

Abstract
DNA supercoiling is an inherent consequence of twisting DNA and is critical for regulating gene expression and DNA replication. However, DNA supercoiling at a genomic scale in human cells is uncharacterized. To map supercoiling we used biotinylated-trimethylpsoralen as a DNA structure probe to show the genome is organized into supercoiling domains. Domains are formed and remodeled by RNA polymerase and topoisomerase activities and are flanked by GC-AT boundaries and CTCF binding sites. Under-wound domains are transcriptionally active, enriched in topoisomerase I, “open” chromatin fibers and DNaseI sites, but are depleted of topoisomerase II. Furthermore DNA supercoiling impacts on additional levels of chromatin compaction as under-wound domains are cytologically decondensed, topologically constrained, and decompacted by transcription of short RNAs. We suggest that supercoiling domains create a topological environment that facilitates gene activation providing an evolutionary purpose for clustering genes along chromosomes.
DNA has a helical structure that is influenced by the localized sequence context and remodeling by cellular machines, but at the level of DNA the net state of the genome is torsionally relaxed1. However, negative DNA supercoils are detected in localized regions2, 3 where they can alter the topology of the chromatin fibre4. This altered structure is maintained by transcription that generates both positive (over-wound) and negative (under-wound) supercoils ahead of and behind the RNA polymerase5, 6, respectively, and in its absence the chromatin returns to a ground state7. In general, genes that are supercoiled are more efficiently transcribed8, 9 and as tension may be a prerequisite for transcription initiation10-13 supercoiling domains might influence the gene expression of surrounding genes. However, as excessive supercoiling inhibits elongation14, 15 topoisomerases are preferentially associated with transcribed rather than non-transcribed regions where they can relieve tension16, 17. It is unclear how negative supercoils are introduced into loci prior to transcriptional activation but certain inducible genes are probably organized into a poised chromatin environment17, 18. At other genes, the transcription initiation complex transcribes short stretches of RNA and previous studies showed that the polymerase synthesizes both sense and antisense RNAs19-21. Furthermore, it is suggested that transcription of short upstream RNAs might be important for DNA supercoiling19.
The human genome is organized into GC-AT-rich sequence regions termed isochores22, originally defined by cesium chloride ultracentrifugation. Genes are preferentially found in GC rich isochores and it has been suggested that gene clustering reflects an impact of some higher-order level of chromatin organization, influencing gene expression23-25. In support of this genome-wide chromatin fiber analysis demonstrated that gene rich domains of the human genome are enriched in disrupted (“open”) chromatin fibers and these regions have a decompacted large-scale chromatin structure26, that is remodeled by transcription27. Furthermore, within gene clusters open chromatin can spread to surrounding regions27, 28, implying an ability to propagate chromatin states. In mammalian cells nucleosome arrays29 fold to form chromatin fibers that are subsequently organized into large-scale chromatin structures in an unknown manner. In metaphase chromosomes, after gentle lysis, chromatin loops can be seen to emanate from a proteinaceous scaffold-like structure30 that might persist in interphase31-33. Consistent with this, in interphase, chromosome loops tens to hundreds of kilobases in length have been seen to form thick “chromonema” fibres34 and nuclease digestion of cells indicates the chromatin is organized into 50 kb loops, forming higher order 300 kb structures35. In contrast the random walk or giant loop model of chromatin organization predicts a flexible backbone to which giant loops, each comprising several Mb of DNA are attached36. To date much of our understanding of genome organization has been dependent on cytological approaches but recently molecular techniques such as Hi-C have been used to show that the interphase genome is organized into self-interacting topological domains31-33 and these may correspond to cytologically visible large-scale chromatin structures. However, the factors that regulate or determine these structural domains and the relationship to function are poorly understood.
To investigate DNA supercoiling on a genomic scale and to study its impact on higher levels of chromatin organization, we developed a novel approach using biotinylated tri-methyl psoralen as a probe to map DNA supercoiling in cells. We found that transcription and topoisomerase activity altered DNA supercoiling around transcription start sites and this is propagated along the chromatin fiber creating heterogeneously sized supercoiling domains. These domains are delimited by GC-AT boundaries and correspond to “under-wound”, “over-wound” or topologically “stable” regions. Under-wound domains are GC-rich, highly transcribed and enriched in “open” chromatin fibers, transcription start sites, RNA polymerase and topoisomerase I binding, but depleted in topoisomerase II. Under-wound domains also have a decompacted large-scale chromatin structure that is maintained by a balance between transcription and topoisomerase activities, suggesting that changes in DNA topology can be propagated through the chromatin fiber affecting further levels of chromatin organization.
Results
Mapping DNA supercoiling at chromosomal loci
Despite its role in transcriptional regulation and DNA replication the organization, remodeling and impact of DNA supercoiling across the genome is unknown. As the introduction of negative DNA supercoils untwists the DNA strands, 4,5,8-trimethylpsoralen (TMP) intercalation has been used to monitor superhelical tension37-39 on a global scale1, 39 and at specific genes2, 3. We further developed this approach to monitor changes in DNA supercoiling at genomic loci, around genes and at regulatory elements in vivo, by attaching a biotin, via a linker, to TMP (bTMP) for use as a DNA structure probe (Fig. 1a). bTMP is cell permeant and preferentially intercalates into regions of DNA enriched in negative supercoils (Fig. 1a)39. After intercalation the bTMP is cross-linked by 360 nm light to form monoadducts between its 3,4 pyrone bond and the 5,6 double bond of pyrimidine bases. Monitoring bTMP incorporation in retinal pigment epithelial (RPE1) cells by using a NeutrAvidin-fluorescein conjugate showed that it is evenly bound across the nucleus with no pronounced enrichment or depletion at heterochromatin or in the vicinity of nucleoli (Fig. 1b). Torsional stress can be rapidly released by treatment of cells with bleomycin7, a glycopeptide antibiotic that introduces DNA strand breaks (Supplementary Fig. 1). Treatment of cells with bleomycin, resulted in a substantial decrease in bTMP binding, demonstrating that the bTMP probe can discriminate between different DNA structures.
Figure 1.
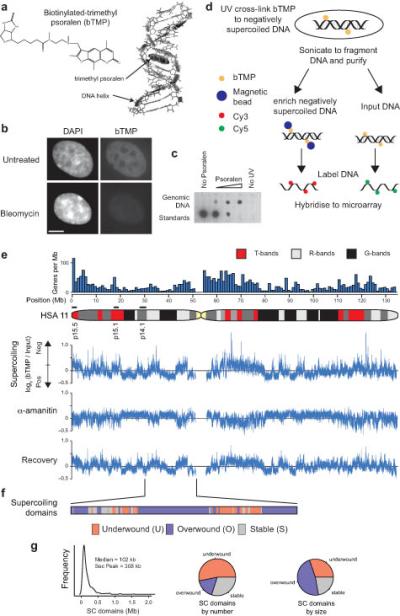
High resolution mapping of DNA supercoiling. (a) Cartoon showing biotinylated-trimethyl psoralen (bTMP)51 as a DNA structure probe that intercalates into under-wound regions of the DNA helix1, 39. (b) Immunofluorescence analysis in RPE1 cells showing the distribution UV cross-linked bTMP detected using NeutraAvidin-Fluorescein before and after bleomycin treatment. Cells were counterstained with DAPI (Bar is 5 μm). (c) Dotblot showing bTMP incorporated into genomic DNA detected by streptavidin conjugated horseradish peroxidase and chemiluminescence. (d) Experimental strategy for high resolution mapping of DNA supercoiling. Cells were treated with bTMP for 20 mins and the bTMP was cross-linked into the DNA helix with 360 nm UV light. DNA was purified, enriched for biotin-TMP using streptavidin coated magnetic beads, amplified, labeled and hybridized to genomic microarrays versus input control. (e) Microarray data showing bTMP binding across human (HSA) chromosome 11 as log2(bTMP binding/Input DNA) revealing pronounced differences in drug binding representative of differences in DNA supercoiling. Transcription inhibition with α-amanitin (5 hours) showed substantial remodeling of DNA supercoiling that was reversed upon drug washout (2 hours recovery). (f) Diagram showing supercoiling domains across 20 Mb of HSA Chr11p identified as regions that were remodeled after transcription inhibition and categorized as “under-wound”, “over-wound” or “stable”. (g) Size distribution of supercoiling domains across regions studied and pie charts showing size and frequency distribution of different domain categories.
For mapping DNA supercoiling at high resolution RPE1 cells were briefly (20 min) treated with bTMP and exposed to UV light (10 min). bTMP is quantitatively incorporated into DNA (Fig. 1c) and bound in a UV dependent manner. DNA bound to bTMP was fragmented by sonication, purified, enriched using streptavidin coated magnetic beads, labeled and hybridized to custom made tiling microarrays covering large regions of the human genome (Fig. 1d; Supplementary Table 1). The concentration of bTMP used introduced approximately one cross-linked psoralen molecule every 1-5 kb. To correct for topology independent effects of the bTMP drug, bTMP binding was analyzed on naked genomic DNA. bTMP binding to genomic DNA was subtracted from bTMP binding in cells i.e. log2(bTMPcells/Input) – log2(bTMPgenomic DNA/Input) giving the normalized log2(bTMP/Input binding) shown.
To investigate DNA supercoiling on a genomic scale bTMP binding was analyzed across human chromosome 11 using a custom-designed Nimblegen tiling array (Fig. 1e). Chromosome 11 is representative of the genome as it covers 135 Mb (5% of the genome) and is composed of regions of differing gene densities and base compositions. For example 11p15.5 and 11p15.1 are gene rich whilst 11p14.1 is gene poor. Control cells showed differential binding of bTMP across the chromosome with gene rich T-bands being more negatively supercoiled than gene poor G-bands and pronounced peaks of negative supercoiling were found at highly expressed genes such as NEAT1 and MALAT1 at 11q13.1 (Supplementary Fig. 2). To investigate the factors responsible for regulating supercoiling, cells were treated with α-amanitin, an inhibitor of transcription (Supplementary Fig. 3). After 5hrs treatment α-amanitin was washed out and the cells were allowed to recover for 2hrs. Transcription inhibition substantially altered bTMP binding at all chromosomal loci (Fig. 1e), indicative of considerable DNA remodeling and after drug washout bTMP binding was rapidly altered back to its starting state, demonstrating that DNA supercoiling was remodeled by transcription.
Examination of chromosomal loci clearly revealed that DNA supercoiling was organized into domains (Fig 1e). We reasoned that supercoiling domains would be regions that could change their structure independently of surrounding regions. We therefore calculated a difference profile between the supercoiling signal for control and α-amanitin treated cells and used an edge filter to identify supercoiling boundaries (Fig. 1f; see Methods). From this we identified 606 domains across chromosome 11 with a median size of approximately 100 kb (Fig. 1g) and characterized them as being “under-wound” (U), “over-wound” (O) or “stable” (S) based on how their supercoiling changed between control cells and cells treated with α-amanitin. Under-wound domains corresponded to 52% of the total domains, but by size only corresponded to 30% of the domains, indicating they were generally smaller than over-wound domains.
Organization of DNA supercoiling domains
To better understand the arrangement of supercoiling domains we examined large chromosomal regions. 11p15.4 (Fig. 2a) was enriched and depleted in bTMP binding corresponding to under-wound and over-wound domains. Under-wound domains were more often enriched in DNaseI binding sites (data from Encode project) e.g. around 6.5 Mb but there were also stable and over-wound regions enriched in DNaseI sensitive sites, which showed there was not a simple relationship between chromatin structure and DNA supercoiling. Recently it has been shown by Hi-C31 that the human genome is organized into large topological domains. These domains are large (approx. 900 kb), conserved between cell types and species and might correspond to a higher level of genome organization. Our analysis revealed that approximately 30% of topological boundaries31 lay within +/−20 kb of supercoiling domain boundaries (P < 0.01 by simulation; Fig. 2b). We noticed that many of these supercoiling boundaries were located at a transition between GC rich and poor regions. To confirm this, we aligned the boundaries and analyzed the base composition across the regions to show that supercoiling domains were flanked by GC-AT boundaries (Fig. 2c). Topological domain boundaries have been shown to be associated with the CTCF insulator protein31. Similarly 25% of CTCF binding sites were found within +/−20 kb of a supercoiling boundary (Fig. 2d), compared to 15% by chance, with a pronounced peak of CTCF binding sites at the boundaries (Fig. 2e; simulated data gives a flat line at 0.32 below the data shown, KS test compared to randomly generated data is P < 2.2 × 10−16). This analysis indicates that large topological domains31 are subdivided into dynamic supercoiling domains flanked by CTCF proteins, revealing an additional level of transcription-dependent genome organization.
Figure 2.
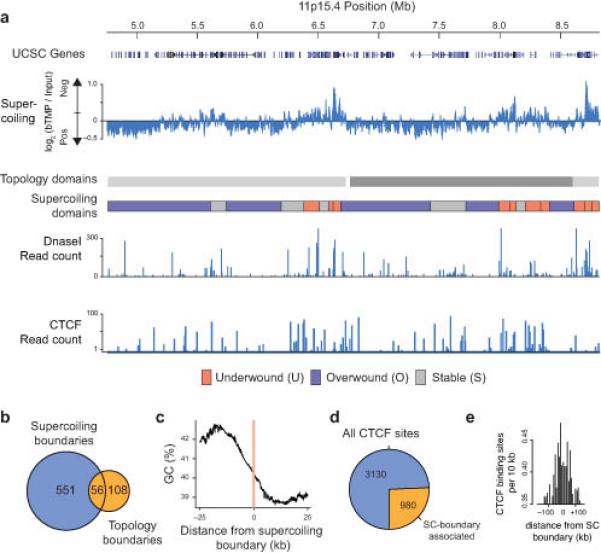
Organization and boundaries of supercoiling domains. (a) Microarray data showing bTMP binding, indicative of DNA supercoiling, at HSA 11p15.4 spanning two topological domains31. Distribution of DNaseI sensitive sites and CTCF binding sites in RPE1 cells obtained from the ENCODE project. (b) Venn diagram showing the overlap between topological domain boundaries31 and supercoiling (SC) boundaries across HSA 11. The overlap was determined by taking a +/− 20 kb window at each topological boundary and assessing whether this overlapped with a SC boundary (P < 0.01 by random permutation). (c) Graph showing the base composition around SC boundaries. (d) Pie chart showing the number of CTCF sites on HSA11 located near to a SC-boundary. The overlap was determined by counting the number of CTCF binding sites within a +/− 20 kb window at each SC-boundary. (e) Bar chart showing the number of CTCF binding sites surrounding a SC-boundary (KS test compared to randomly generated data, P < 2.2 × 10−16).
Remodeling of DNA supercoiling domains
To investigate the formation and remodeling of supercoiling domains we examined 17.5 Mb of the human genome across 7 loci at high resolution using Agilent tiling path arrays (Supplementary Table 1). The Xq13.1 locus (Fig. 3a) has a GC rich cluster of genes around 69.5 Mb that was negatively supercoiled. In contrast, two large gene bodies (EDA (expressed) and TEX11 (not expressed) located near 69 and 70 Mb were AT rich and positively supercoiled, indicating there might be a relationship between GC content and supercoiling. However, this result was not due to a localized (<100 bp) GC effect as we found no relationship between localized GC content and DNA supercoiling (data not shown) and mapping DNA supercoiling using GC rich or AT rich probes gave similar results. Similarly there were very pronounced changes at the 11p15.1 locus (Fig. 3b). This region is gene rich and predominantly consists of under-wound domains with transcription inhibition promoting a rapid change in supercoiling e.g. around 17.5 Mb, that is reversed upon drug washout. Generally genes were located within single domains, however there were some genes, such as SERGEF located at 18 Mb, that was broken into both under-wound and over-wound regions. Supercoiling generated by transcription of one gene maybe cancelled out by transcription of a neighboring gene transcribed in the same direction or enhanced by convergent or divergent transcription of neighboring genes9. The active SCYL1 and LTBP3 genes at 11q13.1 are convergent (Supplementary Fig. 2) but there were no pronounced changes in DNA supercoiling suggesting there were mechanisms to remove extraneous supercoils.
Figure 3.
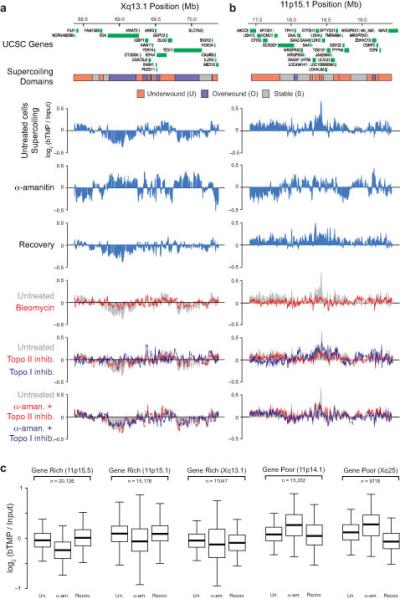
Transcription and topoisomerase dependent remodeling of DNA supercoiling. Diagrams showing the arrangement of genes and supercoiling domains at HSA Xq13.1 (a) and HSA 11p15.1 (b) and microarray data showing bTMP binding presented as log2(bTMP binding/ Input), indicative of DNA supercoiling. The effect of transcription and topoisomerases on supercoiling was investigated using inhibitors. Cells were either treated with bleomycin (10 min) or transcriptionally blocked using α-amanitin (α-aman.) for 5 hours in the presence or absence of topoisomerase inhibitors (camptothecin, topoisomerase I inhibitor (topo I inhib.); and ICRF193, topoisomerase II inhibitor (topo II inhib.), as shown. (c) Boxplots showing the quantification of DNA supercoiling at gene rich and poor genomic loci. (Un., untreated; α-am., α-amanitin; Recov., Recovery). For the boxplots, the bottom and top of the box are the 25th and 75th percentile and the band near the middle is the median whilst the whiskers show 1.5 x the interquartile range. n corresponds to the number of probes analyzed in the locus. For all transitions P < 2.2 × 10−16 (t-test).
Bleomycin treatment of cells introduced DNA nicks (Supplementary Fig. 1) and diminished differences in DNA supercoiling across chromosomal loci demonstrating that different supercoiling structures observed were DNA topology dependent. Furthermore, treatment of cells with topoisomerase inhibitors camptothecin (topoisomerase I) and ICRF193 (topoisomerase II) gave similar results showing that topoisomerases were also required to maintain the supercoiling state. However, treatment of cells with both α-amanitin and topoisomerase inhibitors had a limited effect demonstrating that changes in supercoiling seen upon transcription inhibition required topoisomerase activity.
To quantify changes in supercoiling at both gene rich and gene poor chromosomal regions we summed the amounts of bTMP binding across the loci (Fig. 3c). Gene rich regions (11p15.5, Xq13.1, 11p15.1, Enr312, Enr332) were significantly more negatively supercoiled (under-wound) than gene poor regions (11p14.1, Xq25) (P < 2.2 × 10−16; t test). At gene rich loci there was a decrease in bTMP binding after transcription inhibition that was followed by an increase in bTMP binding, consistent with an increase in negative supercoiling, after transcription recovery. Interestingly, the gene poor regions behaved in the opposite manner with an increase in positive supercoiling after transcription initiation. As total bTMP binding in cells does not change with transcription inhibition (data not shown) this suggests that supercoiling may be propagated between chromosomal regions.
Properties of supercoiling domains
It is possible that supercoiling domain boundaries are physical structures enriched in CTCF insulator proteins (Fig. 2) and topoisomerase II binding to a proteinaceous scaffold or, alternatively, the domains themselves might be enriched in enzymatic activities which facilitate domain formation. To analyze these possibilities we mapped transcription, RNA polymerase and topoisomerase binding across the chromosomal loci (Fig. 4a). Under-wound (U) domains were highly transcribed and there was a strong relationship between transcription and supercoiling (Fig. 4b). U-domains were significantly enriched in DNaseI sensitive sites, disrupted (“open”) chromatin fibres27, transcription start sites, and were GC-rich (Fig. 4c). U-domains were also enriched in RNA polymerase and topoisomerase I which correlated well to each other (Fig. 4d). Furthermore, U-domains were enriched in SINES whereas O-domains were enriched in LINES (data not shown). In contrast, U-domains were significantly depleted in topoisomerase IIα and IIβ (Fig. 4a, c). O-domains generally had the opposite organization to U-domains, whilst S-domains have properties intermediate to both. This data suggests that the polymerases and topoisomerases located in the domains determine their topology allowing supercoiling to propagate between domains but being controlled by the underlying organization of genes, base composition and being flanked by GC-AT boundaries and CTCF insulator proteins (Fig. 2). We therefore suggest that domain structure will vary depending on the transcriptional environment and indicates that supercoiling domains are a dynamic property of the chromatin fiber, rather than being a static, structural feature.
Figure 4.
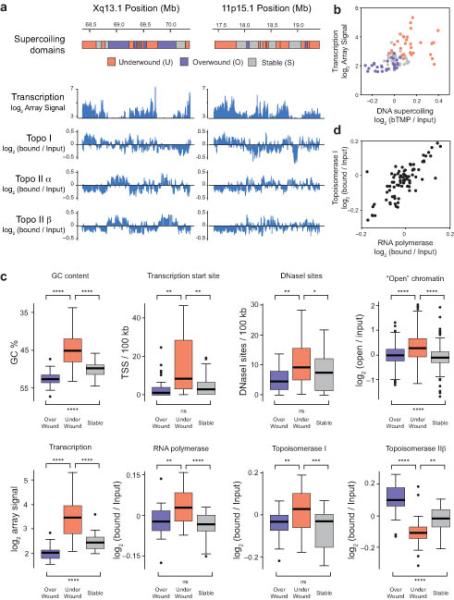
RNA polymerase and topoisomerases define supercoiling domains. Diagrams showing the arrangement of supercoiling domains at HSA Xq13.1 (a) and HSA 11p15.1 (b) and microarray data showing the distribution of RNA transcripts and ChIP for topoisomerase (topo) I, topoisomerase IIα and IIβ binding across the genomic loci. Transcription is presented as log2 (array signal) whilst ChIP data is shown as log2 (bound/input). B. Scatter plot showing the relationship between transcription and DNA supercoiling in under-wound, over-wound and stable domains. (c) Boxplots showing GC content, transcription start site density, DNaseI site frequency, “open chromatin”, RNA transcription, RNA polymerase II binding, topoisomerase I and topoisomerase IIβ binding at “over-wound”, “under-wound” or “stable” supercoiling domains. Boxplots are as described in Fig. 3 and outliers are shown as black dots. Significance was assessed by t-test and asterisks correspond to the P-value (no *, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001). (d) Scatterplot showing the relationship between topoisomerase I and RNA polymerase II binding within different supercoiling domains.
DNA supercoiling at TSSs and regulatory elements
As some gene promoters are known to be negatively supercoiled2, 3 we analyzed DNA supercoiling upstream and downstream of transcription start sites (TSSs) of coding and non-coding genes (Fig. 5a). TSSs were negatively supercoiled in a region that extends approximately 20 kb into the body of the gene and 10 kb upstream. DNA nicking with bleomycin reduces psoralen binding returning the DNA to a relaxed state demonstrating that TSSs were under topological strain. Active genes were also more negatively supercoiled than inactive genes and showed a persistence of negative supercoiling into the body of the gene. Transcription inhibition caused remodeling of TSS DNA to a more positive supercoiled state and subsequent reactivation of transcription, promoted a change back to a more negatively supercoiled state (Fig. 5b). This demonstrated that DNA supercoiling at TSSs is dynamic, reversible and supercoiled regions are formed by transcription. To investigate other enzymatic activities required for remodeling DNA supercoiling at TSSs, cells were treated with inhibitors of topoisomerases I and II (Fig. 5b). Changes in TSS supercoiling were seen with topoisomerase treatment but this was abrogated when cells were treated with both transcription and topoisomerase inhibitors demonstrating that topoisomerase activity is required for remodeling DNA supercoiling at promoters, as well as chromosomal domains (Fig. 3). To further investigate DNA supercoiling at other features we analyzed bTMP binding at DNaseI sites and showed these were negatively supercoiled in a transcription-dependent manner (Fig. 5c). In contrast randomly generated DNA shows no peaks around genomic features (data not shown). Furthermore, CTCF sites that can act as insulators and facilitate chromatin-chromatin interactions40 are negatively supercoiled as are p300-CBP enhancer binding sites (data from A549 cells) (Fig. 5d), indicating that factor binding might be influenced by the underlying DNA topology, facilitated by topoisomerase I activity (Fig. 5e).
Figure 5.
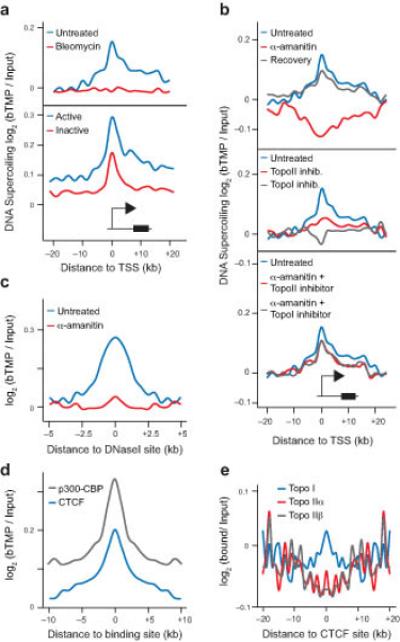
DNA supercoiling around TSSs and regulatory elements. (a) Graph showing bTMP binding, indicative of DNA supercoiling, +/− 20 kb around transcription start sites (TSSs) in the presence or absence of bleomycin for active and inactive genes. (b) Profiles showing the changes in DNA supercoiling around TSSs after inhibiting RNA polymerases and topoisomerases. (c) Graph showing distribution of DNA supercoiling +/−5 kb of DNaseI sensitive sites before and after transcription inhibition. (d) Profile of DNA supercoiling +/− 10 kb around CTCF and p300-CBP binding sites. CTCF binding sites for RPE1 cells were obtained from the ENCODE project. p300-CBP binding sites are for A549 cells from the ENCODE project. (e) Graph showing topoisomerase binding +/− 20 kb around CTCF binding sites as determined by ChIP-microarray. T-tests were used to show the peak signal was significantly different to randomly generated background data, P < 2.2 × 10−16.
Large-scale chromatin compaction of supercoiling domains
The mechanisms responsible for folding large-scale chromatin structures are poorly characterized. As packaging of large-scale chromatin structures can be transcription dependent27, 41, 42 we hypothesized that an alteration in DNA supercoiling may affect higher levels of chromatin organization. To investigate a relationship between supercoiling and compaction of chromatin structures we analyzed under-wound (U) (11p15.5, 11p15.1) and over-wound (O) regions (11p14.1) (Fig. 6a, b) and then used 3D DNA FISH to study large-scale chromatin compaction of these genomic loci. Labeled fosmid probes (Supplementary Table 2) separated by approximately 1.5 Mb were differently labeled and hybridized to RPE1 cells (Fig. 6c). The compaction of the different regions were analyzed by measuring the distance between the probes. The gene-rich under-wound 11p15.5 and 11p15.1 loci (Fig. 6d) were more cytologically decompacted than the over-wound 11p14.1 locus showing a relationship between DNA supercoiling and compaction. To test whether large-scale chromatin structures are under tension cells were treated with bleomycin. 11p15.1 (1.5 Mb) and Xq13.1 (2Mb) were compacted by bleomycin treatment (Fig. 6e, f), demonstrating that the cytological structure of chromosomal regions were under topological strain.
Figure 6.
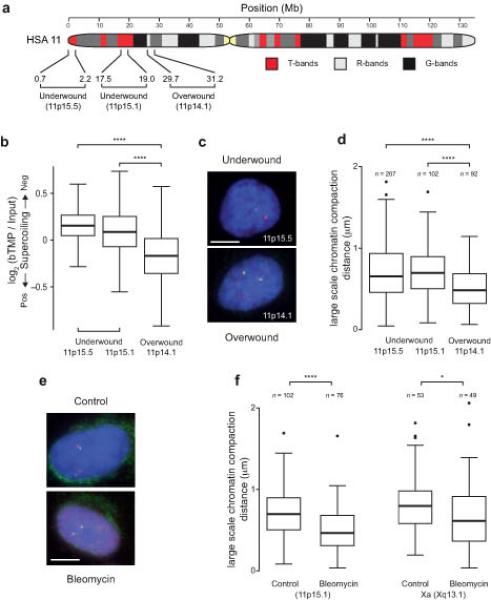
Under-wound domains are cytologically decondensed and torsionally constrained. (a) Ideogram of human (HSA) chromosome 11 showing T-bands, R-bands, G-bands and probe positions at the under and over-wound 1.5 Mb chromosomal loci studied. (b) Boxplot showing the DNA supercoiling at the under-wound 11p15.5 and 15.1 loci and at the over-wound 11p14.1 loci, as measured by bTMP binding. (c) Representative images showing 3D DNA FISH of pairs of labeled fosmid probes (red and green spots) positioned 1.5 Mb apart either side of the loci to measure large-scale chromatin compaction. Nuclei are counterstained with DAPI. Bar is 5 μm. (d) Boxplots showing the distance between pairs of fosmid probes as a measure of chromatin compaction at under-wound and over-wound genomic loci. (e) Representative images showing 3D DNA FISH of pairs of labeled fosmid probes (red and green spots) positioned 1.5 Mb apart at 11p15.1 in the presence and absence of bleomycin. Nuclei are counterstained with DAPI. Bar is 5 μm. (f) Boxplot showing the change in large-scale chromatin compaction at the 11p15.1 and Xq13.1 loci after treatment with bleomycin. Boxplots are as described in Fig. 4 and asterisks correspond to P-values determined by Wilcoxon test, also as described in Fig. 4. n is the number of separate probe pairs examined.
Remodeling of large-scale chromatin structures
To assess the effect of transcription inhibition on large-scale chromatin structures we analyzed the compaction of the 11p15.5 and 11p15.1 loci. After α-amanitin treatment the loci become positively supercoiled (over-wound) (Fig. 3c) and the chromosomal structures concomitantly become compacted (Fig. 7a) suggesting that transcription can affect large-scale chromatin structures by altering the DNA topology.
Figure 7.
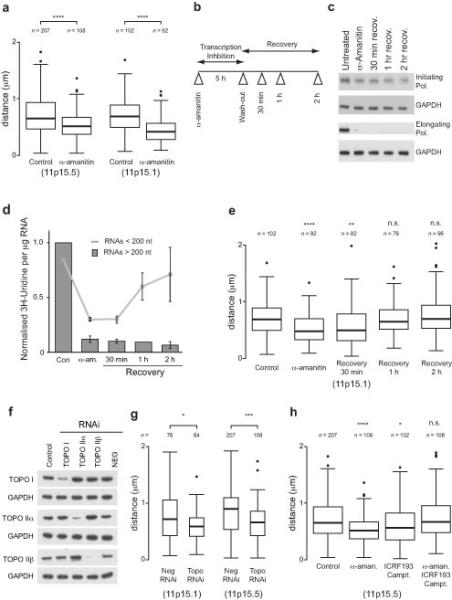
Transcription and topoisomerase dependence of large-scale chromatin structures. (a) Boxplot showing the change in large-scale chromatin compaction using 3D DNA FISH at the 11p15.5 and 11p15.1 loci after 5 hrs α-amanitin treatment to inhibit transcription. (b) Schematic showing the experimental approach used to investigate changes in chromatin structure after transcription inhibition and recovery. (c) Western blot showing the global levels of RNA polymerase using antibodies against differently activated forms of the polymerase after transcription inhibition. GAPDH is shown as a loading control. (d) Graph showing incorporation of 3H-Uridine into short (< 200 nt) and long (> 200 nt) RNA after 30 min pulse labeling to measure RNA synthesis after inhibition of transcription by α-amanitin followed by recovery. Error bars are s.d. (n = 3). (e) Boxplots showing the compaction of the 11p15.1 locus after transcription inhibition and drug washout. (f) Western blot showing the loss of topoisomerase (topo) I and II proteins after topoisomerase RNAi. GAPDH is shown as a loading control. (g) Boxplot showing distance between pairs of fosmid probes at 11p15.1 and 11p15.5 loci after topoisomerase RNAi (Neg., negative; topo, topoisomerase). (h) Boxplot showing distance between pairs of fosmid probes at the 11p15.1 locus after transcription inhibition by α-amanitin in the presence or absence of topoisomerase inhibitors, ICRF193 or camptothecin. Boxplots are as described in Fig. 4 and asterisks correspond to P-values determined by Wilcoxon test, also as described in Fig. 4. n = the number of separate probe pairs examined. In panels (e) and (h), P-values are calculated compared to control.
To investigate the kinetics of transcription responsible for altering DNA supercoiling we inhibited transcription with α-amanitin, washed the drug out and monitored changes in RNA polymerase and RNA transcription during recovery (Fig. 7b). Western blots showed a substantial, proteasome dependent (data not shown), loss in elongating RNA polymerase that persisted through recovery whilst the initiating form of the polymerase was more resistant to degradation (Fig. 7c). After transcriptional activation we reasoned that elongating RNA polymerase would produce large RNA fragments consistent with gene transcription, whilst a polymerase engaged in non-productive transcription will produce shorter transcripts43, 44. Pulse labeling of the cells with 3H-Uridine showed that production of long RNAs (> 200 not) was substantially reduced after 5 hours α-amanitin treatment and continued to decrease after α-amanitin wash out (Fig. 7d). In contrast, short RNAs (< 200 nt) were produced in abundance by 1 hour recovery and continued to increase at 2 hours. These results demonstrated that changes in DNA supercoiling precedes transcriptional elongation, at a time when the cell is synthesizing short RNAs and only the initiating form of RNA polymerase is present. From this, we hypothesized that the initiating form of RNA polymerase might be synthesizing short RNAs. To further investigate this we inhibited transcription elongation with flavopiridol, a kinase inhibitor that blocks phosphorylation of the C-terminal domain of RNA polymerase II leaving it in a non-processive state45. There is a substantial reduction in elongating polymerase (Supplementary Fig. 4a) and monitoring RNA synthesis (Supplementary Fig. 4b) showed a rapid reduction in long RNA production, but a lag before a drop in short RNA synthesis, consistent with initiating RNA polymerase producing short RNAs. To assess how large scale decompacted chromatin structures were formed, cells were treated with α-amanitin followed by drug wash-out and the compaction of 11p15.1 was monitored by FISH. Upon transcription inhibition the locus was rapidly compacted but was decompacted after α-amanitin wash-out with the same kinetics (Fig. 7e) as the remodeling of DNA supercoiling (Fig. 3) and the transcription of short RNAs (Fig. 7d).
Our previous experiments showed that topoisomerase inhibition caused a change in DNA supercoiling (Fig. 3). Therefore to further test the relationship between changes in DNA supercoiling and large-scale chromatin structures we ablated topoisomerases with RNAi (Fig. 7f). siRNA transfected cells, co-transfected with a fluorescent oligo, were FACS sorted onto slides (Supplementary Fig. 5a, b) and chromatin compaction was measured at the 11p15.1 and 11p15.5 loci. 48 hours depletion of topoisomerases did not affect transcription (Supplementary Fig. 5c) but as with α-amanitin treatment (Fig. 7a) the loci were compacted, showing that large-scale chromatin structures are also topoisomerase dependent (Fig. 7g). To confirm this result topoisomerases were inhibited using drugs that also promoted the compaction of large-scale chromatin structures (Fig. 7h). Treatment of cells with both α-amanitin and topoisomerase inhibitors together abrogates any change in supercoiling (Fig. 3) or a change in large-scale chromatin structure (Fig. 7f) showing that compaction of these structures by transcription inhibition requires topoisomerase activity. Taken together, this data supports a surprising model that links transcription to the alteration of supercoiling domains and the subsequent decompaction of large-scale chromatin structures (Fig. 8).
Figure 8. Relationship between transcription, DNA supercoiling and large-scale chromatin structures.
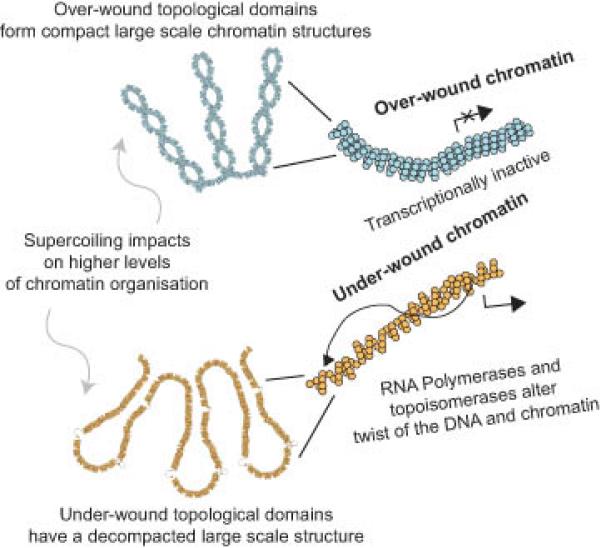
Transcriptionally inactive chromatin is topologically over-wound and has a cytologically compact large scale chromatin structure. In contrast transcriptionally active regions or transcriptional activation alters DNA supercoiling, remodeling supercoiling domains and is accompanied by a decompaction of large scale chromatin structures. Therefore, large structural domains, for example as described by Hi-C31, are subdivided into smaller transcription dependent supercoiling domains providing an additional level of functional organization within the human genome.
Discussion
Formation and functional role of supercoiling domains
This study shows that RNA polymerase and topoisomerase activities remodel DNA supercoiling, creating supercoiling domains (Fig. 1-4) and impact on the folding of large-scale chromatin structures (Fig. 6-7). As transcription is more efficient on a supercoiled template genes located within under-wound domains will be more efficiently activated than genes that are located in a less transcriptionally permissive chromatin environment. Supercoiling introduced at a transcriptionally active loci could be transmitted to adjacent gene regions, creating an environment which promotes the transcriptional activation of surrounding genes9. This could provide an additional level of gene regulation and create an evolutionary constraint for maintaining genes with similar properties together24, 28. However, it is unclear how genes initially adopt this under-wound configuration. Some genes, such as HSP70, are in a transcriptionally poised state with the polymerase already engaged at the locus18 whilst other genes are probably in a torsionally relaxed state that is less able to form an initiation complex11, 12. Our data suggests that DNA supercoiling is initially introduced at these loci by the activity of the initiating form of RNA polymerase generating short RNA transcripts (Fig. 7), creating an environment for the generation of a stable initiation complex10. After transcription initiation, polymerases in collaboration with topoisomerase and helicases can then maintain the supercoiling state of the locus in a regulated manner4. Genes like pS2 cyclically recruit factors for transcription46 and these bursts of transcriptional activity will generate supercoiling that needs to be dissipated before the next round of transcription. This implies there must be mechanisms for monitoring the levels of DNA supercoiling, so the recruitment and regulation of topoisomerases will be essential, to maintain a suitable level of torsion, ensuring optimal transcription. Furthermore, our results suggest that topoisomerases are actively required for remodeling DNA supercoiling, supercoiling domains (Fig. 3, 5) and large-scale chromatin structures (Fig. 7). A surprising result is the pronounced changes in supercoiling that occur after transcription inhibition and to a lesser extent for topoisomerase inhibition (Fig. 3a, b). As the DNA microarrays show relative bTMP binding it is necessary to consider these changes as relative to each other. It is therefore possible that most changes in supercoiling are occurring in the gene-rich under-wound domains whilst the structure of the over-wound domains remains relatively constant.
Supercoiling domains and large-scale chromatin structures
Polymerases introduce supercoils into DNA increasing the free energy available in the chromatin fiber. Therefore releasing supercoils is thermodynamically favored, releasing energy that can be used by transcription, DNA replication and chromatin remodeling. Unconstrained DNA, found between nucleosomes, will partially unwind in response to negative supercoiling whilst nucleosomal DNA will behave differently. As this DNA is tightly associated with nucleosomes, untwisting of the DNA will cause unconstrained chromatin fibers to rotate, enabling superhelical turns to be transmitted through the domain. However, it is unknown how DNA supercoiling affects chromatin fiber structures; the fiber can probably absorb a certain amount of supercoiling but at some point it would become distorted, influencing its conformation.
Previously, we showed that gene rich regions of the human genome are enriched in disrupted (“open”) chromatin fibers but these do not correspond directly to gene transcription as inactive genes can be located in “open” domains, whilst active genes can be located in compact domains26. Recently, we also showed that “open” chromatin can be propagated from one region to adjacent regions and that disrupted chromatin from neighboring active genes has an additive effect27. As there is a net change in supercoiling across chromosomal loci (Fig. 3c) we suggest that superhelical tension can be propagated from one region to another. This is further supported by the data, as supercoiling domains cover larger regions than just the promoters of genes, suggesting that supercoils must be able to “flow” along the chromatin fiber. This would explain why transcriptionally inactive genes can be found in “open” chromatin, if surrounding genes are also active26 and provides an explanation for the propagation of “open” chromatin states27.
Previous studies showed that large-scale chromatin domains are approximately 150 kb in size34, 35 and these might correspond to structures constrained by topoisomerases or by an attachment to an underlying matrix. Our study indicates that supercoiling domains are heterogeneous, are approximately 100 kb in size and are generated by polymerase and topoisomerase activities. We suggest that supercoiling domains also have a structural role, as “under-wound” domains have a decompact large-scale chromatin structure and “over-wound” domains have a compact structure, suggesting they impact on higher levels of chromatin organization. Although supercoiling domains are formed by enzymatic activities (Fig. 3) it is possible that other proteins e.g. HMGA1 are required to stabilize these structures. Alternatively, as an underlying nuclear matrix has not been well defined, domain boundaries may interact with each other, generating a “chromatin matrix”. These dynamic interactions could then stabilize the domains but at the same time provide sufficient flexibility to allow domain remodeling as the transcription profiles change.
The genome is organized into isochores, with a mean size of 0.9 Mb47. GC rich isochores are gene rich and are also more negatively supercoiled than GC poor regions, but what is the important determinant of the supercoiling state, genes or GC? These two properties have evolved hand in glove but as GC rich sequences position nucleosomes more tightly48 these strongly positioned nucleosomes will create a fiber that is relatively irregular, which may in turn facilitate negative supercoiling, providing a permissive environment for genes and gene activation. In contrast, regions that bind nucleosomes less tightly will enable nucleosomes to slide or move, to evenly spaced positions, forming a regular chromatin fiber, that is less readily supercoiled. Therefore, different isochores would be expected to have different properties, affecting the behavior of genes located within them. As gene rich (and GC rich) regions of the genome are also more likely to contain chromosomal aberrations and genes mis-regulated in cancer are more often found in GC rich, “open” chromatin (49, data not shown) it is possible that alterations in DNA supercoiling and changes in supercoiling domains could influence genome stability. In addition to the fundamental role DNA supercoiling plays in influencing gene transcription and DNA replication in normal cells, it may be particularly important in disease as many tumors over express topoisomerases. Therefore topoisomerase mis-regulation might affect supercoiling giving rise to chromosomal aberrations or increase mutation rate50 increasing cellular heterogeneity providing a driving force for tumor evolution.
Methods
Cell Culture
RPE1 cells (ATCC CRL-4000) were cultured in DMEM-F12, 3mM Glutamine, 15 mM HEPES supplemented with 0.34% Sodium Bicarbonate, 10% FBS, penicillin (100 U.ml−1), streptomycin (100 μg.ml−1) and phenol red (8.1 mg.L−1). Transcription was blocked by adding flavopiridol (100 μM) or α-amanitin (50 μg.ml−1) to cells for the times indicated. α-amanitin treatment was reversed by three PBS washes and the addition of fresh medium. Topoisomerase I and II were inhibited using Camptothecin (5 μM) or ICRF-193 (35 μM) (Biomol), respectively.
3D DNA FISH
Cells were grown overnight on glass slides. Slides were rinsed with PBS and fixed in 4% paraformaldehyde for 10 minutes. Slides were rinsed with PBS and cells were permeabilized for 10 minutes on ice with PBS supplemented with 0.2% triton. After rinsing slides were stored in 70% EtOH at 4°C.
For processing, slides were dehydrated through an ethanol series and incubated with 2×SSC supplemented with 100 μg.ml−1 RNase A (Invitrogen) at 37°C for 60 minutes. Slides were then rinsed briefly with 2×SSC, dehydrated through an ethanol series and air-dried. Slides were warmed by incubating in a 70°C oven for 5 minutes before being denatured for 1 minute in 70% formamide in 2×SSC pH 7.5 at 70°C. Slides were then transferred to 70% ethanol on ice, dehydrated through an ethanol series and air dried before overnight hybridization at 37°C with pairs of probes (listed in Table S2) labeled in digoxigenin-11-dUTP or biotin-16-dUTP. 150 ng of each labeled probe was hybridized with 5μg of Salmon Sperm and 10 μg of human Cot1 DNA. Slides were washed: 4× 3 mins 2×SSC at 45°C, 4× 3 mins 0.1×SSC at 60°C before transferring to 4×SSC with 0.1% Tween 20 at room temperature. Digoxigenin-labeled probes were detected by using one layer of rhodamine conjugated sheep anti-digoxigenin and a second layer of Texas red conjugated anti-sheep (Vector Laboratories). Biotin-labeled probes were detected by using one layer of FITC conjugated streptavidin followed by a layer of biotin conjugated anti-avidin and a second layer of FITC conjugated streptavidin (Vector Laboratories). Slides were counterstained with 0.5 μg.ml−1 DAPI.
Image Capture and Analysis
Four color 3D RNA/DNA FISH slides were imaged using a Hamamatsu Orca AG CCD camera (Hamamatsu Photonics (UK) Ltd, Selwyn Garden City, UK) Zeiss Axioplan II fluorescence microscope with Plan-neofluar objectives, a 100W Hg source (Carl Zeiss, Welwyn Garden City, UK) and Chroma #86000v2 quadruple band pass filter set (Chroma Technology Corp., Rockingham, VT) with the single excitation and emission filters installed in motorized filter wheels (Prior Scientific Instruments, Cambridge, UK). Image capture and analysis were performed using in-house scripts written for Iola Spectrum (Scanalytics Corp, Fairfax, VA). For three color imaging the microscope was the same but a Chroma #83000 triple band pass filter set was used with single excitation filters installed. For FISH, images were collected from at least 50 randomly selected nuclei for each experiment and then analyzed by using custom Iola scripts that calculate the distance between two probe signals. The statistical significance of compaction between pairs of probes was tested using the nonparametric Wilcoxon test for paired samples whilst p values between chromosomal loci was calculated by using the nonparametric Mann–Whitney U test (using R programming). P < 0.05 has been taken as statistically significant.
RNA extraction and analysis
Total RNA (large RNAs (> 200 nt) and small RNAs (< 200 nt)) was extracted from cells using Tri Reagent (Sigma) and purified on a silica matrix (Qiagen miRNeasy kit). Separate large and small RNAs were extracted by selective binding to a silica matrix (Qiagen miRNeasy kit). Residual DNA was removed by on-column DNaseI (Roche) treatment. RNA size fractionation was monitored by analyzing RNA species on 10 or 15% TBE-Urea gels (Invitrogen).
3H Uridine Incorporation
Global transcription was determined by measuring [5-3H]Uridine incorporation for a 30 min time period. Cold dA, dG, dC, dT and C (37 nM final) were added to cells, to suppress label incorporation into DNA, with 185nM [5-3H] Uridine. After 30 minutes incubation, cells were rinsed with PBS and RNA was extracted by selective binding to a silica matrix (miRNeasy Kit, Qiagen). Residual DNA was removed by on-column DNaseI treatment. RNA was quantified using a Nanodrop and 3H incorporation was measured by scintillation counting.
Western blotting
Cells were suspended in 2 × SDS lysis buffer, incubated at 100°C for 5 minutes and sonicated briefly. For RNA polymerase blots protein samples were resolved on 8% bis-tris gels and transferred to PVDF membrane by wet-transfer. For histone blots protein samples were resolved on 12% bis-tris gels and transferred to PVDF membrane by semi-dry transfer. Membranes were probed with antibodies using standard techniques and detected by enhanced chemiluminescence. RNA polymerase II antibodies: H-224, 1:200 (Santa Cruz, sc-9001); 8WG16, 1:200 (Covance, MMS-126R); H5, 1:500 (Covance, MMS-129R); H14, 1:500 (Covance, MMS-134R); CTD4H8, 1:200,000 (Upstate 15-623) as described previously (Stock et al., 2007). Other antibodies were GAPDH, 1:1000 (Cell Signaling, #2118).
Chromatin nicking and analysis
Cells in 6 well plates (for DNA extraction) or on slides (for FISH analysis) were treated with bleomycin (100 μM) for 10 mins. For DNA extraction cells were lysed in 150 mM NaCl, 1% SDS, 10 mM EDTA and incubated for 1 hr at 55°C with 100 μg/ml Proteinase K. DNA was purified by phenol chloroform extraction and recovered by ethanol precipitation. DNA was suspended in 50 mM NaOH, 1 mM EDTA, 3% Ficoll, 0.02% bromocresol green, 0.04% xylene cyanol and fractionated on a 0.7% agarose gel in 50 mM NaOH, 1 mM EDTA at 2 V/cm with buffer circulation for 20 hrs. The gel was stained with EtBr in 1 × TBE and imaged on a FLA5100 laser scanner (Fuji).
Chromatin immuno-precipitation
ChIP was performed as described previously52 except that magnetic Protein A beads (Invitrogen) were used for rabbit antibodies and Protein L beads were used for mouse IgM antibodies. Antibodies were Topoisomerase I (Abcam, ab3825), Topoisomerase IIα (Santa Cruz, sc-5347) and Topoisomerase IIβ (BD Transduction Laboratories, 611493), and total RNA polymerase H-224 (Santa Cruz, sc-9001). All antibodies were characterized in Western blots and ChIP was optimized using qPCR assays. For microarray hybridization immunoprecipitated DNA was amplified using whole genome amplification (Sigma).
Short Interfering (siRNA) Transfections
Cells in 6 well plates (for protein extraction) or on slides (for FISH analysis) were transfected with Topoisomerase siRNA (ON-TARGETplus SMARTpool siRNA, Dharmacon) using Lipofectamine 2000 reagent (Invitrogen) for 48 hours with a fluorescent marker oligo (BLOCK-iT fluorescent oligo, Invitrogen). siRNA sequences were: TOP1: GAAAAUGGCUUCUCUAGUC, GAUUUCCGAUUGAAUGAUU, GCACAUCAAUCUACACCCA, and CGAAGAAGGUAGUAGAGUC. TOP2A: CGAAAGGAAUGGUUAACUA, GAUGAACUCUGCAGGCUAA, GGAGAAGAUUAUACAUGUA and GGUAACUCCUUGAAAGUAA. TOP2B: GAAGUUGUCUGUUGAGAGA, CGAAAGACCUAAAUACACA, GAUCAUAUGGGAUGUCUGA and GGUGUAUGAUFAAGAUGUA. NEG: ON-TARGETplus Non-targeting Pool D-001810-10-05. All siRNA were used at 100 nM.
Biotinylated Psoralen
Biotinylated trimethylpsoralen (bTMP) was synthesized according to Saffron et al.,51 and purified by HPLC giving >95% by TLC and ELSD (evaporative light scattering). The structure was confirmed by 1H NMR.
Analyzing changes in DNA supercoiling
Cells or control genomic DNA were treated with various concentrations (50-500 μg.ml−1) of Biotinylated-TMP for 20 mins at RT in the dark. Biotinylated-TMP was UV cross-linked to DNA at 360 nm for 10 mins. DNA was purified from the cells using SDS/PK digestion followed by phenol:chloroform:isoamylaclohol extraction. DNA was fragmented by sonication (10 × 20s at 2 μ). Biotin incorporation into the DNA was detected by dot blotting using an alkaline phosphatase conjugated avidin as a probe. The Biotinylated-psoralen DNA complex in TE was immunoprecipitated using avidin conjugated to magnetic beads for 2 hours at room temperature and then overnight at 4°C. Beads were washed sequentially for 5 min each at room temperature with TSE I (20 mM Tris pH 8.1, 2 mM EDTA, 150 mM NaCl, 1% Triton X-100 and 0.1% SDS), TSE II (20 mM Tris pH 8.1, 2 mM EDTA, 500 mM NaCl, 1% Triton X-100 and 0.1% SDS) and buffer III (10 mM Tris pH 8.1, 0.25 M LiCl, 1 mM EDTA, 1% NP40 and 1% deoxycholate). Beads were then washed twice with TE buffer for 5 min. To extract the DNA and to release psoralen adducts the samples were boiled for 10 mins at 90°C in 50 μl of 95% Formamide with 10 mM EDTA. Samples were than made up to 200 μl with water and the DNA was purified using a Qiagen PCR purification Kit. To release torsional stress in the chromatin fiber the DNA was cleaved by treatment of cells with 100 μM bleomycin (Sigma B2434) for 10 minutes at 37°C. DNA was extracted and DNA damage analyzed by non denaturing gel electrophoresis.
Biotinylated trimethyl psoralen Immuno
Cells were grown overnight on glass slides. Slides were rinsed with 3 x PBS and then treated with Biotinylated-TMP (1mg.ml−1) for 20 mins at RT in the dark. Biotinylated-TMP was UV cross-linked to the DNA at 360 nm for 10 mins. Slides were rinsed 3 x with PBS and fixed in 4% paraformaldehyde. Slides were rinsed 3 x with PBS and cells were permeabilized for 10 minutes with PBS supplemented with 0.2% triton. After rinsing with PBS, slides were blocked 5% horse serum in PBS for 15minutes at room temperature in a humidified chamber. The Biotinylated-TMP was detected by incubation overnight with NeutrAvidin FITC (25μg.ml−1) (Invitrogen).
Microarray hybridization, data processing and analysis
Supplementary Material
Acknowledgements
We would like to thank J. Reek for help with bioinformatic analysis, F. Rustenburg, D. Israeli for microarray hybridizations, B. Ramsahoye, J. Allan, W. Bickmore, J. Caceres and N. Hastie for helpful discussions. This work was funded by the Wellcome Trust 078219/Z/05/Z (N.G.) and Breakthrough Breast Cancer (N.G.). N.G. is a recipient of a MRC Senior Fellowship (MR/J00913X/1).
Footnotes
Accession codes. Gene Expression Omnibus database: GSE*** (bTMP binding, RNA polymerase ChIP, Topoisomerase ChIP and RNA expression)
Author contributions C.N., S.C., M.B., B.Y. and N.G. conceived, designed and interpreted experiments. C.N., S.C. and N.G. performed biological experiments. N.A., L.M., N.G. carried out chemical synthesis and P.E. performed microarray experiments. J.P., S.C., and N.G. performed all bioinformatic analysis. N.G. supervised the project and all authors wrote the manuscript.
The authors declare no competing financial interests.
References
- 1.Sinden RR, Carlson JO, Pettijohn DE. Torsional tension in the DNA double helix measured with trimethylpsoralen in living E. coli cells: analogous measurements in insect and human cells. Cell. 1980;21:773–783. doi: 10.1016/0092-8674(80)90440-7. [DOI] [PubMed] [Google Scholar]
- 2.Ljungman M, Hanawalt PC. Localized torsional tension in the DNA of human cells. Proc. Natl. Acad. Sci. U. S. A. 1992;89:6055–6059. doi: 10.1073/pnas.89.13.6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ljungman M, Hanawalt PC. Presence of negative torsional tension in the promoter region of the transcriptionally poised dihydrofolate reductase gene in vivo. Nucleic Acids Res. 1995;23:1782–1789. doi: 10.1093/nar/23.10.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Villeponteau B, Lundell M, Martinson H. Torsional stress promotes the DNAase I sensitivity of active genes. Cell. 1984;39:469–478. doi: 10.1016/0092-8674(84)90454-9. [DOI] [PubMed] [Google Scholar]
- 5.Liu LF, Wang JC. Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. U. S. A. 1987;84:7024–7027. doi: 10.1073/pnas.84.20.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giaever GN, Wang JC. Supercoiling of intracellular DNA can occur in eukaryotic cells. Cell. 1988;55:849–856. doi: 10.1016/0092-8674(88)90140-7. [DOI] [PubMed] [Google Scholar]
- 7.Villeponteau B, Martinson HG. Gamma rays and bleomycin nick DNA and reverse the DNase I sensitivity of beta-globin gene chromatin in vivo. Mol. Cell Biol. 1987;7:1917–1924. doi: 10.1128/mcb.7.5.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weintraub H, Cheng PF, Conrad K. Expression of transfected DNA depends on DNA topology. Cell. 1986;46:115–122. doi: 10.1016/0092-8674(86)90865-2. [DOI] [PubMed] [Google Scholar]
- 9.Dunaway M, Ostrander EA. Local domains of supercoiling activate a eukaryotic promoter in vivo. Nature. 1993;361:746–748. doi: 10.1038/361746a0. [DOI] [PubMed] [Google Scholar]
- 10.Mizutani M, Ohta T, Watanabe H, Handa H, Hirose S. Negative supercoiling of DNA facilitates an interaction between transcription factor IID and the fibroin gene promoter. Proc. Natl. Acad. Sci. U. S. A. 1991;88:718–722. doi: 10.1073/pnas.88.3.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schultz MC, Brill SJ, Ju Q, Sternglanz R, Reeder RH. Topoisomerases and yeast rRNA transcription: negative supercoiling stimulates initiation and topoisomerase activity is required for elongation. Genes Dev. 1992;6:1332–1341. doi: 10.1101/gad.6.7.1332. [DOI] [PubMed] [Google Scholar]
- 12.Tabuchi H, Hirose S. DNA supercoiling facilitates formation of the transcription initiation complex on the fibroin gene promoter. J. Biol. Chem. 1988;263:15282–15287. [PubMed] [Google Scholar]
- 13.Mizutani M, Ura K, Hirose S. DNA superhelicity affects the formation of transcription preinitiation complex on eukaryotic genes differently. Nucleic Acids Res. 1991;19:2907–2911. doi: 10.1093/nar/19.11.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pedone F, Filetici P, Ballario P. Yeast RNA polymerase II transcription of circular DNA at different degrees of supercoiling. Nucleic Acids Res. 1982;10:5197–5208. doi: 10.1093/nar/10.17.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joshi RS, Pina B, Roca J. Positional dependence of transcriptional inhibition by DNA torsional stress in yeast chromosomes. EMBO J. 2010 doi: 10.1038/emboj.2009.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gilmour DS, Pflugfelder G, Wang JC, Lis JT. Topoisomerase I interacts with transcribed regions in Drosophila cells. Cell. 1986;44:401–407. doi: 10.1016/0092-8674(86)90461-7. [DOI] [PubMed] [Google Scholar]
- 17.Stewart AF, Herrera RE, Nordheim A. Rapid induction of c-fos transcription reveals quantitative linkage of RNA polymerase II and DNA topoisomerase I enzyme activities. Cell. 1990;60:141–149. doi: 10.1016/0092-8674(90)90724-s. [DOI] [PubMed] [Google Scholar]
- 18.Gilmour DS, Lis JT. RNA polymerase II interacts with the promoter region of the noninduced hsp70 gene in Drosophila melanogaster cells. Mol. Cell Biol. 1986;6:3984–3989. doi: 10.1128/mcb.6.11.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seila AC, et al. Divergent transcription from active promoters. Science. 2008;322:1849–1851. doi: 10.1126/science.1162253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Preker P, et al. RNA exosome depletion reveals transcription upstream of active human promoters. Science. 2008;322:1851–1854. doi: 10.1126/science.1164096. [DOI] [PubMed] [Google Scholar]
- 21.Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bernardi G. The human genome: organization and evolutionary history. Annu. Rev. Genet. 1995;29:445–76. doi: 10.1146/annurev.ge.29.120195.002305. 445-476. [DOI] [PubMed] [Google Scholar]
- 23.Caron H, et al. The human transcriptome map: clustering of highly expressed genes in chromosomal domains. Science. 2001;291:1289–1292. doi: 10.1126/science.1056794. [DOI] [PubMed] [Google Scholar]
- 24.Lercher MJ, Urrutia AO, Hurst LD. Clustering of housekeeping genes provides a unified model of gene order in the human genome. Nat. Genet. 2002;31:180–183. doi: 10.1038/ng887. [DOI] [PubMed] [Google Scholar]
- 25.Versteeg R, et al. The human transcriptome map reveals extremes in gene density, intron length, GC content, and repeat pattern for domains of highly and weakly expressed genes. Genome Res. 2003;13:1998–2004. doi: 10.1101/gr.1649303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gilbert N, et al. Chromatin architecture of the human genome: gene-rich domains are enriched in open chromatin fibers. Cell. 2004;118:555–566. doi: 10.1016/j.cell.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 27.Naughton C, Sproul D, Hamilton C, Gilbert N. Analysis of active and inactive X chromosome architecture reveals the independent organisation of 30-nm and large scale chromatin structures. Mol. Cell. 2010;40:397–409. doi: 10.1016/j.molcel.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sproul D, Gilbert N, Bickmore WA. The role of chromatin structure in regulating the expression of clustered genes. Nat. Rev. Genet. 2005;6:775–781. doi: 10.1038/nrg1688. [DOI] [PubMed] [Google Scholar]
- 29.Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- 30.Marsden MP, Laemmli UK. Metaphase chromosome structure: evidence for a radial loop model. Cell. 1979;17:849–858. doi: 10.1016/0092-8674(79)90325-8. [DOI] [PubMed] [Google Scholar]
- 31.Dixon JR, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hou C, Li L, Qin ZS, Corces VG. Gene density, transcription, and insulators contribute to the partition of the Drosophila genome into physical domains. Mol. Cell. 2012;48:471–484. doi: 10.1016/j.molcel.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nora EP, et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Belmont AS, Bruce K. Visualization of G1 chromosomes: a folded, twisted, supercoiled chromonema model of interphase chromatid structure. J. Cell Biol. 1994;127:287–302. doi: 10.1083/jcb.127.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Filipski J, Leblanc J, Youdale T, Sikorska M, Walker PR. Periodicity of DNA folding in higher order chromatin structures. EMBO J. 1990;9:1319–1327. doi: 10.1002/j.1460-2075.1990.tb08241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sachs RK, van den EG, Trask B, Yokota H, Hearst JE. A random-walk/giant-loop model for interphase chromosomes. Proc. Natl. Acad. Sci. U. S. A. 1995;92:2710–2714. doi: 10.1073/pnas.92.7.2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cimino GD, Gamper HB, Isaacs ST, Hearst JE. Psoralens as photoactive probes of nucleic acid structure and function: organic chemistry, photochemistry, and biochemistry. Annu. Rev. Biochem. 1985;54:1151–93. doi: 10.1146/annurev.bi.54.070185.005443. 1151-1193. [DOI] [PubMed] [Google Scholar]
- 38.Tomic MT, Wemmer DE, Kim SH. Structure of a psoralen cross-linked DNA in solution by nuclear magnetic resonance. Science. 1987;238:1722–1725. doi: 10.1126/science.3686011. [DOI] [PubMed] [Google Scholar]
- 39.Bermudez I, Garcia-Martinez J, Perez-Ortin JE, Roca J. A method for genome-wide analysis of DNA helical tension by means of psoralen-DNA photobinding. Nucleic Acids Res. 2010;38:e182. doi: 10.1093/nar/gkq687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Merkenschlager M. Cohesin: a global player in chromosome biology with local ties to gene regulation. Curr. Opin. Genet. Dev. 2010;20:555–561. doi: 10.1016/j.gde.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 41.Chambeyron S, Bickmore WA. Chromatin decondensation and nuclear reorganization of the HoxB locus upon induction of transcription. Genes Dev. 2004;18:1119–1130. doi: 10.1101/gad.292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu Y, Kireev I, Plutz M, Ashourian N, Belmont AS. Large-scale chromatin structure of inducible genes: transcription on a condensed, linear template. J. Cell Biol. 2009;185:87–100. doi: 10.1083/jcb.200809196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hay N, Skolnik-David H, Aloni Y. Attenuation in the control of SV40 gene expression. Cell. 1982;29:183–193. doi: 10.1016/0092-8674(82)90102-7. [DOI] [PubMed] [Google Scholar]
- 44.Saunders A, Core LJ, Lis JT. Breaking barriers to transcription elongation. Nat. Rev. Mol. Cell Biol. 2006;7:557–567. doi: 10.1038/nrm1981. [DOI] [PubMed] [Google Scholar]
- 45.Blagosklonny MV. Flavopiridol, an inhibitor of transcription: implications, problems and solutions. Cell Cycle. 2004;3:1537–1542. doi: 10.4161/cc.3.12.1278. [DOI] [PubMed] [Google Scholar]
- 46.Metivier R, et al. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- 47.Costantini M, Clay O, Auletta F, Bernardi G. An isochore map of human chromosomes. Genome Res. 2006;16:536–541. doi: 10.1101/gr.4910606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Locke G, Tolkunov D, Moqtaderi Z, Struhl K, Morozov AV. High-throughput sequencing reveals a simple model of nucleosome energetics. Proc. Natl. Acad. Sci. U. S. A. 2010 doi: 10.1073/pnas.1003838107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou Y, et al. Genome-wide identification of chromosomal regions of increased tumor expression by transcriptome analysis. Cancer Res. 2003;63:5781–5784. [PubMed] [Google Scholar]
- 50.Schuster-Bockler B, Lehner B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature. 2012;488:504–507. doi: 10.1038/nature11273. [DOI] [PubMed] [Google Scholar]
- 51.Saffran WA, et al. Preparation and characterization of biotinylated psoralen. Nucleic Acids Res. 1988;16:7221–7231. doi: 10.1093/nar/16.15.7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Reference for online methods
- 52.Naughton C, et al. Progressive loss of estrogen receptor alpha cofactor recruitment in endocrine resistance. Mol. Endocrinol. 2007;21:2615–2626. doi: 10.1210/me.2007-0110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.