

Abstract
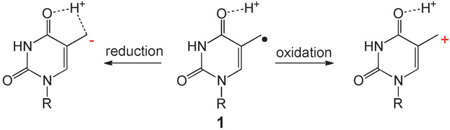
Sleeping beauty: The 5-(2’-Deoxyuridinyl) methyl radical 1 is a key intermediate in the thymine oxidative reaction mediated by reactive oxygen species. Evidence is presented that 1 is prone to both oxidation and reduction reactions at the absence of O2. These results question the current paradigm and suggest that the redox chemistry of 1, which has been largely overlooked in the past, may play a major role in determining the fate of 1.
Keywords: DNA, photochemistry, radicals, reaction mechanisms, thymine
Thymine is susceptible toward ionizing radiation, a typical source of reactive oxygen species (ROS). The 5-(2’-deoxyuridinyl) methyl radical (1) is a key intermediate of thymine radical reactions involving either a hydroxyl radical-mediated H atom abstraction at the methyl moiety[1] or a one-electron oxidation followed by deprotonation process.[2] Species 1 was implied to possess neither reducing nor oxidizing properties.[3] It can add to atom C8 of a vicinal guanine or adenine under anaerobic conditions.[4] A similar 5-(2’-deoxycytidinyl)methyl radical gave rise to an intrastrand cross-link lesion in dinucleotides d(mCpG) and d(GpmC).[5] Further studies of 1 by Greenberg et al. found that it also reacts with the opposing 2’-deoxyadenosine in duplex DNA to yield a crosslinked TpA dimer with the thymine methyl group added to the adenine N6.[6] Such a lesion is readily detected in γ-irradiated DNA and accounts for at least 25% of the DNA interstrand cross-links.[6b]
These cross-linked base dimers are suggested to block replication and transcription and are quite toxic to cells. It is thus of great significance to understand the reactivity of 1. O2 reacts with 1, forming a peroxyl radical intermediate, which then decomposes to yield 5-(hydroxymethyl)-2’-deoxyuridine (HmdU), 5-(hydroperoxy methyl)-2’-deoxyuridine (HpmdU), and 5-formyl-2’-deoxyuridine (fdU).[1e, 7] Specifically, the formation of HpmdU requires one electron to reduce the peroxyl radical precursor, which may be provided by a superoxide radical (O2•−)[8] or a thiol compound.[7a] The reaction between 1 and O2 is reversible;[9] however, it is still two orders of magnitude faster than its reaction with thiol (H abstraction),[7a] suggesting that O2 may prevent the nucleobase cross-linking reaction. However, a recent study revealed that after complete consumption of the radical precursor, only 25% of the generated 1 were involved in the crosslinking reaction with adjacent nucleobases. The yield of the crosslinking product remains the same under degassed or aerobic conditions.[6a] The fact that the presence of O2 did not further lower the reaction yield implies that other routes may also be involved in the quenching process of 1.
We thus re-examined the reactivity of 1 generated by photolytic cleavage of the C–S bond in 5-(phenylthiomethyl)-2’-deoxyuridine (PhSmdU, Scheme 1). The PhSmdU was linked to another thymidine by phosphoramidite chemistry to yield the dinucleotide TpPhSmdU (2).[10] The photoreaction of 2 was allowed to proceed for 5 min under 254 nm UV light in the presence or absence of O2 (Figure 1), which usually consumed about 15% of 2. The reaction was then analyzed by HPLC; the yields for the major products in the anaerobic reactions are shown in Table 1. Our results suggest that 1 can undergo both oxidative and reductive reactions in the absence of O2 ; the resulting anion and cation are quenched by the general acid and base in solution respectively. Under an oxidizing environment, the anion formation is inhibited. Under a reducing environment, the cation formation is suppressed.
Scheme 1.

Generation of 1 by photolytic cleavage of the C–S bond in 5-(phenylthiomethyl)-2’-deoxyuridine (PhSmdU) as well as thiol addition products in the dinucleotide context.
Figure 1.

HPLC chromatograph of TpPhSmdU (2, 200 µm) photoreaction under 254 nm UV light. A) In H2O in air; B) in degassed H2O in a Coy anaerobic chamber; C) in degassed H2O in a Coy anaerobic chamber and supplemented by 1 mm sodium dithionite.
Table 1.
Product yields in photoreactions of 2 at the absence of O2.
| Reactions | Yield [%][a] | Mass balance |
||
|---|---|---|---|---|
| Anion TpT |
Adduct X | Cation Other thiol adduct |
||
| none | 44.5 ± 2.3 | 43.2 ± 4.1 | – | 89.2 ± 5.9[b] |
| Na2S2O4 | 77.5 ± 3.4 | – | – | 91.3 ± 4.3[c] |
| PhSH | 85.4± 5.7 | – | – | 87.3 ± 7.0 |
| 4-OHC6H4SH | 39.0 ± 2.5 | – | 46.6 ± 1.9 | 86.2 ± 3.0 |
| DTT(1 mm) | 48.5 ± 2.1 | 37.4 ± 3.9 | 5.1 ± 0.7 | 88.7 ± 5.4 |
| DTT (10 mm) | 44.0 ± 1.9 | 26.1 ± 1.5 | 20.7 ± 1.4 | 89.1 ± 4.2 |
| DTT (40 mm) | 36.3 ± 4.0 | 13.8 ±1.9 | 33.8 ± 3.3 | 86.5 ± 7.5 |
The yields were based upon the amount of 2 reacted and were calculated by HPLC peak integrations (See the Supporting Information).
The yield of TpHmdU was about 2.5%.
The yield of TpmdUSO2H was 12.1 ± 2.8%.
The aerobic reaction generated TpHmdU, TpHpmdU, and TpfdU (Figure 1 A), in agreement with the previous reports that 1 is prone to O2 oxidation.[1e, 7a] Surprisingly, an unknown product X was also generated. In contrast, the anaerobic reaction yields TpT and X as the major products and TpHmdU as the minor product. The TpT formation is puzzling as it was suggested to be formed by an H-abstraction mechanism, with thiols such as glutathione or thiophenol serving as the H-donor.[1e, 7a] However, no thiol was added to our reaction. One possibility is that the PhS· generated may be reduced to PhSH and subsequently donates an H atom to 1 to yield TpT. To exclude this possibility, 1 mm PhSH was added to the solution before 2 was irradiated. Such a large excess of PhSH only improved the TpT yield by about 20% (Figure 3B), proving that PhSH can serve as the H donor; however, such an H donation process is not the major reaction pathway to quench 1.
Figure 3.

HPLC chromatograph of anaerobic TpPhSmdU (2, 200 µm) photoreaction under 254 nm UV light. A) In H2O; B) with 1 mm PhSH; C) with 1 mm 4-OH-C6H4SH; D) with 1 mm DTT; E) with 10 mm DTT; and F) with 40 mm DTT. As the DTT utilized was a DL mixture, the resulting adducts 6 were a mixture as well, as indicated by the doublet peak in the HPLC chromatograph. The two peaks marked by * correspond to DTT and DTT disulfide, respectively, which already exist in the DTT used.[13]
Besides TpT, species X is another major product, whose nature needs to be revealed before the reactivity of 1 can be understood. X exhibits a mass of 1199.26 in the positive-ion mode, corresponding to a formula of [TpT+2-2H]. Such a compound can be obtained either by TpTradical addition to 2 followed by elimination of an H atom or by TpT cation addition followed by loss of a proton. 1H NMR analysis suggests X to be a mixture of products with the thymine methyl group added to the phenyl ring of 2 (Figure 2A); however structural details cannot be obtained for the compounds that comprise the spectrum owing to signal overlap. This addition pattern was further supported by the MS/MS spectrum of X, in which three major fragments were found (Figure 2B), with the corresponding chemical structures shown in Figure 2A. To reveal the structure of X, we examined the PhSmdU photoreaction as a model system.[10] As expected, the reaction produced thymidine and HmdU. More importantly, two species X’1 and X’2 were formed and separated by HPLC, both of which possess a mass of [T+PhSmdU-2H]. 1H NMR analyses confirm that the thymine methyl moiety is added to the para position of the phenyl ring in X’1 and to the ortho position in X’2 (Scheme 2). The yield of isolated X’2 is about 50% higher than that of X’1. As the ring possesses two positions ortho and one position para to the SR substituent, our data suggest that the ortho positions are slightly less active toward the addition reaction probably due to the steric hindrance of the SR moiety.
Figure 2.

A) Proposed structure of product X, suggesting that a TpT moiety is added to the phenyl ring of -SPh. B) LC-MS/MS analysis of ion [X+H]+ at 1199.26. The structures of the three fragmentation ions (f1, f2, and f3) are indicated in (a).
Scheme 2.

Addition of the thymine methyl cation to the para position of the phenyl ring in X’1 and to the ortho position in X’2.
Such a reaction pattern suggests that X is formed by a cation addition mechanism. If a radical addition is involved, three products, with the thymine methyl group added randomly to any of the five positions at the phenyl ring, are expected. The thymine cation is still formed in the presence of O2, as indicated by the generation of X in Figure 1A. Its formation may be explained by competitive elimination of the superoxide radical (O2−) from the peroxyl radical precursor, similar to what was observed in the addition of O2 to 2’- deoxyuridin-1’-yl radical.[11] Alternatively, the peroxyl radical may undergo a reverse process,[9] resulting in 1 and O2 with 1 being subsequently oxidized to the cation. Although HmdU is typically formed by O2 oxidation to 1,[1e, 7a, 12] the TpHmdU formed in the absence of O2 likely results from the H2O addition to the cation followed by loss of a proton. After diluting 2 by tenfold, the TpHmdU yield is improved at the expense of X,[10] further supporting this mechanism. Moreover, if the reaction is conducted in methanol, the corresponding methoxy adduct TpMeOmdU is produced.[10
The presence of thiol compounds barely enhances the TpT formation, contrasting with the fourfold enhancement induced by 1 mm sodium dithionite in Figure 1C. Photoreaction of 2 in the presence of 1 mm PhSH results in the disappearance of X (Figure 3 B). As PhSH is an excellent nucleophile, it likely quenches the thymine cation, reproducing 2, which escapes from the HPLC detection. To test this hypothesis, we repeated the photoreaction in the presence of 1 mm 4-hydroxythiophenol (4-OH-C6H4-SH). As expected, formation of X was suppressed and a new species 5 was generated (Figure 3C). NMR and MS analyses confirm that 5 is the addition product Tp(4-OH)C6H4SmdU formed between the TpT cation and 4-OH-C6H4-S−(Scheme 1).[10] Equal amounts of 5 and X were produced as shown in Figure 3A and C, indicating that 5 is formed at the expense of X (Table 1). The cation can also be trapped by other thiols, such as DTT. Addition of 1–40 mm DTT did not markedly increase the yield of TpT (Figure 3 D–F), again suggesting that H-abstraction is unlikely to play a major role. Under 1 mm DTT, formation of X decreased by only about 10%, contrasting to the nearly complete quenching by 1 mm PhSH. With 40 mm DTT, formation of X decreased by 50%. These results are in line with the rationale that DTT is a better reductant, but a much worse nucleophile than PhSH. The reactions also yield the expected TpmdU-DTT adduct 6 (Scheme 1).[10] 6 exist as a pair of DL isomers, as indicated by the doublet peak in HPLC chromatograph, which result from the DL mixture of DTT used.
Formation of thymine cation from 1 is a one-electron oxidation process. As no obvious electron acceptor can be identified in the anaerobic reaction, it is intriguing to suggest that another molecule of 1 accepts the electron to yield a methyl anion, which then obtains a proton from water, yielding TpT. This hypothesis is supported by the reaction in D2O. The bond dissociation energy (BDE) for the O–H bond in water is 119 kcalmol−1,[14] which is about 30 kcalmol−1 higher than the C–H bond in the thymine CH3 group.[15] It is highly unlikely that 1 would abstract a deuterium atom from D2O. However, more than 95% of the TpTs formed contain one deuterium, which is located on the methyl group, as shown by 1H NMR spectroscopy.[10] When 2 was irradiated in [D1]methanol (CH3OD), more than 95% of the TpTs are [D1]TpTs as well,[10] showing that the stronger O–D bond in CH3OD is involved in TpT formation. As 2-deoxyribose is prone for H-abstraction reactions,[16] these labeling studies also allow us to exclude the possible involvement of the 2-deoxyribose. Collectively, our data discriminate against the H-abstraction mechanism, but support the anion reaction. The formed anion takes a deuteron from solvent to yield [D1]TpT.
Should 1 be reduced by another thymine radical, it would be reduced by a stronger reductant. We thus examined the reactivity of 1 in the presence of 1 mm sodium dithionite. As expected, the reducing environment totally abolished the cation-related products; the radical reduction product TpT was increased about fourfold (Figure 1C), confirming that the presence of a strong reductant facilitates the thymine anion formation. The reaction also produced TpmdUSO2− as a minor product (Figure 1C),[17] with its yield roughly 1/6 of that for TpT. TpmdUSO2− is likely formed by a radical recombination mechanism. The dithionite dianion has a dissociation constant Kd≈10−6 mm in water,[18] which results in the formation of •SO2−. UV irradiation may promote the S–S bond cleavage in dithionite dianion, making the concentration of •SO2− even higher. It should be fairly favorable for 1 to recombine with •SO2−, yielding TpmdUSO2− Although the TpmdUSO2− formation rate cannot be obtained owing to the difficulty to determine the exact concentrations of 1 and •SO2−, the lower yield of TpmdUSO2− indicates that the radical reduction reaction to produce TpT likely occurs with a rate that is at least comparable to that of the radical recombination reaction.
These experiments clearly indicate that 1 can undergo both oxidation and reduction reactions (Scheme 3A); it disproportionates in the absence of stronger redox reagents. Pyrimidine radicals were known to disproportionate under anaerobic conditions.[3, 19] However, the reaction of 1 is different as the disproportionations of previous pyrimidine radicals and traditional alkyl radicals require an H atom β to the radical center and the reactions proceed by a radical mediated H-abstraction mechanism.[20] We tentatively ascribe the difference to the polar solvents used here, which stabilize the charged reaction intermediates by electrostatic interactions. Moreover, although 1 can be reduced, a direct oneelectron reduction followed by protonation to yield thymine is unfavorable owing to the poor stability of the thymine methyl anion intermediate. Compound 1 is likely to be protonated at the C4=O bond to a radical cation before the reduction occurs (Scheme 3A).
Scheme 3.

Oxidation and reduction reactions of 1 and PhS•.
The mechanistic discussions above only involve the carbon-based radical 1. Considering the similar electronegativity between C (2.544) and S (2.589),[21] the PhSC generated after the C–S bond cleavage in 2 is likely involved in the redox reactions as well. We thus carefully quantified all of the PhSC related redox products under varying pH. The photoreaction produces two minor products 3 and 4 (Figure 1A,B and Supporting Information), both of which exhibit a mass of 761.14 at the negative-ion mode,[10] corresponding to a formula of [PhS•+2-H]. Limited by the low yields, we were unable to isolate enough compound for NMR spectroscopy characterization. However, 3 and 4 are likely to be formed either by PhS+ addition to a double bond in 2 followed by loss of a H+, similar to the generation of X, or by PhS• addition to 2 followed by loss of a H• (e+H+). The released H• can combine with PhS• or 1, resulting in the reducing products PhSH and TpT, respectively. Both routes indicate that 3 or 4 can be treated as a cation product of PhSC (Scheme 3B). Furthermore, the formed PhSH, the anion (reducing) product of PhS•, was extracted with hexane and quantified by GC-MS spectroscopy (Table 2).
Table 2.
Products isolated [nmol] in photoreactions of 2 [40 nmol].
| Anion products | Cation products | Dimer of 1 | ||||
|---|---|---|---|---|---|---|
| pH | TpT | PhSH | TpTOH | X | 3 +4 | |
| 3–7 | 2.73 ± 0.22 | 0.78 ± 0.05 | 0.25 ± 0.03 | 2.50 ± 0.20 | 0.55 ± 0.03 | <0.02 |
| 9 | 2.88 ± 0.10 | 0.94 ± 0.06 | 0.29 ± 0.02 | 2.83 ± 0.17 | 0.66 ± 0.07 | 0.05 ± 0.01 |
| 11 | 0.38 ± 0.05 | 2.59 ± 0.12 | 0.85 ± 0.07 | 2.18 ± 0.14 | 0.20 ± 0.05 | 0.39 ± 0.04 |
Excellent mass balance was obtained for the redox process in all of the reactions in Table 2. When pH≤7, the amounts of cationic and anionic products of 1 are roughly equal. Assuming PhS• and 1 do not interact with each other, disproportionation of 1 becomes a simple interpretation for our data. In contrast, under basic pH where [H+] is low, the thymine anion formation is unfavored. Species 1 is still oxidized to a cation; however, the preferred electron acceptor becomes PhS•, yielding PhS−. Formation of PhS− was implied by a 5,6-dihydro-5-hydroxythymidin-6-yl radical study.[22] Furthermore, we observed the dimer of 1 in our reaction, the formation of which was suggested in previous studies.[3,16b,19b,c] The dimer yield is very low under neutral or acidic pH, which is consistent with the low yield obtained in TpmdUSO2− formation. These results re-confirm that the radical recombination pathway is unfavorable; dimer formation increases only when the disproportionation reaction slows down at pH 11. As PhS• is the electron acceptor in this case, this result also indicates the electron transfer between 1 and PhSC to be slightly slower than that between two molecules of 1, which likely rationalizes why disproportionation of 1 is favored in most of our reactions.
It is worth pointing out that the electron transfer process between two 1 molecules (radical disproportionation reaction) is unlikely to happen in vivo as it is implausible to have two thymine radicals close to each other in duplex DNA. Our data should be interpreted as the indication that 1 possesses both oxidizing and reducing properties; thus the current paradigm in DNA biochemistry may not be correct.[3] Formation of cation or anion from 1 may dominate in vivo given a different local redox environment. Also, without O2, 1 was proposed to be quenched by an H-abstraction reaction with the H atom provided by thiol compounds.[1e, 7a] Our report suggests that this conclusion may need to be reexamined.
Supplementary Material
Footnotes
We thank all of the referees for their insightful suggestions and comments. We thank Professor Steven Rokita at the Johns Hopkins University for helpful discussions on the TpT radical reduction. We thank the National Institutes of Health (R00ES017177) as well as the IUPUI startup fund for financial support. The NMR and MS facilities are supported by the National Science Foundation MRI grants CHE-0619254 and DBI-0821661.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201209454.
References
- 1.a) Box HC, Dawidzik JB, Budzinski EE. Free Radical Biol. Med. 2001;31:856. doi: 10.1016/s0891-5849(01)00653-0. [DOI] [PubMed] [Google Scholar]; b) Box HC, Patrzyc HB, Dawidzik JB, Wallace JC, Freund HG, Iijima H, Budzinski EE. Radiat. Res. 2000;153:442. doi: 10.1667/0033-7587(2000)153[0442:dblidx]2.0.co;2. [DOI] [PubMed] [Google Scholar]; c) Box HC, Budzinski EE, Dawidzik JB, Gobey JS, Freund HG. Free Radical Biol. Med. 1997;23:1021. doi: 10.1016/s0891-5849(97)00166-4. [DOI] [PubMed] [Google Scholar]; d) Box HC, Budzinski EE, Dawidzik JD, Wallace JC, Evans MS, Gobey JS. Radiat. Res. 1996;145:641. [PubMed] [Google Scholar]; e) Romieu A, Bellon S, Gasparutto D, Cadet J. Org. Lett. 2000;2:1085. doi: 10.1021/ol005643y. [DOI] [PubMed] [Google Scholar]
- 2.Cadet J, Douki T, Ravanat J-L. Free Radical Biol. Med. 2010;49:9. doi: 10.1016/j.freeradbiomed.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 3.von Sonntag C. Free-radical-induced DNA damage and its repair: A chemical perspective. Berlin: Springer; 2006. [Google Scholar]
- 4.a) Bellon S, Ravanat JL, Gasparutto D, Cadet J. Chem. Res. Toxicol. 2002;15:598. doi: 10.1021/tx015594d. [DOI] [PubMed] [Google Scholar]; b) Budzinski EE, Dawidzik JB, Rajecki MJ, Wallace JC, Schroder EA, Box HC. Int. J. Radiat. Biol. 1997;71:327. doi: 10.1080/095530097144210. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Q, Wang Y. J. Am. Chem. Soc. 2003;125:12795. doi: 10.1021/ja034866r. [DOI] [PubMed] [Google Scholar]
- 6.a) Hong IS, Greenberg MM. J. Am. Chem. Soc. 2005;127:3692. doi: 10.1021/ja042434q. [DOI] [PubMed] [Google Scholar]; b) Ding H, Greenberg MM. Chem. Res. Toxicol. 2007;20:1623. doi: 10.1021/tx7002307. [DOI] [PubMed] [Google Scholar]; c) Ding H, Majumdar A, Tolman JR, Greenberg MM. J. Am. Chem. Soc. 2008;130:17981. doi: 10.1021/ja807845n. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Peng XH, Pigli YZ, Rice PA, Greenberg MM. J. Am. Chem. Soc. 2008;130:12890. doi: 10.1021/ja805440v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Hong IS, Greenberg MM. Org. Lett. 2004;6:5011. doi: 10.1021/ol047754t. [DOI] [PubMed] [Google Scholar]; b) Decarroz C, Wagner JR, van Lier JE, Krishna CM, Riesz P, Cadet J. Int. J. Radiat. Biol. 1986;50:491. doi: 10.1080/09553008614550901. [DOI] [PubMed] [Google Scholar]; c) Wagner JR, van Lier JE, Berger M, Cadet J. Am. Chem. Soc. 1994;116:2235. [Google Scholar]
- 8.Wagner JR, van Lier JE, Johnston LJ. Photochem. Photobiol. 1990;52:333. doi: 10.1111/j.1751-1097.1990.tb04189.x. [DOI] [PubMed] [Google Scholar]
- 9.Hong IS, Ding H, Greenberg MM. J. Am. Chem. Soc. 2006;128:485. doi: 10.1021/ja0563657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. See the Supporting Information.
- 11.Emanuel CJ, Newcomb M, Ferreri C, Chatgilialoglu C. J. Am. Chem. Soc. 1999;121:2927. [Google Scholar]
- 12.Steenken S. Free Radical Res. 1992;16:349. doi: 10.3109/10715769209049187. [DOI] [PubMed] [Google Scholar]
- 13.Cindrić M, Čepo T, Marinc S, Paškvan I, Mijić I, Bindila L, Peter-Katalinić J. J. Sep. Sci. 2008;31:3489. doi: 10.1002/jssc.200800207. [DOI] [PubMed] [Google Scholar]
- 14.Stubbe J, van der Donk WA. Chem. Rev. 1998;98:705. doi: 10.1021/cr980059c. [DOI] [PubMed] [Google Scholar]
- 15.a) Hioe J, Zipse H. Faraday Discuss. 2010;145:301. [Google Scholar]; b) Hioe J, Zipse H. Org. Biomol. Chem. 2010;8:3609. doi: 10.1039/c004166a. [DOI] [PubMed] [Google Scholar]; c) Hioe J, Zipse H. Chem. Eur. J. 2012;18:16463. doi: 10.1002/chem.201202869. [DOI] [PubMed] [Google Scholar]
- 16.a) Pogozelski WK, Tullius TD. Chem. Rev. 1998;98:1089. doi: 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]; b) Anderson AS, Hwang JT, Greenberg MM. J. Org. Chem. 2000;65:4648. doi: 10.1021/jo000271s. [DOI] [PubMed] [Google Scholar]
- 17.Chandor-Proust A, Berteau O, Douki T, Gasparutto D, Ollagnier-de-Choudens S, Fontecave M, Atta M. J. Biol. Chem. 2008;283:36361. doi: 10.1074/jbc.M806503200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Lynn S, Rinker RG, Corcoran WH. J. Phys. Chem. 1964;68:2363. [Google Scholar]; b) Rinker RG, Gordon TP, Mason DM, Corcoran WH. J. Phys. Chem. 1959;63:302. [Google Scholar]; c) Rinker RG, Lynn S, Mason DM, Corcoran WH. Ind. Eng. Chem. Fund. 1965;4:282. [Google Scholar]; d) Lambeth DO, Palmer G. J. Biol. Chem. 1973;248:6095. [PubMed] [Google Scholar]
- 19.a) Haysom HR, Phillips JM, Scholes G. J. Chem. Soc. Chem. Commun. 1972:1082. [Google Scholar]; b) Shragge PC, Varghese AJ, Hunt JW, Greenstock CL. Radiat. Res. 1974;60:250. [Google Scholar]; c) Cadet J, Balland A, Berger M. Int. J. Radiat. Biol. 1981;39:119. doi: 10.1080/09553008114550141. [DOI] [PubMed] [Google Scholar]
- 20.Gibian MJ, Corley RC. Chem. Rev. 1973;73:441. [Google Scholar]
- 21.Miessler GL, Fischer PJ, Tarr DA. Inorganic chemistry. 5th ed. Upper Saddle River: Pearson Education; 2013. [Google Scholar]
- 22.San Pedro JMN, Greenberg MM. Org. Lett. 2012;14:2866. doi: 10.1021/ol301109z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.