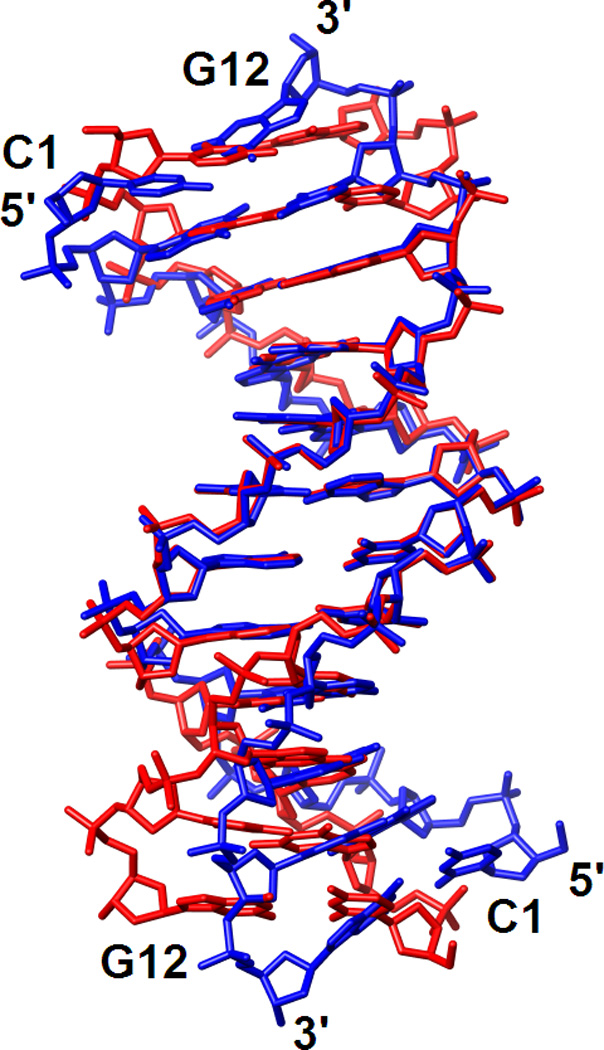

Conformations of the DDD in the free state and bound to RNase H. (A) Superimposition of the crystal structures of the DDD bound to Bh-RNase HC (blue) and the DDD alone (red; PDB ID code 436d37). Only phosphorus atoms of the central A-tract region were used for generating the overlay. (B) Comparison between the minor groove widths (solid lines) and depths (dashed lines) of the DDD bound to Bh-RNase HC (blue), the free DDD (red), and the RNA/DNA hybrid bound to Bh-RNase HC (green18).
Discussion
The complex between a DNA inhibitor duplex and RNase H reveals several key differences to the complex with RNA/DNA substrate but also some shared features. The differences concern mainly interactions with the RNA strand in the latter, which is perhaps not unexpected as the DNA strand assuming the position of RNA in the complex lacks 2′-hydroxyl groups. Two of the amino acids tracking the ribose 2′-hydroxyls of five consecutive nucleotides in the substrate complex instead interact with bridging or non-bridging phosphate oxygens in the complex with the DNA duplex (Table 2). More importantly the steric and electrostatic discrepancies between the 2′-deoxyribose and ribose sugars in the two backbones contribute to the absence of Mg2+ ions from the enzyme active site with the DNA duplex bound (Fig. 5). Different curvatures of the RNA (hybrid) and the corresponding DNA strand (DDD) result in a considerably less intimate binding mode with the latter. This is borne out by a higher number of water-mediated interactions in the complex with the DDD in place of direct RNA-protein interactions. Therefore, our structural data are consistent with the reduced binding affinity of RNase H for dsDNA compared with RNA/DNA hybrids. The interactions of the enzyme with the DNA strand of the hybrid duplex are virtually matched in the complex with dsDNA (Fig. 3, Table 2), corroborating the conclusion that the DNA strand in hybrids adopts a B-like conformation.18 To use an analogy, the binding mode to dsDNA observed here is not unlike climbing up a ladder while holding on to just one rail. By comparison, RNase H holds on to both the DNA and RNA rails when interacting with hybrids. This difference may allow the enzyme to more efficiently scan duplexes in order to locate hybrid targets enabling a higher turnover rate.6 This scenario is somewhat reminiscent of the hemispecific binding state of the endonuclease BstYI bound to noncognate DNA.34
Although the minor groove width of the duplex is clearly important for RNase H substrate recognition and binding,18,23 it cannot account for the enzyme’s inability to cleave dsDNA. The minor groove widths of the DDD and RNA/DNA duplexes in complex with Bh-RNase HC are very similar (Fig. 6B). However, the hybrid RNA strand and the corresponding DNA strand in the DDD exhibit very different curvatures (Fig. 3A), unlike the hybrid DNA strand and the corresponding strand in the DDD (Fig. 3B). Therefore, minor groove width and depth, strand curvature and chemistry (2′-OH) are all being used by the enzyme as identity elements. Overall, the binding modes of RNase H to RNA/DNA and dsDNA are not fundamentally different, given the facts that (i) both duplexes are bound in the minor groove (Fig. 1A), (ii) the orientations of the duplexes as judged by the alignment of their helix axes are similar (Fig. 3), and (iii) phosphates from opposite strands of the DNA duplex that are separated by two base pairs are lodged at the phosphate binding site and the active site as seen in the substrate complex (Fig. 5A). Further it is remarkable that the enzyme induces specific changes in the conformation of the DDD that include bending into the major groove and minor groove widening. The hybrid duplex exhibits a strong kink into the major groove at the U7pG8 step at the border of the binding interface. However, in the absence of a structure of the RNA/DNA alone it remains unclear whether this feature is caused by RNase H binding. The structure of the complex with a dsDNA inhibitor duplex provides a refined understanding of RNase H substrate recognition and processing. Similar insights into the enzyme’s inability to cleave dsRNA and the protein-RNA interactions underlying the enzyme’s comparable binding affinities for dsRNA and RNA/DNA have to await the structure determination of an RNase H:dsRNA complex.
Materials and Methods
Protein Expression and Purification
B. halodurans genomic DNA was purchased from American Type Culture Collection (ATCC, Manassas, VA). The Asp132→Asn mutant of Bh-RNase HC (Met58 to Lys196) was expressed in E. coli and purified as described previously.18 The protein solution was concentrated to 35 mg/mL.
DNA Synthesis and Purification
The dodecamer 5′-d(CGCGAATTCGCG) was synthesized on a 1 µmole scale on an ABI 381A DNA synthesizer, using commercial phosphoramidite building blocks (ChemGenes Corp., MA). Following deprotection and cleavage from the CPG solid support, the oligonucleotide was purified using anion exchange HPLC and the concentration of the stock solution was adjusted to 2.4 mM.
Crystallization and Structure Determination
Protein and DNA solutions were mixed in a 1:1 molar ratio in the presence of 5 mM MgCl2 and crystallization experiments were performed by the sitting drop vapor diffusion technique using the sparse matrix screen.41 Briefly, 1 µL complex solution were mixed with 1 µL of reservoir solution and equilibrated against 50 µL reservoir wells. Crystals appeared in droplets that were mixed and equilibrated with 0.1 M NaOAc•3H2O (pH 4.6), 8% (w/v) PEG 4000 in about 3–4 days. Crystals were mounted in nylon loops, cryo-protected in 20% glycerol containing reservoir solution and frozen in liquid nitrogen. Diffraction data were collected with a Mar225 CCD on the 21-ID-F beam line of the Life Sciences Collaborative Access Team (LS-CAT) at the Advanced Photon Source, Argonne National Laboratory (Argonne, IL). Diffraction data were integrated and scaled with the program HKL2000.42 The structure was determined with the Molecular Replacement technique using the program MOLREP43,44 and the Bh-RNase HC structure with PDB ID 1zbi18 as the search model. Initial refinement was carried out with the program CNS45 and the DNA duplex was gradually built into the electron density, two to three base pairs at a time, followed by refinement. Manual rebuilding was performed with the program TURBO.46 Water molecules were added gradually and isotropic/TLS refinement was continued with the program REFMAC.47,48 A summary of crystallographic parameters is provided in Table 1.
DNA Helical Analysis and Illustrations
Geometric parameters for the DNA duplex were calculated with the program CURVES.49 All illustrations were generated with the program CHIMERA.50
Acknowledgments
This work was supported by National Institutes of Health grant R01 GM055237. We are grateful to Dr. Z. Wawrzak, Northwestern University, for assistance with X-ray diffraction data collection. Vanderbilt University is a member institution of the Life Sciences Collaborative Access Team (LS-CAT) at sector 21 of the Advanced Photon Source, Argonne, IL. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under Contract No. W-31-109-Eng-38.
Footnotes
Author contributions: P.S.P. and M.E. designed research; P.S.P. performed research; and P.S.P. and M.E. wrote the paper.
The authors declare no conflict of interest.
Data deposition: Structure factors and final coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 3d0p).
References
- 1.Stein H, Hausen P. Enzyme from calf thymus degrading the RNA moiety of DNA-RNA hybrids: effect on DNA-dependent RNA polymerase. Science. 1969;166:393–395. doi: 10.1126/science.166.3903.393. [DOI] [PubMed] [Google Scholar]
- 2.Hostomsky Z, Hostomska Z, Matthews DA. Ribonucleases H. In: Linn SM, Lloyd SR, Roberts RJ, editors. Nucleases. 2nd edition. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1993. pp. 341–376. [Google Scholar]
- 3.Ohtani N, Haruki M, Morikawa M, Kanaya S. Molecular diversities of RNases H. J Biosci Bioeng. 1999;88:12–19. doi: 10.1016/s1389-1723(99)80168-6. [DOI] [PubMed] [Google Scholar]
- 4.Chapados BR, Chai Q, Hosfield DJ, Qiu J, Shen B, Tainer JA. Structural biochemistry of a type 2 RNase H: RNA primer recognition and removal during DNA replication. J Mol Biol. 2001;307:541–556. doi: 10.1006/jmbi.2001.4494. [DOI] [PubMed] [Google Scholar]
- 5.Kanaya S, Crouch RJ. DNA sequence of the gene coding for Escherichia coli ribonuclease H. J Biol Chem. 1983;258:1276–1281. [PubMed] [Google Scholar]
- 6.Lima WF, Crooke ST. Binding affinity and specificity of Escherichia coli RNase H1: impact on the kinetics of catalysis of antisense oligonucleotide-RNA hybrids. Biochemistry. 1997;36:390–398. doi: 10.1021/bi962230p. [DOI] [PubMed] [Google Scholar]
- 7.Lima WF, Mohan V, Crooke ST. The influence of antisense oligonucleotide-induced RNA structure on E. coli RNase H1 activity. J Biol Chem. 1997;272:18191–18199. doi: 10.1074/jbc.272.29.18191. [DOI] [PubMed] [Google Scholar]
- 8.Lima WF, Nichols JG, Wu H, Prakash TP, Migawa MT, Wyrzykiewicz TK, Bhat B, Crooke ST. Structural requirements at the catalytic site of the heteroduplex substrate for human RNase H1 catalysis. Biol Chem. 2004;9:36317–36326. doi: 10.1074/jbc.M405035200. [DOI] [PubMed] [Google Scholar]
- 9.Walder RT, Walder JA. Role of RNase H in hybrid-arrested translation by antisense oligonucleotides. Proc Natl Acad Sci USA. 1988;85:5011–5015. doi: 10.1073/pnas.85.14.5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crooke ST. Basic principles of antisense therapeutics. In: Crooke ST, editor. Antisense Research and Application. vol. 131. Berlin: Springer; 1998. pp. 1–50. [Google Scholar]
- 11.Crooke ST. Phosphorothioate oligonucleotides. In: Crooke ST, editor. Therapeutic Applications of Oligonucleotides. Austin, TX: R. G. Landes; 1995. pp. 63–79. [Google Scholar]
- 12.Damha MJ, Wilds CJ, Noronha A, Brukner I, Borkow G, Arion D, Parniak MA. Hybrids of RNA and arabinonucleic acids (ANA and 2′F-ANA) are substrates of ribonuclease H. J Am Chem Soc. 1998;120:12976–12977. [Google Scholar]
- 13.Cook PD. Second generation antisense oligonucleotides: 2′-modifications. Annu Rep Med Chem. 1998;33:313–325. [Google Scholar]
- 14.Manoharan M. 2′-Carbohydrate modifications in antisense oligonucleotide therapy: importance of conformation, configuration and conjugation. Biochim Biophys Acta. 1999;1489:117–130. doi: 10.1016/s0167-4781(99)00138-4. [DOI] [PubMed] [Google Scholar]
- 15.Katayanagi K, Miyagawa M, Matsushima M, Ishikawa M, Kanaya S, Ikehara M, Matsuzaki T, Morikawa K. Three-dimensional structure of ribonuclease H from E. coli. Nature. 1990;347:306–309. doi: 10.1038/347306a0. [DOI] [PubMed] [Google Scholar]
- 16.Yang W, Hendrickson WA, Crouch RJ, Satow Y. Structure of ribonuclease H phased at 2 Å resolution by MAD analysis of the selenomethionyl protein. Science. 1990;249:1398–1405. doi: 10.1126/science.2169648. [DOI] [PubMed] [Google Scholar]
- 17.Ishikawa M, Oda Y, Katayanagi K, Iwai S, Ohtsuka E, Morikawa K. Cocrystallization of Escherichia coli RNase H with synthetic DNA/RNA hybrid oligomers. Nucleic Acids Res Symp Ser. 1991;24:253. [PubMed] [Google Scholar]
- 18.Nowotny M, Gaidamakov SA, Crouch RJ, Yang W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 2005;1:1005–1016. doi: 10.1016/j.cell.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 19.Loukachevitch LV, Egli M. Crystallization and preliminary X-ray analysis of Escherichia coli RNaseHI-dsRNA complexes. Acta Cryst F. 2007;63:84–88. doi: 10.1107/S1744309106055461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nowotny M, Gaidamakov SA, Ghirlando R, Cerritelli SM, Crouch RJ, Yang W. Structure of human RNase H1 complexed with an RNA/DNA hybrid: insight into HIV reverse transcription. Mol Cell. 2007;28:264–276. doi: 10.1016/j.molcel.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 21.Nowotny M, Yang W. Stepwise analyses of metal ions in RNase H catalysis from substrate destabilization to product release. EMBO J. 2006;25:1924–1933. doi: 10.1038/sj.emboj.7601076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakamura H, Oda Y, Iwai S, Inoue H, Ohtsuka E, Kanaya S, Kimura S, Katsuda C, Katayanagi K, Morikawa K, Miyashiro H, Ikehara M. How does RNase H recognize a DNA•RNA hybrid? Proc Natl Acad Sci USA. 1991;88:11535–11539. doi: 10.1073/pnas.88.24.11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fedoroff OY, Salazar M, Reid BR. Structure of a DNA:RNA hybrid duplex. Why RNase H does not cleave pure RNA. J Mol Biol. 1993;233:509–523. doi: 10.1006/jmbi.1993.1528. [DOI] [PubMed] [Google Scholar]
- 24.Minasov G, Teplova M, Nielsen P, Wengel J, Egli M. Structural basis of cleavage by RNase H of hybrids of arabinonucleic acids and RNA. Biochemistry. 2000;39:3525–3532. doi: 10.1021/bi992792j. [DOI] [PubMed] [Google Scholar]
- 25.Sarafianos SG, Das K, Tantillo C, Clark AD, Jr, Ding J, Whitcomb JM, Boyer PL, Hughes SH, Arnold E. Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J. 2001;20:1449–1461. doi: 10.1093/emboj/20.6.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yazbeck DR, Min K-L, Dahma MJ. Molecular requirements for degradation of a modified sense RNA strand by Escherichia coliribonuclease H1. Nucleic Acids Res. 2002;30:3015–3025. doi: 10.1093/nar/gkf429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li F, Sarkhel S, Wilds CJ, Wawrzak Z, Prakash TP, Manoharan M, Egli M. 2′-Fluoroarabino- and arabinonucleic acid show different conformations, resulting in deviating RNA affinities and processing of their heteroduplexes with RNA by RNase H. Biochemistry. 2006;45:4141–4152. doi: 10.1021/bi052322r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Albright RA, Mossing MC, Matthews BW. Crystal structure of an engineered Cro monomer bound nonspecifically to DNA: possible implications for nonspecific binding by the wild-type protein. Protein Sci. 1998;7:1485–1494. doi: 10.1002/pro.5560070701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuo C-F, McRee DE, Fisher CL, O’Handley SF, Cunningham RP, Tainer JA. Atomic structure of the DNA repair [4Fe-4S] enzyme endonuclease III. Science. 1992;258:434–440. doi: 10.1126/science.1411536. [DOI] [PubMed] [Google Scholar]
- 30.Dodson ML, Michaels ML, Lloyd RS. Unified catalytic mechanism for DNA glycosylases. J Biol Chem. 1994;269:32709–32711. [PubMed] [Google Scholar]
- 31.Winkler FK, Banner DW, Oefner C, Tsernoglou D, Brown RS, Heathman SP, Bryan RK, Martin PD, Petratos K, Wilson KS. The crystal structure of EcoRV endonuclease and of its complexes with cognate and non-cognate DNA fragments. EMBO J. 1993;12:1781–1795. doi: 10.2210/pdb4rve/pdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Viadiu H, Aggarwal AK. Structure of BamHI bound to nonspecific DNA: a model for DNA sliding. Mol Cell. 2000;5:889–895. doi: 10.1016/s1097-2765(00)80329-9. [DOI] [PubMed] [Google Scholar]
- 33.Halford SE, Marko JF. How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res. 2000;32:3040–3052. doi: 10.1093/nar/gkh624. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Townson SA, Samuelson JC, Bao Y, Xu S, Aggarwal AK. BstYI bound to noncognate DNA reveals a “hemispecific” complex: implications for DNA scanning. Structure. 2007;15:449–459. doi: 10.1016/j.str.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 35.Wing R, Drew H, Takano T, Broka C, Takano S, Itakura K, Dickerson RE. Crystal structure analysis of a complete turn of B-DNA. Nature. 1980;287:755–758. doi: 10.1038/287755a0. [DOI] [PubMed] [Google Scholar]
- 36.Drew H, Wing R, Takano T, Broka C, Tanaka S, Itakura K, Dickerson RE. Structure of a B-DNA dodecamer: conformation and dynamics. Proc Natl Acad Sci USA. 1981;78:2179–2183. doi: 10.1073/pnas.78.4.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tereshko V, Minasov G, Egli M. The Dickerson-Drew B-DNA dodecamer revisited - at atomic resolution. J Am Chem Soc. 1999;121:470–471. [Google Scholar]
- 38.Minasov G, Tereshko V, Egli M. Atomic-resolution crystal structures of B-DNA reveal specific influences of divalent metal ions on conformation and packing. J Mol Biol. 1999;291:83–99. doi: 10.1006/jmbi.1999.2934. [DOI] [PubMed] [Google Scholar]
- 39.Egli M, Tereshko V. Lattice- and sequence-dependent binding of Mg2+ in the crystal structure of a B-DNA dodecamer. In: Stellwagen N, Mohanty U, editors. Curvature and Deformation of Nucleic Acids: Recent Advances, New Paradigms. Vol. 884. ACS Symp Ser; 2004. pp. 87–109. [Google Scholar]
- 40.Allemann RK, Egli M. DNA recognition and bending. Chem Biol. 1997;4:643–650. doi: 10.1016/s1074-5521(97)90218-0. [DOI] [PubMed] [Google Scholar]
- 41.Berger I, Kang CH, Sinha N, Wolters M, Rich A. A highly efficient 24-condition matrix for the crystallization of nucleic acid fragments. Acta Cryst D. 1996;52:465–468. doi: 10.1107/s0907444995013564. [DOI] [PubMed] [Google Scholar]
- 42.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Meth Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 43.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 44.Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Cryst D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 45.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR System: a new software suite for macromolecular structure determination. Acta Cryst D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 46.Cambillau C, Roussel A. Turbo Frodo. Version OpenGL.1. 1997 [Google Scholar]
- 47.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 48.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Cryst D. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 49.Lavery R, Sklenar H. Defining the structure of irregular nucleic acids: conventions and principles. J Biomol Struct Dyn. 1989;6:655–667. doi: 10.1080/07391102.1989.10507728. [DOI] [PubMed] [Google Scholar]
- 50.Pettersen EF. UCSF Chimera - a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]