

Abstract
Phospholipase A2 (PLA2), isolated from Daboia russelli pulchella (Russell’s viper), is enzymatically active as well as induces several pharmacological disorders including neurotoxicity, myotoxicity, cardiotoxicity, anti-coagulant, hemolytic, and platelet effects. Indomethacin reduces the effects of anti-coagulant and pro-inflammatory actions of PLA2. Pyrazolo[3,4-d]pyrimidines constitute a class of naturally occurring fused uracils that posses diverse biological activities. The in-silico docking studies of nine pyrazolo[3,4-d]pyrimidine molecules have been carried out with the X-ray crystal structure of Russell’s viper PLA2 (PDB ID: 3H1X) to predict the binding affinity, molecular recognition, and to explicate the binding modes, using AUTODOCK and GLIDE (Standard precision and Extra precision) modules, respectively. Docking results through each method make obvious that pyrazolo[3,4-d]pyrimidine molecules with trimethylene linker can bind with both anti-coagulation and enzymatic regions of PLA2.
Electronic supplementary material The online version of this article (doi: 10.1007/s10867-013-9299-7) contains supplementary material, which is available to authorized users.
Keywords: Pyrazolo[3,4-d]pyrimidines; PLA2; Molecular docking; AUTODOCK4.2; Schrödinger; ADME calculatioin
Introduction
Intra- and inter-molecular aromatic π − π (arene) interactions play a vital role in biological systems in determining their conformations and activities [1–7]. Being attractive in nature, they have a wide application in the areas of crystal packing [8, 9], structural variations, and molecular recognition processes [10]. Scientists of various disciplines have made intensive investigations in these areas for over four decades both using experimental [11] as well as theoretical techniques [12]. Intermolecular interactions minimize the energy of the complexes and play an important role in drug–receptor, protein–DNA, protein–protein interactions, etc., while intramolecular interactions minimize the energy of a molecule and are responsible for tertiary structure of proteins, DNA, RNA etc. [13–16]. In the literature, to facilitate arene interactions, several models have been proposed [17]. The trimethylene linker connecting two aromatic moieties has played a significant role in understanding the nature of intramolecular interactions and has been successfully employed to analyze the interactions between nucleic acid bases and other compounds [18, 19]. Since 1995, studies have been going on and have resulted in the development of several novel ‘polymethylene linker’ compounds based on pyrazolo[3,4-d]pyrimidine (PP) core, which is isomeric with biologically important purine system, as new models [20]. Pyrazolo[3,4-d]pyrimidines constitute a class of naturally occurring fused uracils that posses diverse biological activities [21]. These compounds are reported as potential anti-inflammatory agents [22], anti-coagulation inhibitor[23], xanthine oxidase inhibitor [24], antiproliferative and proapoptotic agents in several tumor types [25, 26], SRC kinase inhibitors [27, 28], treatment of human cancers sustaining oncogenic activation of RET [29] etc.
Phospholipase A2 (PLA2), isolated from Daboia russelli pulchella (Russell’s viper), is enzymatically active and induces several pharmacological disorders including neurotoxicity, myotoxicity, cardiotoxicity, anti-coagulant, hemolytic, and platelet effects [30]. It has been reported that the pharmacological effects of venom PLA2 are not necessarily dependent on their catalytic function and that is might be based on the protein–protein interactions involving a specific site on the protein surface [31]. These pharmacological sites are neither defined clearly nor their modes of interactions characterized. Therefore, the potent chemical compounds have not been designed so far for preventing the interactions of these sites so as to control the harmful pharmacological effects. The three-dimensional structures of various isomorphs of PLA2 from a number of sources have been determined and the catalytic residues and substrate binding sites are well characterized in PLA2 enzymes [32, 33]. Various ligands have been designed to inhibit their enzymatic function [34–36]. Non-steroidal anti-inflammatory drugs have been attributed to their binding to PLA2 [37]. The anti-coagulant activity was detected in snake venom some decades ago [38]. Although the precise nature of the anti-coagulant loop is not yet clearly understood, it is suggested that the presence of basic residues at specific positions in the loop 54–77 contributes the strong anti-coagulant action of PLA2. However, the mode of ligand binding for inhibiting the anti-coagulant action of PLA2 is not understood and hence the molecules could not be designed to stop the anti-coagulant effect of PLA2. Indomethacin reduces the effects of both anti-coagulant and pro-inflammatory actions of PLA2 [39]. One of the carboxylic group oxygen atoms of indomethacin interacts with Asp49 and His48, which are essential for catalysis while the second carboxylic oxygen atom forms an ionic interaction with the side chain of Lys69, which is a part of the anti-coagulant loop (residues 54–77). In order to predict binding affinity, molecular recognition and to explicate the binding modes, the in-silico docking study of the trimethylene linker pyrazolo[3,4-d]pyrimidine derivatives has been undertaken with PLA2. Like indomethacin, pyrazolo[3,4-d]pyrimidine molecules have planar moieties and have various pharmacological properties. The pyrazolo[3,4-d]pyrimidine molecules considered for docking studies are 1,3-bis(4,6-diethylthio-1Hpyrazolo[3,4-d]pyrimidin-1-yl)propane [40] (1); 1,3-bis(4-ethoxy-6-methyl-sulfanyl-1H-pyrazolo[3,4-d] pyrimidin-1-yl)propane [41] (2); 1,10-(1,3-propanediyl)bis(5-methyl-6-methylthio-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-4-one) [42] (3); 1,10-(1,3-propanediyl)bis(5-ethyl-6-methylthio-4,5-dihydro-1H-pyrazolo[3,4-d] pyrimidin-4-one) [43] (4); 1,10-(1,3- propanediyl)bis(5-benzyl-6-methylsulfanyl-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-4- one) [44] (5); 1-(4,6-dimethylsulfanyl-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-3-(5-methyl- 6-methylsulfanyl-4-oxo-1,5-dihydropyrazolo[3,4-d]pyrimidin-1-yl)propane [45] (6); 4, 6-bis(methylsulfanyl)-1-phthalimidopropyl-1H-pyrazolo[3,4-d]-pyrimidine [46] (7); 6- methylsulfanyl-1-phthalimidopropyl-4(pyrrolidin-1-yl)-1H-pyrazolo[3,4-d]pyrimidine [46] (8); and 6-methylsulfanyl-1-(3-phenylpropyl)-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-4-one [47] (9). All these molecules have either two PP moieties or one PP moiety and one other aromatic moiety connected through a trimethyline linker (Fig. 1), which are flexible models for inter- and intra-molecular π − π interactions studies [11, 12]. Because of the similarity of these compounds with indomethacin, molecular docking studies have been performed with the native receptor of IMN, i.e., 3H1X. In order to ensure success at clinical testing level [48, 49], the prediction of ADME (absorption, distribution, metabolism and excretion) and other pharmaceutically important properties of these compounds has also been carried out.
Fig. 1.

Chemical diagram of ligands (molecules 1–9 and IMN)
Materials and methods
The three-dimensional crystal structure of Russell’s viper PLA2 complexed with indomethacin (PDB ID: 3H1X), determined by X-ray crystallography was retrieved from the Protein Databank Bank (http://www.rcsb.org/). PDB files have a variety of potential problems, e.g., missing atoms, added waters, more than one molecule, chain breaks, alternate locations etc., that need to be corrected before use. Before docking, the preparations of protein and ligands were carried out and subsequently electron affinity grids were generated. The docking of ligands with the receptor has been carried out using AUTODOCK4.2 and GLIDE5.8 modules (Standard precision and Extra precision modes), respectively.
AUTODOCK docking
The AUTODOCK4.2 program starts with a ligand molecule in an arbitrary conformation, orientation and position and finds favorable docking in a protein-binding site using both simulating annealing and genetic algorithms [50]. The program ADT(Autodock tools), which has been released as an extension suite to the Python Molecular Viewer, was used to prepare the protein and the ligands. For the preparation of macromolecule, polar hydrogens were added in the crystal structure of PLA2, and then Kollman United Atom charges and atomic solvation parameters were assigned. The grid maps of docking studies were computed using the AutoGrid4 included in the Autodock4.2 distribution. Grid center that was centered on the active site (IMN binding site) was obtained using the AutoGrid4 algorithm [51], and 60×60×60 points with grid spacing of 0.375 Å and distance-dependent dielectric constant were calculated. The affinity and electrostatic potential grid were calculated for each type of atoms in the ligands. The GA–LS method was adopted to perform the molecular docking. The parameters for Genetic Algorithm (GA) were defined as follows: a maximum number of 250,000 energy evaluations; a maximum number of generations of 27,000; mutation and crossover rates of 0.02 and 0.8, respectively. Pseudo-Solis &Wets parameters were used for local search (LS), and 300 iterations of Solis & Wets local search were imposed. The number of docking runs was set to 100. After docking, all the structures generated were assigned to clusters based on a tolerance of 1 Å all-atom RMSD from the lowest-energy structure. Hydrogen bonding and hydrophobic interactions between docked potent agents and macromolecule were analyzed using ADT [50]. The best docking result can be considered to be the conformation with the lowest (docked) energy and lowest RMSD.
GLIDE docking
Further, docking studies have also been performed using GLIDE5.8 (Grid-based Ligand Docking with Energetics) [52, 53] in standard precision (SP) and extra precision (XP) modes, respectively, as implemented in the Schrödinger Suite. Glide carries out an exhaustive conformational search, augmented by a heuristic screen that rapidly eliminates conformations deemed not to be suitable for binding to a receptor, such as conformations that have long-range internal hydrogen bonds. In SP and XP both dockings, the receptor was kept rigid while ligands were treated as flexible, which enables to dock the ligand at the receptor’s binding site to generate multiple poses of the receptor–ligand complex, each including unique structural conformations of the ligands to fit in the binding site of receptor and ranks them by Glide score (G-score) to find the best structure of the docked complex. The G-score takes into account a number of parameters like hydrogen bonds (H-bond), hydrophobic contacts (Lipo), van der Waals (vdW), columbic (Coul), polar interactions in the binding site (Site), metal binding term (Metal) and penalty for buried polar group (BuryP) & freezing rotatable bonds (RotB).
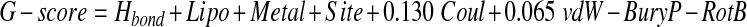 |
In the Extra Precision (XP) docking protocol, in addition to unique water desolvation energy terms, protein–ligand structural motifs leading to enhanced binding affinity are included: (i) hydrophobic enclosure where groups of lipophilic ligand atoms are enclosed on opposite faces by lipophilic protein atoms, (ii) neutral-neutral single or correlated hydrogen bonds in a hydrophobically enclosed environment, and (iii) five categories of charged–charged hydrogen bonds. The XP Glide score contains the following terms [54]:
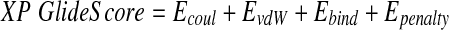 |
where,
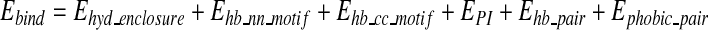 |
and
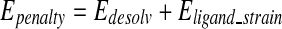 |
The protein–ligand complex was prepared using the protein-preparation wizard of the Schrödinger suite where hydrogens were added and subsequent refinement of structure was carried out. It was observed that no water molecules were involved in the hydrogen bonding of ligand with protein therefore all the co-crystallized water molecules were removed and bond orders were reassigned. Further, the whole system was minimized to an RMSD of 0.30 Å. The missing residues were added through the prime application. The coordinate of the indomethacin (IMN) was extracted from the protein complex while the coordinate of pyrazolo[3,4-d]pyrimidine ligands were obtained from the reported entries [40–47].
All the ligands (molecules 1–9 and IMN) were prepared using the LIGPREP module of the Schrödinger Suite. The bond orders were modified according to their data and different conformers were generated. Each generated conformer was subjected to a full minimization in the gas phase with the OPLS (Optimized Potential for liquid Simulations) force field [55] to eliminate the bond length and bond angles biased from the crystal structure. Ligprep produced the structures with various ionization states, tautomers, stereo-chemistries, and ring conformations. The receptor grid was generated using 1.0 van der Waals radius scaling factor and 0.25 partial charge cut-off. Prepared ligands and the receptor were used as the initial coordinates for docking purposes. The first stage for ligand docking is the receptor grid generation for which the PLA2 structure complexed with IMN was considered. During the grid generation, no van der Waals radius sampling was done and the partial charge cut-off was taken as 0.25 and no constraint was applied [56]. The location of indomethacin (IMN) was taken as the binding site for docking of all ligands. The docking simulations were performed in the Standard precision and Extra precision modes, respectively. During the docking process, the receptor was kept fixed while the ligands were treated as flexible. For the minimization of ligands, the distance-dependent dielectric constant with a value of 2.0 and a conjugate gradient algorithm with 100 steps were used. All of the inhibitors were passed through a scaling factor of 0.80 and partial charge cut-off of 0.15. After docking, the post-docking minimization was performed to improve the geometry of the poses. The post-docking minimization specifies a full force-field minimization of those poses, which are considered for the final scoring. After minimization, the results were used for binding energy calculations and docking scores [57].
ADME prediction
The accurate prediction of absorption, distribution, metabolism, and excretion (ADME properties) is often difficult owing to the complexity of the underlying physiological mechanisms. However, these properties have been studied by the ADME scoring using the QikProp3.5 module [58] of Maestro. QikProp is a quick, accurate, easy-to-use absorption, distribution, metabolism, and excretion prediction program. QikProp predicts physically significant descriptors and pharmaceutically relevant properties of organic molecules, either individually or in batches. QikProp settings determine which molecules are flagged as being dissimilar to other 95% known drugs. The compounds were neutralized before being used by QikProp, and the program was processed in normal mode. The neutralizing step is essential, as QikProp is unable to neutralize a structure, and no properties will be generated in the normal mode. The program was processed in normal mode and predicted 44 properties for all the molecules, consisting of principal descriptors and physiochemical properties with a detailed analysis of the log P (octanol / water), QP%, and log HERG, etc. It also evaluates the acceptability of the analogs based on Lipinski’s rule [59, 60] of 5, which is essential for rational drug design. Lipinski’s rule of 5 is a rule of thumb to evaluate drug likeness or to determine whether a chemical compound with a certain pharmacological or biological activity has properties that would make it a likely orally active drug in humans. The rule describes molecular properties important for a drug’s pharmacokinetics in the human body, including its ADME. However, the rule does not predict whether a compound is pharmacologically active [59].
Results and discussion
In this study, nine trimethylene linker compounds based on pyrazolo[3,4-d]pyrimidine (PP) moieties are taken as ligands (Fig. 1). Molecule 1 is a symmetrical compound with two PP moieties connected with trimethylene linker having tetra thioethyl substituents at the 4th and 6th positions of both moieties. Molecule 2 is similar to 1 having ethoxy substituents at the 4th position and thiomethyl substituents at the 6th position. Molecules 3, 4, and 5 are also a symmetrical class of compounds. Molecule 3 has the oxo-group at the 4th position, the methyl-group at 5th position, and thiomethyl-group at the 6th position of both moieties. Molecules 4 and 5 are similar to 3 but have ethyl and benzoyl moieties, respectively, at the 5th positions. Compound 6 is a dissymmetrical class of molecule that has the SMe group in the place of the oxo-group at the 4th position of one PP moiety. Molecules 7 and 8 are obtained by replacing one PP moiety of molecule 1 by phthalimide moiety. Molecule 7 has SMe-groups at 4th and 6th position whereas 8 has SMe at 6th position and pyrrolidin moiety at 4th position of the remaining PP moiety. Molecule 9 also has only one PP moiety but the other moiety is replaced by phenyl group. It has the oxo-group at the 4th position and the SMe group at the 6th position. Similarity of pyrazolo[3,4-d]pyrimidine moieties with indomethacin (IMN) prompted us the present in-silico docking with the native receptor of
IMN, i.e., vPLA2. The docked poses with best scores and lowest RMSD were considered as the best pose [61, 62]. The docking results as obtained from the calculations through AUTODOCK and Standard and Extra Precision modes of GLIDE module are discussed below.
AUTODOCK
The Conformational search and docking studies using AUTODOCK4.2 suggest that electronic interaction plays an important role in ligand–channel interaction. The results are shown in Table 1. For the best rank of the docked poses, compared to the indomethacin binding to PLA2, molecules 3, 5, 7 and 9 have better binding energy values, molecule 8 has the comparable energy while molecules 1, 2, 4 and 6 have poor binding energies. Molecule 7 has the minimum docking energy of −6.91 kcal/mol while in the case of IMN it is −6.33 kcal/mol. It is also clear that the drug that showed the least binding energies with PLA2 was found to have higher minimum-inhibitory-concentration(MIC), i.e., that drug was not showing better efficacy while the drug complexed with enzyme with higher binding energy was showing lower MIC and was considered to be a better drug. It is also clear from Table 1 that molecule 7 demonstrates the lowest MIC value. Molecule 3 has the second lowest inhibitory concentration value. It has been reported that indomethacin forms an ionic interaction with the side chain Lys69 of anticoagulant region of vPLA2 at a distance of 2.6 Å. In addition, indomethacin is also reported to interact effectively with the important residues of the active site including Asp49 and His48, essential for anti-inflammatory activity [39]. The Autodock results demonstrated that indomethacin binds with Gly30 at a distance of 2.047 Å. This may happen because of optimizations of protein and ligands and the medium, in which systems are considered, cannot be exactly same. Hydrogen bonding interactions in the best docking poses are shown in Fig. 2. Molecules 1, 2, 3, 6 and 8 demonstrated hydrogen bonding interactions with Asp49, molecules 3 and IMN with Gly30, molecule 7 and 9 with His48 and molecule 5 with Gly33. Additionally, molecule 7 also shows binding with Lys69. Molecule 4 shows two N-H...O hydrogen bondings with the same residue Lys69 [supplementary information: Table S1]. On account of binding energy and inhibition concentration, molecule 7 exhibits better binding than all the other compounds, including IMN, while on account of hydrogen bondings of ligand with the receptor, molecules 3 and 7 show better binding capabilities.
Table 1.
Inhibition coefficient (KI) and different energy values of the ligands as obtained through the docking with 3H1X using AUTODOCK4.2
| Molecules | Rank | Kl (μM) | Intramolecular energy (kcal/mol) | Internal energy (kcal/mol) | Torsional energy (kcal/mol) | Binding energy (kcal/mol) |
|---|---|---|---|---|---|---|
| 1 | 1_1 | 74.69 | − 9.21 | 0.19 | 3.58 | − 5.63 |
| 2_1 | 131.66 | − 8.87 | − 0.83 | 3.58 | − 5.29 | |
| 3_1 | 160.42 | − 8.76 | − 0.55 | 3.58 | − 5.18 | |
| 3_2 | 388.60 | − 8.23 | − 0.60 | 3.58 | − 4.65 | |
| 4_1 | 249.63 | − 8.49 | − 0.80 | 3.58 | − 4.91 | |
| 2 | 1_1 | 290.50 | − 7.80 | − 0.59 | 2.98 | − 4.83 |
| 2_1 | 295.25 | − 7.80 | − 0.23 | 2.98 | − 4.82 | |
| 3_1 | 368.95 | − 7.67 | − 0.35 | 2.98 | − 4.68 | |
| 3_2 | 375.14 | − 7.66 | − 0.66 | 2.98 | − 4.67 | |
| 4_1 | 504.85 | − 7.48 | − 0.70 | 2.98 | − 4.50 | |
| 3 | 1_1 | 9.21 | − 8.66 | − 0.12 | 1.79 | − 6.87 |
| 1_2 | 10.95 | − 8.56 | − 0.14 | 1.79 | − 6.77 | |
| 1_3 | 20.45 | − 8.19 | − 0.20 | 1.79 | − 6.40 | |
| 2_1 | 10.76 | − 8.57 | 0.30 | 1.79 | − 6.78 | |
| 2_2 | 32.88 | − 7.91 | − 0.25 | 1.79 | − 6.12 | |
| 4 | 1_1 | 91.64 | − 7.90 | − 0.50 | 2.39 | − 5.51 |
| 2_1 | 115.41 | − 7.76 | − 0.54 | 2.39 | − 5.37 | |
| 2_2 | 178.43 | − 7.50 | − 0.25 | 2.39 | − 5.11 | |
| 3_1 | 233.02 | − 7.34 | − 0.49 | 2.39 | − 4.96 | |
| 4_1 | 282.48 | − 7.23 | − 0.73 | 2.39 | − 4.84 | |
| 5 | 1_1 | 18.62 | − 9.44 | − 0.74 | 2.98 | − 6.45 |
| 2_1 | 34.87 | − 9.06 | − 1.44 | 2.98 | − 6.08 | |
| 3_1 | 79.36 | − 8.58 | − 1.11 | 2.98 | − 5.59 | |
| 4_1 | 84.09 | − 8.54 | − 1.29 | 2.98 | − 5.56 | |
| 5_1 | 142.00 | − 8.23 | − 1.11 | 2.98 | − 5.25 | |
| 6 | 1_1 | 81.41 | − 7.67 | − 0.18 | 2.09 | − 5.58 |
| 2_1 | 333.24 | − 6.83 | − 0.88 | 2.09 | − 4.74 | |
| 3_1 | 349.47 | − 6.80 | − 0.61 | 2.09 | − 4.72 | |
| 4_1 | 456.35 | − 6.65 | − 0.16 | 2.09 | − 4.56 | |
| 5_1 | 480.47 | − 6.62 | − 0.60 | 2.09 | − 4.53 | |
| 7 | 1_1 | 8.56 | − 8.70 | − 0.29 | 1.79 | − 6.91 |
| 1_2 | 11.47 | − 8.53 | − 0.31 | 1.79 | − 6.74 | |
| 1_3 | 13.87 | − 8.42 | − 0.15 | 1.79 | − 6.63 | |
| 2_1 | 69.00 | − 7.47 | − 0.38 | 1.79 | − 5.68 | |
| 3_1 | 74.44 | − 7.42 | − 1.23 | 1.79 | − 5.63 | |
| 8 | 1_1 | 27.26 | − 8.02 | − 0.63 | 1.79 | − 6.23 |
| 1_2 | 38.61 | − 7.81 | − 0.82 | 1.79 | − 6.02 | |
| 2_1 | 118.32 | − 7.15 | − 1.50 | 1.79 | − 5.36 | |
| 3_1 | 128.14 | − 7.10 | − 1.36 | 1.79 | − 5.31 | |
| 4_1 | 171.61 | − 6.93 | − 1.29 | 1.79 | − 5.14 | |
| 9 | 1_1 | 13.88 | − 8.12 | − 0.6 | 1.49 | − 6.63 |
| 2_1 | 45.30 | − 7.42 | − 0.32 | 1.49 | − 5.93 | |
| 2_2 | 58.17 | − 7.27 | − 0.32 | 1.49 | − 5.78 | |
| 3_1 | 51.53 | − 7.34 | − 0.51 | 1.49 | − 5.85 | |
| 3_2 | 58.36 | − 7.27 | − 0.48 | 1.49 | − 5.78 | |
| IMN | 1_1 | 22.90 | − 7.52 | − 0.66 | 1.19 | − 6.33 |
| 2_1 | 24.65 | − 7.48 | − 0.70 | 1.19 | − 6.29 | |
| 2_2 | 27.00 | − 7.43 | − 0.68 | 1.19 | − 6.23 | |
| 3_1 | 27.26 | − 7.42 | − 0.66 | 1.19 | − 6.23 | |
| 3_2 | 32.31 | − 7.32 | − 0.69 | 1.19 | − 6.13 |
Fig. 2.

Hydrogen bonding interactions in the best docking poses as obtained through AUTODOCK (H-bonds are shown by dotted lines)
Glide docking
In GLIDE docking, the number of conformers of ligands were generated which were utilized for the docking purposes. The number of conformers, as calculated using CONFGEN were 14, 5, 12, 18, 20, 21, 12, 8, 8, and 4 in the case of molecules 1, 2, 3, 4, 5, 6, 7, 8, 9 and IMN, respectively. After the ligand preparation, protein preparation was carried out and subsequently the grid was generated at the IMN-binding site. GLIDE docking of all the conformers was carried out using Standard Precision (SP) and Extra Precision (XP) modules. The results of SP and XP docking are discussed below.
Standard precision (SP)
The results of Glide docking in Standard Precision mode are summarized in Table 2. Docking of pyrazolo[3,4-d]pyrimidine ligands and IMN revealed a great variation in their binding energy. Initially, five poses of each molecule were saved from which the best pose with best G-score and lowest docking energy were chosen. Although the predicted free energy of binding is a useful descriptor of ligand–receptor complementarity, the choice of the ‘best’ docking model was ultimately dictated by various parameters of ADME study. These docked complexes were considered for further analysis and the results are compared. It has been observed that molecule 4 shows a better Glide score (−6.7051) and better Docking energy (−54.784 kcal/mol) compared to all other ligands (including IMN) considered for docking. However, the docking energy of molecule 4 is comparable to that of molecules 3 and 6. Since the molecules 3, 4 and 5 have similar moieties with a small difference in their substituents, it has been observed that ethyl analog 4 is more suitable for binding than methyl analog 3 but the more bulky substituent benzyl group, molecule 5 does not show good results. In other words long elkyl substituents will give better results than aromatic moieties (ring systems) probably due to steric hindrance. These results are important from the molecular recognition point of view [10]. SP docking of molecules 7 and 8 show similar glide scores (~ −5.8) as they are very similar in structure. Because of the presence of the non-aromatic pyrrolidin ring in molecule 8 the glide score of molecule 8 is slightly higher than 7. Molecule 9 is a low molecular weight molecule having one phenyl group at the place of one PP moiety; interact weakly with the viper PLA2. Its Glide score (−4.828) is the third lowest. The lowest Glide score of best docking pose is −4.636 exhibited by molecule 5 while IMN binding shows a somewhat better Glide score −4.709, but the binding energy of molecule 5 is much higher than that of 9 and originally crystallized molecule IMN. Molecules 1, 2 & 5 have more rotatable bonds, thereby paying more rotatable penalty and hence their Glide scores are poorer. It has also been observed that all the pyrazolo[3,4-d]pyrimidine molecules, except for molecule 5, show better docking scores than IMN. However, on account of Glide energy consideration, IMN has the poorer energy than all other molecules. The glide energy for the best docked pose of IMN is −32.814 kcal/mole and is comparable to docked energy of 9 (−34.5539 kcal/mol).
Table 2.
Glide scores and average electrostatic (coul), van der Waals (vdw), site energy (site) and Glide energy obtained through Glide SP docking
| Molecules (SP) | Entry ID | Glide score | Ecoul (kcal/mol) | Evdw (kcal/mol) | Esite (kcal/mol) | Glide energy (kcal/mol) |
|---|---|---|---|---|---|---|
| 1 | 32 | − 5.565 | − 3.270 | − 48.226 | − 0.157 | − 51.496 |
| 40 | − 5.367 | − 0.968 | − 49.591 | − 0.167 | − 50.559 | |
| 44 | − 5.307 | − 2.830 | − 48.543 | − 0.185 | − 51.372 | |
| 47 | − 5.247 | − 2.134 | − 49.201 | − 0.180 | − 51.335 | |
| 56 | − 5.187 | − 1.967 | − 48.731 | − 0.183 | − 50.698 | |
| 2 | 69 | − 4.856 | − 2.183 | − 43.014 | − 0.163 | − 45.197 |
| 76 | − 4.791 | − 3.310 | − 42.334 | − 0.193 | − 45.644 | |
| 86 | − 4.694 | − 2.842 | − 42.245 | − 0.204 | − 45.087 | |
| 93 | − 4.565 | − 3.323 | − 44.350 | − 0.142 | − 47.673 | |
| 110 | − 4.073 | − 4.202 | − 43.388 | − 0.170 | − 47.589 | |
| 3 | 8 | − 6.417 | − 4.752 | − 48.490 | − 0.017 | − 53.243 |
| 13 | − 5.887 | − 5.426 | − 47.934 | − 0.111 | − 53.360 | |
| 14 | − 5.884 | − 5.442 | − 47.828 | − 0.017 | − 53.270 | |
| 24 | − 5.787 | − 5.894 | − 43.697 | − 0.091 | − 49.592 | |
| 25 | − 5.757 | − 5.637 | − 44.583 | − 0.090 | − 50.221 | |
| 4 | 2 | − 6.705 | − 4.248 | − 50.535 | − 0.010 | − 54.783 |
| 3 | − 6.688 | − 4.189 | − 50.629 | − 0.010 | − 54.819 | |
| 4 | − 6.668 | − 4.200 | − 50.783 | − 0.012 | − 54.983 | |
| 5 | − 6.493 | − 4.304 | − 50.249 | − 0.010 | − 54.554 | |
| 6 | − 6.435 | − 0.814 | − 52.373 | 0.0000 | − 53.187 | |
| 5 | 89 | − 4.636 | − 2.338 | − 46.642 | 0.000 | − 48.980 |
| 100 | − 4.318 | − 7.057 | − 42.092 | − 0.042 | − 49.149 | |
| 102 | − 4.283 | − 2.417 | − 43.022 | − 0.046 | − 45.439 | |
| 103 | − 4.273 | − 3.476 | − 42.849 | − 0.002 | − 46.325 | |
| 105 | − 4.216 | − 6.989 | − 39.835 | − 0.045 | − 46.823 | |
| 6 | 7 | − 6.433 | − 1.882 | − 52.926 | 0.0000 | − 54.809 |
| 36 | − 5.429 | − 3.008 | − 43.402 | − 0.162 | − 46.410 | |
| 37 | − 5.420 | − 2.630 | − 43.489 | − 0.173 | − 46.119 | |
| 38 | − 5.392 | − 2.871 | − 43.764 | − 0.177 | − 46.635 | |
| 42 | − 5.380 | − 3.307 | − 41.347 | − 0.168 | − 44.655 | |
| 7 | 17 | − 5.822 | − 3.086 | − 44.766 | − 0.018 | − 47.852 |
| 19 | − 5.818 | − 2.983 | − 45.060 | − 0.018 | − 48.043 | |
| 20 | − 5.817 | − 2.871 | − 45.000 | − 0.017 | − 47.872 | |
| 21 | − 5.815 | − 2.970 | − 45.107 | − 0.018 | − 48.077 | |
| 22 | − 5.812 | − 2.854 | − 45.048 | − 0.017 | − 47.902 | |
| 8 | 15 | − 5.866 | − 2.921 | − 46.758 | − 0.017 | − 49.679 |
| 34 | − 5.489 | − 1.316 | − 44.899 | − 0.043 | − 46.215 | |
| 92 | − 4.591 | − 0.780 | − 41.055 | − 0.029 | − 41.836 | |
| 94 | − 4.543 | − 1.039 | − 41.186 | − 0.045 | − 42.225 | |
| 97 | − 4.396 | − 0.641 | − 40.911 | − 0.032 | − 41.553 | |
| 9 | 71 | − 4.828 | − 3.552 | − 31.002 | 0.000 | − 34.554 |
| 74 | − 4.819 | − 3.611 | − 30.910 | 0.000 | − 34.521 | |
| 75 | − 4.809 | − 3.538 | − 30.922 | 0.000 | − 34.460 | |
| 77 | − 4.786 | − 3.340 | − 31.093 | 0.000 | − 34.432 | |
| 78 | − 4.776 | − 3.606 | − 30.638 | 0.000 | − 34.244 | |
| IMN | 84 | − 4.709 | − 8.409 | − 24.405 | − 0.087 | − 32.814 |
| 87 | − 4.672 | − 8.357 | − 24.488 | − 0.088 | − 32.846 | |
| 111 | − 4.064 | − 6.586 | − 21.505 | − 0.100 | − 28.091 | |
| 121 | − 3.243 | − 5.559 | − 23.459 | − 0.100 | − 29.018 |
The anti-coagulation region of the PLA2 protein lies between residues 54 and 77 and it is positively charged with the residues such as Lys69, Arg72, Arg74, Lys76, and Arg77 [63]. The hydrogen bonding interactions of the best poses, as obtained through SP docking, are shown in Fig. 3. Almost all the ligands considered for docking make hydrogen bonding interactions with Lys69, which is the anticoagulation region. Hydrogen bonding distances and angles are much favorable. These molecules also exhibit either hydrogen bonding or electrostatic interactions with His48, responsible for the catalytic activity. Aromatic π − π stacking interactions are exhibited by ligands with residues Tyr52 and His48. There also exist cation-π interaction in the docking complex of 5 with 3H1X. Also, all the molecules are well positioned within the hydrophobic channel of the binding site, which is effective in blocking the exposure of the amino acid residues present in the anti-coagulant region and also the catalytic region to bind the substrate, thereby arresting the enzymatic activity of vPLA2). However, IMN shows the least hydrophobic interaction. It has been observed that IMN exhibit hydrogen bonding interaction only with Lys69 [Supplementary information: Figure S1 and Table S2].
Fig. 3.


Hydrogen bonding interactions in the best docking complexes as obtained through GLIDE SP docking (H-bonds are shown by dotted lines)
Extra precision (XP)
In the Extra Precision protocol of the Glide module, protein-ligand structural motifs leading to enhanced binding affinity are included, in addition to unique water desolvation energy terms. The Glide docking results out of extra precision docking protocol has been summarized in Tables 3. It has been observed that the Glide scores and docking energies differ from that of standard precision mode. Molecule 5 has the best Glide score (−7.09) and best docking energy (−60.90 Kcal/mol), which had very poor Glide scores within SP docking. Molecule 4, which had the best Glide score with SP, received the second-best Glide score. The docking energies of molecules 3, 4 and 6 are almost of the same order, as were found with SP mode simulation.
Table 3.
Glide scores and average electrostatic (coul), van der Waals (vdw), site energy (site) and Glide energy through GLIDE (XP) docking
| Molecules (XP) | Entry ID | Glide score | Ecoul (kcal/mol) | Evdw (kcal/mol) | Esite (kcal/mol) | Glide energy (kcal/mol) |
|---|---|---|---|---|---|---|
| 1 | 43 | − 4.440 | − 2.534 | − 48.777 | − 0.367 | − 51.311 |
| 50 | − 4.385 | − 2.727 | − 49.192 | − 0.200 | − 51.919 | |
| 58 | − 4.268 | − 2.050 | − 49.442 | − 0.353 | − 51.492 | |
| 68 | − 4.136 | − 2.626 | − 46.726 | − 0.287 | − 49.353 | |
| 75 | − 4.022 | − 2.872 | − 50.935 | − 0.323 | − 53.807 | |
| 2 | 79 | − 4.001 | − 3.226 | − 40.419 | − 0.526 | − 43.645 |
| 83 | − 3.905 | 1.504 | − 44.889 | − 0.382 | − 43.385 | |
| 87 | − 3.877 | − 1.527 | − 43.303 | − 0.304 | − 44.830 | |
| 103 | − 3.699 | − 2.153 | − 39.876 | − 0.203 | − 42.029 | |
| 112 | − 3.453 | − 2.172 | − 44.035 | − 0.044 | − 46.207 | |
| 3 | 23 | − 4.637 | − 7.677 | − 45.699 | − 0.480 | − 53.376 |
| 24 | − 4.631 | − 8.040 | − 44.812 | − 0.476 | − 52.852 | |
| 25 | − 4.590 | − 6.927 | − 47.990 | − 0.479 | − 54.917 | |
| 27 | − 4.543 | − 6.807 | − 48.252 | − 0.474 | − 55.059 | |
| 29 | − 4.534 | − 6.939 | − 45.279 | − 0.477 | − 52.218 | |
| 4 | 22 | − 4.689 | − 6.739 | − 46.328 | − 0.450 | − 53.067 |
| 26 | − 4.554 | − 7.097 | − 43.261 | − 0.459 | − 50.358 | |
| 32 | − 4.507 | − 6.337 | − 47.700 | − 0.433 | − 54.037 | |
| 34 | − 4.498 | − 6.562 | − 42.893 | − 0.461 | − 49.456 | |
| 35 | − 4.496 | − 5.791 | − 44.389 | − 0.465 | − 50.180 | |
| 5 | 2 | − 7.092 | − 3.946 | − 56.957 | − 0.339 | − 60.903 |
| 3 | − 6.840 | − 2.530 | − 55.586 | − 0.324 | − 58.116 | |
| 4 | − 6.805 | − 3.289 | − 54.978 | − 0.324 | − 58.267 | |
| 5 | − 6.763 | − 1.975 | − 42.393 | − 0.336 | − 44.368 | |
| 6 | 28 | − 4.539 | − 5.690 | − 48.480 | − 0.469 | − 54.170 |
| 40 | − 4.474 | − 3.748 | − 51.528 | − 0.735 | − 55.276 | |
| 44 | − 4.434 | − 3.155 | − 51.707 | − 0.756 | − 54.863 | |
| 49 | − 4.386 | − 1.243 | − 53.653 | − 0.455 | − 54.897 | |
| 52 | − 4.366 | − 5.624 | − 47.438 | − 0.471 | − 53.062 | |
| 7 | 39 | − 4.488 | − 0.795 | − 45.049 | − 0.426 | − 45.845 |
| 55 | − 4.335 | − 2.860 | − 44.400 | −0.451 | − 47.260 | |
| 67 | − 4.155 | − 0.790 | − 44.331 | − 0.399 | − 45.121 | |
| 73 | − 4.081 | − 2.446 | − 39.649 | − 0.373 | − 42.095 | |
| 85 | − 3.884 | − 2.941 | − 44.896 | − 0.496 | − 47.837 | |
| 8 | 61 | − 4.217 | − 2.512 | − 46.652 | − 0.481 | − 49.164 |
| 72 | − 4.083 | − 2.112 | − 47.312 | − 0.460 | − 49.424 | |
| 81 | − 3.969 | − 2.028 | − 45.755 | − 0.412 | − 47.783 | |
| 82 | − 3.960 | − 2.941 | − 46.844 | − 0.481 | − 49.785 | |
| 84 | − 3.897 | − 1.915 | − 47.461 | − 0.422 | − 49.376 | |
| 9 | 95 | − 3.792 | − 3.209 | − 27.435 | 0.000 | − 30.644 |
| 104 | − 3.688 | − 3.290 | − 27.338 | 0.000 | − 30.628 | |
| 111 | − 3.455 | − 5.615 | − 26.814 | 0.000 | − 32.429 | |
| 113 | − 3.441 | − 5.120 | − 27.138 | 0.000 | − 32.257 | |
| 118 | − 3.362 | − 6.191 | − 26.100 | 0.000 | − 32.291 | |
| IMN | 63 | − 4.201 | − 6.812 | − 21.909 | 0.000 | − 28.721 |
| 70 | − 4.109 | − 4.885 | − 20.459 | 0.000 | − 25.344 | |
| 94 | − 3.812 | − 5.678 | − 21.258 | 0.000 | − 26.936 | |
| 123 | − 2.734 | − 7.989 | − 23.130 | 0.000 | − 31.119 |
Almost all the ligands exhibit interactions with anti-coagulation and enzymatic both regions (Fig. 4). Docking results show that there exists hydrogen bonding between molecule 5 and His48 with a distance of 1.826 Å. Also, π − π interaction was exhibited by 5 with His48, electrostatic interactions with Lys69 and Glu129 and polar interactions with Ser23. Side chain hydrogen bondings with Lys69 and His48 and backbone hydrogen bonding with Gly30 are exhibited by molecules 3 and 4. Molecules 1, 3, 4, 5 and 6 show π − π stacking interactions with His48 while molecules 1 and 8 with Tyr52 and molecule 2 with Trp31. There exists cation—π interactions exhibited by molecule 2 with Lys69 and molecule 4 withHis48. All the ligands including IMN have electrostatic interactions with His48 and Lys69, and polar interactions with Ser23. In addition to that, all the molecules except 9 and IMN formed an array of hydrophobic interactions with the residues Phe5, Tyr22, Tyr28, Tyr52, Pro56, Cys61, Pro68, Trp31, Cys45, Cys29, Phe106, Ala18, Ile9, Leu2, Ile 19, Cys50 etc., lining the binding region IMN form the hydrophobic interaction with Trp31 and Tyr52 while 9 forms with Tyr52, Cys133, Cys50, Phe46, Leu130, Tyr28, and Trp31 residues [supplementary information: Figure S2 and Table S3].
Fig. 4.


Hydrogen bonding interactions in the best docking complexes as obtained through GLIDE XP docking (H-bonds are shown by dotted lines)
Drug likeliness and bioavailability
The Lipinski rule states, that most “drug-like” molecules have logP ≤ 5, molecular weight ≤ 500, number of hydrogen bond acceptors ≤ 10, and number of hydrogen bond donors ≤ 5. Molecules violating more than one or two of these rules may have problems with bioavailability. This rule is called the “rule of 5”, because the border values are 5, 500, 2*5, and 5. Drug likeliness and bioavailability of the ligand is inspected using the QIKPROP3.5 module. The calculated molecular properties of pyrazolo[3,4-d]pyrimidine molecules and IMN are listed in Table 4. It was found that all the ligand molecules (except molecules 1, 2 and 5) satisfies the ‘rule of 5’, indicating that these molecules can be used as potent drugs/inhibitors. The number of violations of the rule in the case of molecules 1 and 5 is 2 whereas in the case of molecule 2 it is 1, but the values are within the range of 95% drugs.
Table 4.
Some molecular properties of ligands: pryazolo[3,4-d]pyrimidines and indomethacin
| Properties | LogP | M. W. | nON | nOHNH | nRotb | PSA | Volume | Abs. | nViol. |
|---|---|---|---|---|---|---|---|---|---|
| Molecule 1 | 7.248 | 520.746 | 6.0 | 0.0 | 12.0 | 73.653 | 1633.671 | 100 | 2 |
| Molecule 2 | 5.898 | 460.571 | 6.0 | 0.0 | 10.0 | 88.192 | 1448.708 | 100 | 1 |
| Molecule 3 | 2.181 | 432.517 | 10.0 | 0.0 | 6.0 | 116.957 | 1286.759 | 87 | 0 |
| Molecule 4 | 2.898 | 460.571 | 10.0 | 0.0 | 8.0 | 114.181 | 1387.167 | 93 | 0 |
| Molecule 5 | 5.703 | 584.718 | 10.0 | 0.0 | 10.0 | 110.055 | 1749.906 | 85 | 2 |
| Molecule 6 | 3.987 | 448.578 | 8.0 | 0.0 | 7.0 | 93.776 | 1337.749 | 100 | 0 |
| Molecule 7 | 4.114 | 399.485 | 6.0 | 0.0 | 6.0 | 96.680 | 1224.358 | 100 | 0 |
| Molecule 8 | 4.280 | 422.504 | 6.5 | 0.0 | 5.0 | 98.930 | 1316.275 | 100 | 0 |
| Molecule 9 | 3.325 | 300.378 | 4.5 | 1.0 | 5.0 | 69.777 | 988.713 | 100 | 0 |
| IMN | 4.261 | 357.793 | 5.75 | 1.0 | 4.0 | 82.902 | 1061.995 | 92 | 0 |
| Range of 95% drugs | − 2.0 to 6.5 | 130 to 725 | 2.0 to 20 | 0.0 to 6.0 | 0.0 to 15.0 | 7.0 to 200.0 | 500 to 2000 | >25% | 0–4 |
[LogP Octanol-water partition coefficient; MW Molecular weight; nON Number of hydrogen-bond acceptors (O and N atoms); nOHNH Number of hydrogen-bond donors (OH and NH groups); nRotb Number of rotatable bonds; PSA Polar surface area; Volume Molecular volume; Abs. % Human oral absorption in GI (+ −20%); nViol Number of rule of 5 violations]
Conclusions
The molecular docking studies of nine trimethylene pyrazolo[3,4-d]pyrimidine molecules with viper Phospholipase A2 have been performed using Autodock, Glide-SP and Glide-XP docking protocols. Autodock calculations demonstrate that molecules 3, 5, 7 and 9 have better binding capabilities and minimum inhibitory concentrations than indomethacin. Docking simulations through Standard Precision and Extra Precision protocols of Glide reveal that almost all the pyrazolo[3,4-d]pyrimidine molecules have better binding energies than indomethacin. However, molecule 5 through Standard Precision calculation and molecules 2 and 9 through Extra Precision calculation have poorer Glide scores than indomethacin. Almost all the ligand molecules exhibit interactions with both enzymatic and anticoagulation regions of vPLA2 and also satisfy the ADME parameters’ range, essential for designing drugs. However, molecules 1, 2 and 5 seem to violate some filters of the ‘rule of 5’. On the basis of these calculations, from all the methods, molecules 3 and 7 have been proven to have more potential than indomethacin for the inhibition of anti-coagulation and inflammation activities of vPLA2. These compounds can be co-crystallized with PLA2 and the in vitro binding mode and energy with the protein could be studied. It may be a good start to use a novel class of biologically active pyrazolo[3,4-d]pyrimidine compounds in treating inflammatory disorders and snake bites.
Electronic Supplementary Material
Acknowledgements
This work is supported by the Department of Science and Technology, Government of India through the FAST TRACK Scheme awarded to Dr. Umesh Yadava (Ref. No. SR/FT/CS-78/2010). The computational facility provided through FIST scheme to our department by DST, New Delhi, is also gratefully acknowledged.
References
- 1.Grimme S, Lichtenfeld CM, Antony J. Analysis of non-covalent interactions in (bio)organic molecules using orbital-partitioned localized MP2. Phys. Chem. Chem. Phys. 2008;10:3327–3334. doi: 10.1039/b803508c. [DOI] [PubMed] [Google Scholar]
- 2.Lee EC, Kim D, Jurecka P, Tarakeshwar P, Hobza P, Kim KS. Understanding of assembly phenomena by aromatic-aromatic interactions: benzene dimer and the substituted systems. J. Phys. Chem. A. 2007;111:3446–3457. doi: 10.1021/jp068635t. [DOI] [PubMed] [Google Scholar]
- 3.Müller-Dethlefs K, Hobza P. Noncovalent interactions: a challenge for experiment and theory. Chem. Rev. 2000;100:143–167. doi: 10.1021/cr9900331. [DOI] [PubMed] [Google Scholar]
- 4.Sinnokrot MO, Sherrill CD. High-accuracy quantum mechanical studies of π − π interactions in benzene dimers. J. Phys. Chem. A. 2006;110:10656–10668. doi: 10.1021/jp0610416. [DOI] [PubMed] [Google Scholar]
- 5.Cerny J, Hobza P. The X3LYP extended density functional accurately describes H-bonding but fails completely for stacking. Phys. Chem. Chem. Phys. 2005;7:1624–1626. doi: 10.1039/b502769c. [DOI] [PubMed] [Google Scholar]
- 6.Tsuzuki S, Honda K, Uchimaru K, Mikami M, Tanabe K. Origin of attraction and directionality of the π/π interaction: model chemistry calculations of benzene dimer interaction. J. Am. Chem. Soc. 2002;124:104–112. doi: 10.1021/ja0105212. [DOI] [PubMed] [Google Scholar]
- 7.Burley SK, Petsko GA. Aromatic-aromatic interaction: a mechanism of protein structure stabilization. Science. 1985;229:23–28. doi: 10.1126/science.3892686. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka T, Tasaki T, Aoyama Y. Acridinylresorcinol as a self-complementary building block of robust hydrogen-bonded 2D nets with coordinative saturation. Preservation of crystal structures upon guest alteration, guest removal, and host modification. J. Am. Chem. Soc. 2002;124:12453–12462. doi: 10.1021/ja026704l. [DOI] [PubMed] [Google Scholar]
- 9.Desiraju GR. Supramolecular synthons in crystal engineering–a new organic synthesis. Angew. Chem. Int. Ed. Engl. 1995;34:2311–2327. doi: 10.1002/anie.199523111. [DOI] [Google Scholar]
- 10.Hunter CA. The role of aromatic interactions in molecular recognition. Chem. Soc. Rev. 1994;23:101–109. doi: 10.1039/cs9942300101. [DOI] [Google Scholar]
- 11.Avasthi K, Kumar A, Aswal S, Kant R, Raghunandan R, Maulik PR, Khanna RS, Ravikumar K. Role of arene interactions and substituent effects in conformational (syn/anti) control of 1,2-diarylethanes. Cryst. Eng. Comm. 2012;14:383–388. doi: 10.1039/c1ce06001e. [DOI] [Google Scholar]
- 12.Yadava U, Singh M, Roychoudhury M. Gas-phase conformational and intramolecular π–π interaction studies on some pyrazolo[3,4-d]pyrimidine derivatives. Comput. Theo. Chem. 2011;977:134–139. doi: 10.1016/j.comptc.2011.09.025. [DOI] [Google Scholar]
- 13.Gung BW, Emenike BU, Lewis M, Kirschbaum K. Quantification of CH...π interactions: implications on how substituent effects influence aromatic interactions. Chem. Eur. J. 2010;16:12357–12362. doi: 10.1002/chem.201001362. [DOI] [PubMed] [Google Scholar]
- 14.Leonard NJ. Trimethylene bridges as synthetic spacers for the detection of intramolecular interactions. Acc. Chem. Res. 1979;12:423–429. doi: 10.1021/ar50144a001. [DOI] [Google Scholar]
- 15.Watt M, Hardebeck LKE, Kirkpatrick CC, Lewis M. Face-to-face arene-arene binding energies: dominated by dispersion but predicted by electrostatic and dispersion/polarizability substituent constants. J. Am. Chem. Soc. 2011;133:3854–3862. doi: 10.1021/ja105975a. [DOI] [PubMed] [Google Scholar]
- 16.Wheeler SE. Local nature of substituent effects in stacking interactions. J. Am. Chem. Soc. 2011;133:10262–10274. doi: 10.1021/ja202932e. [DOI] [PubMed] [Google Scholar]
- 17.Hunter CA, Lawson KR, Urch CJ. Aromatic interactions. J. Chem. Soc. Perkin Trans. 2001;2:651–669. [Google Scholar]
- 18.Pickering AL, Seeber G, Long DL, Cronin L. The importance of π–π, π–CH and N–CH interactions in the crystal packing of Schiff-base derivatives of cis,cis- and cis,trans-1,3,5-triaminocyclohexane. Cryst. Eng. Comm. 2005;7:504–510. doi: 10.1039/b506718a. [DOI] [Google Scholar]
- 19.Aravinda S, Shamala N, Das C, Sriranjini A, Karle IL, Balaram P. Aromatic-aromatic interactions in crystal structures of helical peptide scaffolds containing projecting phenylalanine residues. J. Am. Chem. Soc. 2003;125:5308–5315. doi: 10.1021/ja0341283. [DOI] [PubMed] [Google Scholar]
- 20.Biswas G, Chandra T, Avasthi K, Maulik PR. 1,3-Bis[4,6-bis(methylthio)-1H-pyrazolo[3,4-d]pyrimidin-1-yl]propane. Acta Crystallogr. 1995;C51:2453–2455. [Google Scholar]
- 21.Sutcliffe EY, Zee-Cheng KY, Cheng CC, Robins RK. Potential purine antagonists. XXXII. The synthesis and antitumor activity of certain compounds related to 4-Aminopyrazolo[3,4-d]pyrimidine. J. Med. Chem. 1962;5:588–607. doi: 10.1021/jm01238a017. [DOI] [PubMed] [Google Scholar]
- 22.Bekhit AA, Abdel-Aziem T. Design, synthesis and biological evaluation of some pyrazole derivatives as anti-inflammatory-antimicrobial agents. Bioorg. Med. Chem. 2004;12:1935–1945. doi: 10.1016/j.bmc.2004.01.037. [DOI] [PubMed] [Google Scholar]
- 23.Varnes JG, Wacker DA, Jacobson IC, Quan ML, Ellis CD, Rossi KA, He MY, Luettgen JM, Knabb RM, Bai S, He K, Lam PYS, Wexler RR. Design, structure–activity relationship, and pharmacokinetic profile of pyrazole-based indoline factor Xa inhibitors. Bioorg. Med. Chem. Lett. 2007;17:6481–6488. doi: 10.1016/j.bmcl.2007.09.091. [DOI] [PubMed] [Google Scholar]
- 24.Elion GB, Callahan SW, Nathan H, Bieber S, Rundles RW, Hilching GH. Potentiation by inhibition of drug degradation: 6-substituted purines and xanthine oxidase. Biochem. Pharmacol. 1963;12:85–93. doi: 10.1016/0006-2952(63)90012-1. [DOI] [Google Scholar]
- 25.Schenone S, Brullo C, Musumeci F, Botta M. Novel dual Src/ Abl inhibitors for hematologic and solid malignancies. Expert Opin. Investig. Drugs. 2010;19:931–945. doi: 10.1517/13543784.2010.499898. [DOI] [PubMed] [Google Scholar]
- 26.Carraro F, Naldini A, Pucci A, Locatelli GA, Maga G, Schenone S, Bruno O, Ranise A, Bondavalli F, Brullo C, Fossa P, Menozzi G, Mosti L, Modugno M, Tintori C, Manetti F, Botta M. Pyrazolo[3,4-d]pyrimidine Inhibitors of c-Src Phosphorylation. J. Med. Chem. 2006;49:1549–1561. doi: 10.1021/jm050603r. [DOI] [PubMed] [Google Scholar]
- 27.Indovina P, Giorgi F, Rizzo V, Khadang B, Schenone S, Marzo D, Forte IM, Tomei V, Mattioli E, Urso VD, Grilli B, Botta M, Giordano A, Pentimalli F. New pyrazolo[3,4-d]pyrimidine SRC inhibitors induce apoptosis in mesothelioma cell lines through p27 nuclear stabilization. Oncogenes. 2012;31(7):929–938. doi: 10.1038/onc.2011.286. [DOI] [PubMed] [Google Scholar]
- 28.Rossi A, Schenone S, Angelucci A, Cozzi M, Caracciolo V, Pentimalli F, Pucca A, Pucci B, Montagna RL, Bologna M, Botta M, Giordano A. New pyrazolo-[3,4-d]-pyrimidine derivative Src kinase inhibitors lead to cell cycle arrest and tumor growth reduction of human medulloblastoma cells. FASEB J. 2010;24:2881–2892. doi: 10.1096/fj.09-148593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carlomagno F, Vitagliano D, Guida T, Basolo F, Castellone MD, Melillo RM, Fusco A, Santoro M. Efficient inhibition of RET/papillary thyroid carcinoma oncogenic kinases by 4-Amino-5-(4-Chloro-Phenyl)-7-(t-Butyl)Pyrazolo[3,4-d]Pyrimidine (PP2) J. Clin. Endocrinol. Metab. 2003;88:1897–1902. doi: 10.1210/jc.2002-021278. [DOI] [PubMed] [Google Scholar]
- 30.Kini RM. Phospholipase A2: a complex multifunctional protein puzzle. In: Kini RM, editor. Venom Phospholipase A2 Enzymes: Structure, Function and Mechanism. Chichester: Wiley; 1997. [Google Scholar]
- 31.Kini RM. Structure–function relationships and mechanism of anticoagulant phospholipase A2 enzymes from snake venoms. Toxicon. 2005;45:1147–1161. doi: 10.1016/j.toxicon.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 32.Singh G, Gourinath S, Sharma S, Paramasivam M, Srinivasan A, Singh TP. Sequence and crystal structure determination of a basic phospholipase A2 from common krait (Bungarus caeruleus) at 2.4A° resolution: identification and characterization of its pharmacological sites. J. Mol. Biol. 2001;307:1049–1059. doi: 10.1006/jmbi.2001.4550. [DOI] [PubMed] [Google Scholar]
- 33.Lok SM, Gao R, Rouault M, Lambeau G, Gopalakrishnakone P, Swaminathan K. Structure and function comparison of Micropechis ikaheka snake venom phospholipase A2 isoenzymes. FEBS J. 2005;272:1211–1220. doi: 10.1111/j.1742-4658.2005.04547.x. [DOI] [PubMed] [Google Scholar]
- 34.Singh N, Jabeen T, Sharma S, Somvanshi RK, Dey S, Srinivasan A, Singh TP. Specific binding of non-steroidal anti-inflammatory drugs (NSAIDs) to phospholipase A2: structure of the complex formed between phospholipase A2 and diclofenac at 2.7A° resolution. Acta Crystallogr. 2006;62:410–416. doi: 10.1107/S0907444906003660. [DOI] [PubMed] [Google Scholar]
- 35.Singh RK, Vikram P, Makker J, Jabeen T, Sharma S, Dey S, Kaur P, Srinivasan A, Singh TP. Design of specific peptide inhibitors for group I phospholipase A2: structure of a complex formed between phospholipase A2 from Naja naja sagittifera (Group I) and a designed peptide inhibitor Val-Ala-Phe-Arg-Ser (VAFRS) at 1.9A° resolution reveals unique features. Biochemistry. 2003;42:11701–11706. doi: 10.1021/bi035076x. [DOI] [PubMed] [Google Scholar]
- 36.Yadava U, Gupta HO, Roychoudhury M. A comparison of crystallographic and DFT optimized geometries on two taxane diterpenoids and docking studies with phospholipase A2. Med. Chem. Res. 2012;21:2162–2168. doi: 10.1007/s00044-011-9724-z. [DOI] [Google Scholar]
- 37.Jabeen T, Singh N, Singh RK, Sharma S, Somvanshi RK, Dey S, Singh TP. Non-steroidal anti-inflammatory drugs as potent inhibitors of phospholipase A2: structure of the complex of phospholipase A2 with niflumi acid at 2.5A° resolution. Acta Crystallogr. 2005;61:1579–1586. doi: 10.1107/S0907444905029604. [DOI] [PubMed] [Google Scholar]
- 38.Boffa MC, Rothen C, Verheij HM, Verger R, DeHaas GH. In: Natural Toxins. Eaker D, Walstrom T, editors. Oxford: Pergamon Press; 1980. pp. 131–138. [Google Scholar]
- 39.Singh N, Kumar RP, Kumar S, Sharma S, Mir R, Kaur P, Srinivasan A, Singh TP. Simultaneous inhibition of anti-coagulation and inflammation: crystal structure of phospholipase A2 complexed with indomethacin at 1.4A° resolution reveals the presence of the new common ligand-binding site. J. Mol. Recognit. 2009;22:437–445. doi: 10.1002/jmr.960. [DOI] [PubMed] [Google Scholar]
- 40.Avasthi K, Rawat DS, Maulik PR, Sarkhel S, Broder CK, Howard JAK. 1H NMR and X-ray crystallographic analysis of 1,2-bis(4,6-diethylthio-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethane and its ‘propylene linker’-analog: molecular recognition versus crystal engineering. Tetrahedron Lett. 2001;42:7115–7117. doi: 10.1016/S0040-4039(01)01478-2. [DOI] [Google Scholar]
- 41.Avasthi K, Aswal S, Kumar R, Yadava U, Rawat DS, Maulik PR. Fine tuning of folded conformation by change of substituents: 1H NMR and crystallographic evidence for folded conformation due to arene interactions in pyrazolo[3,4-d]pyrimidine core-based ‘propylene linker’ compounds. J. Mol. Struct. 2005;750:179–185. doi: 10.1016/j.molstruc.2005.04.033. [DOI] [Google Scholar]
- 42.Maulik PR, Avasthi K, Biswas G, Biswas S, Rawat DS, Sarkhel S, Chandra T, Bhakuni DS. A stacked pyrazolo[3,4-d]pyrimidine-based flexible molecule. Acta Crystallogr. 1998;C54:275–277. [Google Scholar]
- 43.Avasthi K, Aswal S, Maulik PR. A stacked pyrazolo[3,4-d]pyrimidine-based flexible molecule: the effect on stacking of an ethyl group in comparison with a methyl group. Acta Crystallogr. 2001;C57:1324–1325. doi: 10.1107/s0108270101013518. [DOI] [PubMed] [Google Scholar]
- 44.Avasthi K, Tewari A, Rawat DS, Sharon A, Maulik PR. A stacked pyrazolo[3,4-d]pyrimidine-based flexible molecule: the effect of a bulky benzyl group on intermolecular stacking in comparison with methyl and ethyl groups. Acta Crystallogr. 2002;C58:494–495. doi: 10.1107/s0108270102012039. [DOI] [PubMed] [Google Scholar]
- 45.Avasthi K, Farooq SM, Aswal S, Raghunandan R, Maulik PR. 1H NMR and crystallographic evidence for tolerance of bulky electron withdrawing methanesulfonyl group on robustness of the U-motif in pyrazolo[3,4-d]pyrimidine core-based ‘Leonard linker’ compounds and formation of plus (+) motif. J. Mol. Struct. 2007;827:88–94. doi: 10.1016/j.molstruc.2006.05.022. [DOI] [Google Scholar]
- 46.Avasthi K, Bhagat D, Bal C, Sharon A, Yadava U, Maulik PR. Unusual molecular conformation in dissymmetric propylene-linker compounds containing pyrazolo[3,4-d]pyrimidine and phthalimide moieties. Acta Crystallogr. 2003;C59:409–412. doi: 10.1107/s0108270103011442. [DOI] [PubMed] [Google Scholar]
- 47.Avasthi K, Rawat DS, Sarkhel S, Maulik PR. A dimeric layered structure of a 4-oxo-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidine compound. Acta Crystallogr. 2002;C58:325–327. doi: 10.1107/s0108270102006649. [DOI] [PubMed] [Google Scholar]
- 48.Kennedy T. Managing the discovery/development interface. Drug Discov. Today. 1997;2:436–444. doi: 10.1016/S1359-6446(97)01099-4. [DOI] [Google Scholar]
- 49.DiMasi JA. Success rates for new drugs entering clinical testing in the United States. Clin. Pharmacol. Ther. 1995;58:1–14. doi: 10.1016/0009-9236(95)90066-7. [DOI] [PubMed] [Google Scholar]
- 50.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. Autodock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998;19:1639–1662. doi: 10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B. [DOI] [Google Scholar]
- 52.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 53.Glide: version 5.8 Schrödinger, LLC, New York (2012)
- 54.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 55.Jorgenson WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all atom force field on conformational energetic and properties of organic liquids. J. Am. Chem. Soc. 1996;118:11225–11236. doi: 10.1021/ja9621760. [DOI] [Google Scholar]
- 56.Sherman W, Day T, Jacobson MP, Friesner RA. Novel procedure for modelling ligand/receptor induced fit effects. J. Med. Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 57.Bissantz C, Folkers G, Rognan D. Protein-based virtual screening of chemical database. 1. Evaluation of different docking /scoring combinations. J. Med. Chem. 2000;43:4759–4767. doi: 10.1021/jm001044l. [DOI] [PubMed] [Google Scholar]
- 58.QikProp: version 3.5 Schrödinger, LLC, New York (2012)
- 59.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997;23:3–25. doi: 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- 60.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods. 2000;44:235–249. doi: 10.1016/S1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 61.Srivastava HK, Chourasia M, Kumar D, Sastry GN. Comparison of computational methods to model DNA minor groove binders. J. Chem. Inf. Model. 2011;51:558–571. doi: 10.1021/ci100474n. [DOI] [PubMed] [Google Scholar]
- 62.Srivastava HK, Sastry GN. Molecular dynamics investigation on a series of HIV protease inhibitors: assessing the performance of MM-PBSA and MM-GBSA approaches. J. Chem. Inf. Model. 2012;52:3088–3098. doi: 10.1021/ci300385h. [DOI] [PubMed] [Google Scholar]
- 63.Kini RM, Evans HJ. Structure–function relationships of phospholipases. The anticoagulant region of phospholipases A2. J. Biol. Chem. 1987;262:14402–14407. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.