

Abstract
PEO-PPO-PEO triblock copolymers have opposing effects on lipid membrane integrity- they can behave either as membrane sealants or as membrane permeabilizers. To gain insights into their biomembrane activities, the fundamental interactions between a series of PEO-based polymers and phospholipid vesicles were investigated. Specifically, the effect of copolymer hydrophobicity on its ability to prevent liposomes from peroxidation was evaluated, and partitioning free energy and coefficient involved in the interactions were derived. Our results show that the high degree of hydrophilicity is a key feature of the copolymers that can effectively protect liposomes from peroxidation and the protective effect of the copolymers stems from their adsorption at the membrane surface without penetrating into the bilayer core. The origin of this protective effect induced by polymer absorption is attributed to the retardation of membrane hydration dynamics, which is further illustrated in the accompany study on dynamic nuclear polarization (DNP)-derived hydration dynamics1.
Keywords: PEO-PPO-PEO copolymer, hydrophobicity, lipid peroxidation, lipid membrane, hydration dynamics
1. Introduction
The triblock copolymers composed of hydrophilic poly(ethylene oxide) (PEO; A) and hydrophobic poly(propylene oxide) (PPO; B) in an A-B-A architecture are a class of nonionic, surface-active amphiphilic macromolecules. They are known under the generic name poloxamer and the trademarks Pluronic® or Synperonic®. Owing to their biocompatible and amphiphilic nature, these copolymers have, over the past several decades, found widespread applications in medicine as detergents or emulsifiers in pharmaceutical formulations2, carriers of poor water-soluble drugs3, sealants for injured cell membranes4 and permeation enhancers in cancer chemotherapies5, etc. In particular, studies have found that P188, a poloxamer in the form of PEO80-PPO27-PEO80 with fairly large hydrophilic PEO blocks compared to its hydrophobic PPO counterpart, is capable of inhibiting thrombosis6 and protecting cell membranes from various physicochemical insults4, 7-9. Conversely, P181, another poloxamer in the form of PEO2-PPO32-PEO2 with a similar length for the PPO block as P188 but much smaller PEO blocks, is incapable of inhibiting thrombosis and sealing wounded cell membranes. It, however, exhibits ionophoric activities10, can facilitate the permeation of relatively large molecules across lipid bilayers11, and can further act as chemosensitizing agents in cancer treatments12. It is intriguing that these seemingly opposite biological effects can come from the interactions of the same family of triblock copolymers with biological cell membranes. However, no clear relationship between the architecture and biomedical functionality of these copolymers has been established thus far, given the lack of a thorough understanding of intermolecular interactions of cell membranes with these copolymers.
Recently, using giant unilamellar vesicles (GUVs) under osmotic stress as model systems, we have proposed a two-state transition mechanism which involves an adsorption (I) state and an insertion (II) state to elucidate the fundamental interactions between poloxamers and phospholipid membranes13. We find that while poloxamers in state I act to protect lipid vesicles from leaking due to the loss of membrane integrity, their insertion into the lipid bilayer (state II) disturbs lipid packing, thus enhancing membrane permeability. Moreover, our results suggest that the transition from surface adsorption to membrane insertion is driven by hydrophobicity of the copolymer, which provides seemingly opposite biomedical applications of PEO-PPO-PEO triblock copolymers, either as cell membrane preservers or as accelerators for drug intake by drug resistance tumor cells.
To further verify this model of interactions between poloxamers and lipid membranes, we investigated the fundamental interactions of lipid membranes with a series of the triblock copolymers having different hydrophobicity by dynamic light scattering (DLS) and isothermal titration calorimetry (ITC). Specifically, we employed DLS to evaluate the effect of poloxamer hydrophobicity on its ability to protect liposomes from peroxidation and ITC to distinct the two models of polymer-membrane interactions—membrane insertion vs. membrane adsorption. It is well-known that lipid peroxidation, in vivo, plays a key role in many pathological processes, including both inflammatory and degenerative diseases, ionizing radiation injury, chemical and drug toxicity, and some metabolic defects.14 It can induce severe biomembrane dysfunctions at the cellular level15 and alter the chemical structures of polyunsaturated lipids at the molecular level, which subsequently leads to changes in lipid bilayer physical properties, such as permeability, integrity, curvature and fluidity16-18. Here using lipid peroxidation—a biological process closely associated with cell membrane disruption19,20—as a stress source to disrupt unilamellar lipid vesicles, we explored the effect of hydrophobicity of a series of poloxamers on their ability to preserve membrane integrity by protecting liposomes from peroxidation. We find that polymers that are effective protectors of liposomes from peroxidation do not share any common structural features other than the fact that they are all highly hydrophilic; highly hydrophobic copolymers, on the other hand, do not show active protection for the membrane. In addition, the results from the ITC measurement reveal that none of the examined hydrophilic polymers exhibits appreciable heat exchange during the titration as the hydrophobic polymers do, indicating that there is no apparent partitioning of hydrophilic polymers to lipid membranes within our experimental timescale. Thus, the combination of DLS and ITC results confirms our hypothesis that the protective effect of poloxamers on the structural integrity of lipid membranes stems from polymer adsorption at the membrane surface.
2. Experimental Details
Materials
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-(phosphor-rac-(1-glycerol)) (sodium salt) (POPG) and 1-palmitoyl-2-linoleoyl-sn-glycero-3-phosphocholine (PLPC) were purchased from Avanti Polar Lipids (Alabaster, AL). The radical generator, 2, 2′-azobis(2-amidinopropane) hydrochloride (AAPH), polyethylene glycol with a molecular weight of 8000 g/mol (PEG-8K) as well as ethylenediamine tetrakis(propoxylate-block-ethoxylate) tetrol (poloxamine 1107) were purchased from Sigma-Aldrich Chemical Co. Poloxamer 188, 181, 333 and 335 were generous gifts of the BASF Corporation. Detailed information of the polymers is listed in Table 1 and Scheme 1. All chemicals were used without further purification. Ultra-pure water (resistivity ≥ 18 MΩ cm) was obtained from a Milli-Q UV Plus system. (Millipore Inc., Bedford, MA).
Table 1. Summary of the examined polymers.
| Polymer | Mw (g/mol) | m | n | EO/PO | HLB* |
|---|---|---|---|---|---|
| P188 | 8400 | 27 | 80 | 8/2 | 29 |
| P1107 | 15000 | 20 | 58 | 7/3 | 24 |
| PEG-8K | 8000 | 0 | 182 | ∞ | Hydrophilic |
| P181 | 1750 | 32 | 2 | 1/9 | 3 |
| P333 | 4950 | 60 | 17 | 3/7 | 9 |
| P335 | 6500 | 56 | 37 | 5/5 | 15 |
HLB: hydrophile-lipophile balance. For PEO-PPO-PEO copolymers, HLB = -36m/(2n+m)+33, where m represents the number of repeat units in the PPO segment and n represents the number of repeat units in the PEO segment.21 In general, the higher the HLB, the higher the hydrophobicity of the copolymer is.
Scheme 1.

Chemical structures of polyethylene glycol, poloxamer and poloxamine.
The copolymer stock solution of 500 μM and PEG-8K stock solution of 1 wt% (∼1250 μM) were prepared by adding polymers and ultra-pure water to plastic tubes containing magnetic stir bars and then leaving them to mix on a stir plate overnight to ensure complete dissolution. Following degassing under vacuum, polymer stock solutions were diluted to desired concentrations. As the rate of AAPH-induced radical formation is constant only for the first few hours, AAPH solutions were freshly prepared before each experiment.
Liposome Preparation
Large unilamellar vesicles (LUVs) made from a 6:3:1 mixture of POPC-PLPC-POPG for studies of lipid peroxidation and LUVs of pure POPC for ITC measurements were prepared via the freeze-thaw extrusion method.22, 23 Initially, phospholipids in chloroform were dried under ultra-pure argon to re-disperse lipids over the wall of the glass vial. After being kept under vacuum for at least 1 h, the dry lipids were rehydrated in ultra-pure water to a desired concentration and then mixed vigorously by vortexing at room temperature for 1 h to emulsify the lipid mixture. Following that, the LUV solution was completely frozen in a dry-ice/ethanol bath for ∼1 min and then gently thawed in warm water (< 50°C) for ∼30 s. This freeze-thaw process was repeated at least five times. Then the solution was extruded 19 times through a polycarbonate filter having a pore size of ∼100 nm in diameter to form LUVs. The typical diameter of the LUVs, determined by DLS, was centered at ∼100 nm with a standard deviation of ∼30 nm. Liposomes were stored at 4 °C in a refrigerator and were good for use within two weeks of their formation.
Polymer-containing POPC liposomes for ITC measurements were prepared as described above except that polymers were present during the formation of lipid vesicles.
Lipid Peroxidation Procedures
Lipid peroxidation of POPC-PLPC-POPG vesicles (25 μM in lipid concentration) was induced by peroxyl radicals, generated by degradation of the azo compound AAPH (15 mM) at 37 °C under UV irradiation (λ = 254 nm) in a quartz cuvette for 30 min. AAPH is a water-soluble free radical generator that undergoes thermal/UV-irradiation decomposition to yield N2 plus two alkyl radicals, which then react with oxygen to form two peroxyl radicals24, 25 (Scheme 2). It has been demonstrated that the intermediate and end products of lipid peroxidation—hydroperoxide lipids and cleaved-lipids— drastically affect the size and structural integrity of lipid vesicles due to changes in the cross-section area of its lipid tails and solubility.18 Given that, here the protective effect of the polymers was evaluated by monitoring changes in size and size distribution of the LUVs in the presence of the polymers (125 μM for the copolymers and 625 μM for PEG-8K) before and after lipid peroxidation.
Scheme 2.
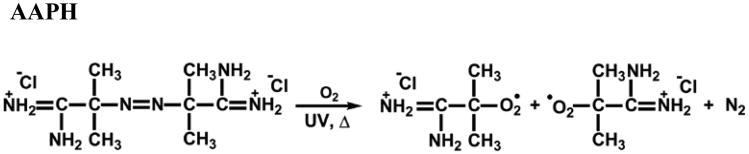
Production of peroxyl radicals from the decomposition of AAPH.
Dynamic Light Scattering
To evaluate the degree of liposome peroxidation, changes in size and size distribution of the LUVs were monitored by DLS (PD2000DLS Precision Detectors, Franklin, MA). Before mixing the LUV dispersion with AAPH and the polymers, the AAPH and polymer solutions were filtered through Whatman syringe filters with pore sizes of ∼200 nm in diameter to remove dust particles. DLS measurements were performed at 25°C with an apparatus of standard design, equipped with a 150-mW He-Ne laser providing light of 800 nm. Scattered light from the sample was analyzed at an angle of 90° from the incident beam. The correlation analysis gave directly diffusion coefficients of the vesicles, from which diameters of the LUVs were calculated using the Stokes-Einstein relationship. The LUVs were diluted 500-fold with water before measurements, and scattering data were taken for 15 min. To check for reproducibility, experiments with different batches of vesicles were repeated at least three times.
Isothermal Titration Calorimetry
ITC was performed using the VP type instrument produced by MicroCal Inc. (Northampton, MA) at 25°C. Typically, 300 μl of the LUV dispersion was loaded into the titration syringe, and 1.4 ml of a polymer solution was loaded into the titration cell. The reference cell was filled with Milli-Q water. Before titration, both the titrant and the sample were degassed using the MicroCal Thermalovac sample degassing equipment. The titration was controlled by VPViewer 2000 ITC software. During the measurements, the first 1 μl LUV dispersion was injected without taking into account the corresponding heat because the initial injection was subject to large errors due to the diffusion of solutions across the syringe tip during the pre-titration equilibration period. Following that, a series of 10 μl of LUV dispersion was injected into the sample cell and the corresponding characteristic heat resulted from the interactions between polymers and lipid membranes was integrated, yielding the heat of partitioning, hi, of each injection.
For ITC measurements, two protocols were used: (1) uptake experiments in which POPC LUV dispersion was injected into a polymer solution; (2) release experiments in which polymer-containing POPC LUV dispersion was injected into water. The combination of these two protocols allows us to obtain thermodynamic parameters of the polymer-liposome interactions, namely, the partition coefficient, K, and the molar enthalpy of partitioning, ΔH. In addition, comparison of the results obtained from the two protocols allows us to gain insights into polymer distribution across lipid membranes and to distinguish whether the polymers permeate through the bilayer membranes to access the liposome interior within our experimental timescale.26 It should be noted that while the ITC measurements used LUVs with different lipid compositions from those used in the DLS experiments, that is, POPC in ITC vs. POPC-PLPC-POPG (6:3:1) in DLS, the control ITC experiment with POPC-PLPC-POPG LUVs indicates that the changes in acyl chain desaturation and the headgroup type do not affect the way of the polymers partition to the lipid membrane. (see Supporting Information S-Figure 2)
To analyze ITC data, the model suggested by Heerklotz for partitioning of surfactants into lipid membranes was used.27 Briefly, the partition coefficient K is defined in terms of mole fractions26:
| (1) |
where W = 55.5 M is the molarity of water, and L and P are the lipid and polymer concentrations, respectively. The subscripts t and b represent the total polymer and the polymer partitioned in the membrane. The normalized heat qobs, resulted from the interaction between the total amount of lipids and polymers during the injection is expressed as:
| (2) |
ΔH is the molar enthalpy of partitioning, that is, the molar heat resulting from transferring the polymer from water to the lipid bilayer: denotes the total polymer mole fraction in the syringe. The term qdil is the molar heat of dilution of the injectant, measured separately in a control experiment.
In the Heerklotz's model, the transmembrane distribution of the polymers can be estimated using a fitting procedure of both permeable and impermeable models. Considering not all of the polymers are
| (3) |
| (4) |
able to redistribute across the bilayer within our ITC experimental timescale, the total lipid and polymer concentrations are replaced by their effective concentrations,
where γL and γp are the fractions of polymers and lipids involved in partitioning, respectively. In the permeable model where all of the polymers are able to redistribute across the bilayer, both γL and γp equal to 1, regardless of the experimental protocols used. In contrast, γL and γp have dissimilar values in the two experimental protocols in the impermeable model, where the polymers cannot cross the lipid bilayer. Using the uptake protocol, the injection of liposomes to polymer solutions results in a scenario where all polymers are free to partition but only lipids in the outer leaflet of liposomes are accessible, therefore, γp= 1 but γL = 0.5. Using the release protocol, on the other hand, since both lipids and polymers are partially trapped inside the inner leaflet of lipid bilayers γp, γL and equal to 0.5, assuming the polymers distribute evenly in the bilayer. Thus, by substituting Eqs. (3) and (4) into the models and simultaneously fitting both the uptake and release data using their γp, γL corresponding and values, the molar enthalpy of partitioning, ΔH, and partition coefficient, K, can be obtained. Comparison of ΔH and K in the uptake and release experiments (they should be similar as the two processes are reversible) therefore allows us to discern whether the lipid membranes are permeable to the polymer.
3. Results
Peroxidation of POPC-PLPC-POPG LUVs
POPC-PLPC-POPG LUVs were used in this study as a simplistic mimic of the cell membranes.27 We deliberately did not include cholesterol, one of the major components of biomembranes, to our lipid system as our rationale for keeping our lipid system as simple as possible is to allow us to reasonably get at the mechanism of intermolecular interactions of lipids with polymers. Because cholesterol can potentially affect both the process of lipid peroxidation28 and membrane structure and properties29, its effect on polymer-membrane interactions is complicated and will be discussed in a separated report.
Lipid peroxidation was accomplished using the water–soluble free radical generator, AAPH, as a peroxidation initiator at 37°C under UV irradiation (λ = 254 nm). Although not a naturally occurring biomolecule, AAPH has been widely employed in studies of lipid peroxidation.30-32 Indeed, this compound undergoes thermal decomposition at 37°C and, upon UV irradiation, generates peroxyl radicals at a fast and, more importantly, constant rate, thereby allowing for reproducible and quantitative analyses.33, 34 In light of the signatures of liposome peroxidation, that is, changes in the vesicle size and membrane integrity18, 27, 35, here we evaluate the possible protective effect of a given polymer on liposome peroxidation by monitoring changes in size and size distribution of the LUVs before and after oxidation by DLS.
Shown in Figure 1 is the DLS analysis of POPC-PLPC-POPG LUVs in the presence of 15 mM AAPH before and after UV-irradiation. The average hydrodynamic radius of the LUVs after extrusion was ∼50±30 nm and no changes in the size and size distribution of the LUVs were observed after mixing the vesicles with the AAPH solution. After exposing the LUV dispersion to UV light for 10 to 15 min, the average hydrodynamic radius of the vesicles increased to ∼65±50 nm. As the first step of the peroxidation of polyunsaturated fatty acid tails (slightly bend chains) leads to the formation of intermediate hydroperoxide lipids, the preferential orientation of the hydrophilic hydroperoxide group to the membrane-water interface leads to the fatty acid tails highly bended and thus to the expansion of the bilayer area.18, 35 Indeed, K. A. Riske et al.35 have found that a 0.5% peroxidation of lipids is enough to cause noticeable size changes of lipid vesicles. Therefore, increases in vesicle size and size distribution here indicate that the peroxyl radical-induced degradation of unsaturated lipids has occurred. Given that the cleavage of the carbon chain near the initial position of the double bound at the final step of lipid peroxidation causes membrane disruption18, the complete loss of scattering signals from the original LUVs upon 20 min irradiation indicates that lipid peroxidation occurs thorough at this experimental condition and all of the original LUVs are disrupted by free radicals.
Figure 1.

DLS analysis of POPC-PLPC-POPG liposome size distribution before and after AAPH-induced oxidation under UV irradiation as a function of time.
Effects of polymers
In our evaluation of the possible protective effects of the various polymers, we used a protocol with a UV exposure of 30 min, 10 min longer than our control experiment, to ensure that adequate time has been given for the peroxyl radical-induced degradation of polyunsaturated lipids to reach completion if the vesicles were in the absence of the polymers but under otherwise identical experimental conditions.
We tested six polymers, which can be roughly divided into two groups based on their hydrophobicity: PEG 8K, P1107 and P188 belong to the hydrophilic group; P335, P333 and P181 belong to the hydrophobic group. Detailed information of each polymer is listed in Table 1. Figure 2 presents the DLS analysis of POPC-PLPC-POPG LUVs in the presence of AAPH and hydrophilic polymers before and after UV exposure. Results from two time points are shown for all polymers examined: immediately after the introduction of the polymers and after 4 h of incubation of lipid vesicles with the polymers; comparison of the two allows us to gauge if possible protective effects can be enhanced with exposure time to polymers. As shown in Figure 2a and c, both PEG-8K and P1107 appear to provide immediate protection for the LUVs from oxidation without incubation; this protective effect remains the same after 4 h of incubation (Figure 2b and 2d), as evidenced by no significant changes in the average size and size distribution of the LUVs. Although P188 did not show any protective effects without incubation (Fig. 2e), it provided similar protective effects as those of P1107 and PEG-8K after 4 h of incubation with the LUVs (Figure 2f).36 It is interesting to note that these three protective polymers are different in architecture, total molecular weight and molecular weight of either the PEO or the PPO block (see Table 1 and scheme 2), but they are all rather hydrophilic. Indeed, many pre-clinical studies have shown that all three polymers are able to seal injured cell membranes7, 37, 38 and protect cells from various physicochemical insults4, 39-42. However, it still remains unclear as to what drives these polymers with rather different architectures to exhibit similar protective effects on cell membranes, and how one can design polymers to be more effective in this role of cell membrane preservation. Our results here suggest that at the molecular level, the overall hydrophilicity of the three polymers might be the driving force for their protective effects on membrane integrity.
Figure 2.

DLS analysis of POPC-PLPC-POPG liposome size distribution (⺷) before and (⋯) after oxidation. Liposomes incubated with (a) PEG-8K for 0 h; (b) PEG-8K for 4 h; (c) P11107 for 0 h; (d) P1107 for 4 h; (e) P188 for 0 h (f) P188 for 4 h. For PEG-8K, the molar ratio of polymer vs. lipid is 25; whereas for the copolymers, the molar ratio of polymer vs. lipid is 5.
It should be noted that all of the aforementioned experiments were performed in the presence of excess polymers, that is, the molar ratios of polymer vs. lipid are 5 and 25 for the copolymers and PEG-8K, respectively. To examine the effect of polymer concentration on their effectiveness in protecting liposomes from peroxidation, reduced molar ratio of 2.4 and 1 for P188 vs. lipid were tested. Figure 3 shows the DLS analysis of POPC-PLPC-POPG LUVs in the presence of AAPH and P188 after 4 h incubation, examined at 3 different polymer-to-lipid molar ratios. As can be seen, P188 provides complete protection at a polymer ratio of 5, much less effective protection at a ratio of 2.4 and no protection at all at a ratio of 1. A similar concentration-dependence protective trend is also observed in the case of P1107 and PEG-8K (see Supporting Information S-Figure 1), indicating that the protective effects of the polymers are indeed concentration-dependent.
Figure 3.

DLS analysis of POPC-PLPC-POPG liposome size distribution (⺷) before and (⋯) after oxidation as a function of P188 concentration after 4 h incubation. The molar ratio of P188 vs. lipid is (a) 5; (b) 2.4; (c) 1.
In our previous GUV study pertaining to osmotic stress13, we have proposed that the more hydrophobic the polymer, the more rapid the polymer is able to insert into the lipid membrane. As PEG-8K, P1107 and P188 are highly hydrophilic polymers (even though P188 and P1107 still have amphiphilic properties), they are unlikely to insert into lipid membranes within our experimental timescale. Therefore, our data suggest that the protective effect of these polymers stems from the accumulation of the polymers at the vesicle surface, but not from their insertion into the bilayer. To test this hypothesis, we compare the protective effect of these three hydrophilic polymers with those of three hydrophobic polymers—P335, P333 and P181—that are known to be able to insert into lipid membranes. Figure 4 shows the DLS analysis of POPC-PLPC-POPG LUVs in the presence of AAPH and P335, P333 and P181 before and after UV exposure. In these cases, after UV irradiation, even though some vesicles might still remain in the solution—with the exception of P181 without incubation where all of the liposomes are completely broken up by free radicals—our DLS analyses depict a significant decrease in scattering intensity, a moderate reduction in the average vesicle sizes, and a remarkable broadening in the vesicle size distribution, indicating that lipid peroxidation actually occurred, but in an incomplete fashion. At present, the exact changes in liposomes due to the presence of these polymers are not completely clear as end products of lipid peroxidation can exist in a variety of forms like micelles, soluble lipid species, or even aggregates with different structures43, and the presence of polymers capable of inserting into lipid membranes further complicates the matter. However, one clear conclusion that can be drawn from these results is that none of hydrophobic copolymers, P335, P333 and P181, can actively protect liposomes from peroxidation as do the hydrophilic copolymers, PEG-8K, P1107 and P188. Our results thus point to the overall hydrophilicity of the polymer being one of the key properties that an effective membrane resealing polymer should possess.
Figure 4.

DLS analysis of POPC-PLPC-POPG liposome size distribution (⺷) before and (⋯) after oxidation. Liposomes incubated with (a) P335 for 0 h; (b) P335 for 4 h; (c) P333 for 0 h; (d) P333 for 4 h; (e) P181 for 0 h (f) P181 for 4 h. The molar ratio of polymer vs. lipid is 5.
ITC measurements
To ascertain that the protective capability of the hydrophilic polymers is due to their association and adsorption at the surface of the vesicles, and the lack thereof for the hydrophobic polymers stems from their insertion into the membrane bilayer within our experimental timescale, we have employed ITC to compare the interactions of these two groups of polymers with lipid vesicles. As is well-known, ITC is a sensitive technique for investigating the binding/partitioning affinity and thermodynamics involved in intermolecular interactions.44 It has been widely used to monitor interactions between surfactants and lipid membranes as the partitioning of surfactant molecules to lipid membranes is generally accompanied by a consumption or release of heat. If there is no apparent partitioning of surfactant molecules to lipid bilayer membranes, no heats of interactions would be expected during titration.45
Shown in Figure 5 are the calorimetric data from the isothermal titration of 15 mM POPC liposome dispersions into 50 μM P188, 50 μM P1107, and 250 μM PEG-8K solutions at 25°C. In all three cases, each titration of the LUVs into the polymer solution only produced a very small exothermal heat flow, and this residual effect was independent of the amount of LUVs injected throughout the entire titration process. Earlier work has shown that such a small, constant exothermal heat flow is the heat of dilution of the liposomes in the polymer solution.46 It has been reported that poloxamers cannot be incorporated into lipid bilayers in the gel phase due to the tight lipid packing.47 Here, as POPC liposomes are in the fluid-phase at 25°C (Tm = -2°C) which principally permits the incorporation of poloxamers, the absence of heat of partitioning of the polymers to lipid membranes confirms that there is no polymer insertion into the membrane over our experimental timescale. This is not too surprising, especially for PEG-8K, as it has been demonstrated that there is no direct collision of PEG homopolymers with phospholipid membranes due to the existence of a depletion layer (∼1 nm) between the surface of lipid vesicles and the bulk polymer aqueous phase.47-49 Since the hydrophobic PPO block in both P188 and P1107 only represents a very small portion of the entire polymer chain, it would be energetically favorable to have the PPO block shielded by the PEO blocks rather than to have it exposed to the water/liposome interface. Therefore, it is reasonable that both P188 and P1107 appear to present a similar behavior at membrane surfaces as PEG does.
Figure 5.

ITC of (a) 50 μM P188 (b) 50 μM P1107 (c) 250 μM PEG-8K with 15 mM POPC liposomes at 25°C. Upper panel: raw data from sequential injections. Lower panel: the integrated heat per injection normalized with respect to the number of moles of POPC injected. The small, constant, exothermic heat flows in all of the three profiles are similar to the heat of dilution, in dicating that there is no partitioning of polymers into the lipid bilayer.
The calorimetric profiles exhibit entirely different features as the proportion of the PPO block in a copolymer chain increases. Figure 6 shows the calorimetric data from the isothermal titration of 15 mM POPC liposome dispersions into 100 μM P181, 50 μM P333 and 50 μM P335 solutions at 25°C. As can be seen, for all three copolymers, each injection generates an endothermic heat flow that decreases with each injection. This decrease in heat flow is due to the gradual depletion of the copolymer in the aqueous phase to partition to the lipid bilayer phase. If the titration process was continued indefinitely, the heat flow would eventually revert to that of the heat of dilution after all of the copolymers were partitioned to the vesicles.46 These decayed features in the calorimetric profiles, qualitatively point to a different mechanism of interaction when hydrophobic polymers are involved. Comparing the measured integrated heat with the one calculated using the equilibrium partitioning model26, assuming that the bilayer is impermeable to all three copolymers, the thermodynamic parameters of ΔH and K are derived, that is, ΔH = 5.8 kcal/mol and K = 2.77 × 104 for P181, ΔH = 10.9 kcal/mol and K = 4.34 × 104 for P333, and ΔH = 9.7 kcal/mol and K = 3.16 × 104 for P335. These results clearly demonstrate that P181, P333 and P335 all insert into the liposomal membrane, rather than merely associated at the vesicle surface. The equilibrium partition model based on the assumption that the bilayer is permeable to all three copolymers was also thoroughly tested, but no consistent results were obtained (see S-table 1 in Supporting Information). Therefore, the consistent fit with the impermeable model but not with the permeable model suggests that the three hydrophobic poloxamers merely insert into the outer leaflet of the membrane rather than access the interior of the vesicles over our experimental timescale.
Figure 6.

ITC of (a) 100 μM P181 (b) 50 μM P333 (c) 50 μM P335 with 15 mM POPC liposomes at 25°C. Upper panel: raw data from sequential injections. Lower panel: the integrated heat per injection normalized with respect to the number of moles of POPC injected. The solid lines are the least-squares fits of the caloricmetric data with Heerklotz's partitioning model where none of the three poloxamers permeates through the membrane on the experimental timescale.
4. Discussion
Lipid peroxidation is deleterious in many ways to biological cells.50 To avoid lipid peroxidation, organisms generally launch both enzymatic and nonenzymatic antioxidant defenses, which work either through neutralization of an oxidant or regeneration of other molecules capable of reacting with the oxidant to block the process or to limit its damage.51 As all poloxamers examined in this work share the same chemical moiety but exhibit opposite effects regarding their ability to protect liposomes from peroxidation mainly due to the difference in hydrophobicity, we argue that the antioxidant effect of the hydrophilic polymers is not primarily because they work as radical scavengers, but rather due to their specific interaction with lipid membranes afforded by their architecture. In fact, PEO-PPO-PEO copolymers are relatively stable under our experimental conditions as the 1H NMR spectra of P188 before and after oxidation (see Supporting Information S-Figure 4) do not show any signature of polymer degradation other than the end hydroxyl group being oxidized to the carboxyl group, which is not significant enough to protect liposomes from peroxidation, considering that 15 mM free radical generators were used with only 50 μM copolymers in our systems.
To understand how polymer adsorption can lead to such a protective effect, the mechanism of lipid peroxidation needs to be elucidated first. Lipid peroxidation is a free radical chain reaction, which includes initiation, propagation and termination. The initiation step dictates the entire oxidation rate and starts by a direct collision between free radicals and hydrogen atoms in the bisallyl position in the acyl side chain of polyunsaturated lipids. As most free radicals are generated in water, these radicals have to diffuse into the lipid bilayer to initiate lipid peroxidation.52 Accordingly, if a substance can affect lipid packing or membrane fluctuation or the hydration shell of the lipid bilayer in a way that suppresses the diffusion of free radicals, it plays the role of an indirect antioxidant to inhibit lipid peroxidation. In fact, it has been demonstrated by Tirosh et al. that the addition of an extended hydration layer by anchoring a PEG layer on the surface of lipid membranes protects the lipid bilayer against oxidation damage.53 In their report, they claimed that the extended hydration layer worked as a radical “freezing” layer as the free radicals “died” before they could pass through the layer by diffusion.
Here, the combination of our DLS and ITC results shows that the mere adsorption of hydrophilic poloxamers at lipid vesicle surfaces can effectively suppress lipid peroxidation, whereas the insertion of the hydrophobic poloxamers cannot. One can argue that perhaps P188 and P1107 work as antioxidants due to their ability to scavenge radicals, rather than their adsorption at the membrane surface. If this were indeed the case, then all other poloxamers, P181, P333 and P335, should be equally effective in protecting lipids against oxidation as they share the identical chemical details. Our findings have indicated otherwise and thus rules out the direct radical scavenger role of these copolymers. Our ITC data have demonstrated that hydrophobic copolymers insert into the lipid bilayer, a process that disturbs lipid packing, and thus enhances membrane permeability. It is therefore not difficult to understand the ineffectiveness of these hydrophobic poloxamers in lipid peroxidation inhibition. Hydrophilic polymers, on the other hand, have been found to adsorb at the vesicle surface and are effective in suppressing lipid peroxidation. One would deduce from these observations that such polymer adsorption should either affect lipid packing or membrane fluctuation in a way that reduces the access of free radicals into the membrane. In fact, evidence provided in our accompanied 1H Overhauser Dynamic Nuclear Polarization (ODNP) study indeed demonstrates that the hydrophilic polymers weakly adsorb at the lipid membrane surface in a way that impede hydration dynamics both at the surface and in the interior of the lipid bilayer, thereby suppressing the diffusion of free radicals into lipid membranes without disrupting the lipid mobility and fluidity.1
5. Conclusions
In summary, the fundamental interactions between PEO-PPO-PEO triblock copolymers and lipid membranes were investigated using DLS and ITC. Specifically, the effect of hydrophobicity of the triblock copolymers on their ability to preserve the structural integrity of lipid membranes by inhibition of lipid peroxidation was evaluated by DLS, and the partitioning free energy and coefficient involved in polymer-lipid membrane interactions were derived from ITC measurements. Our DLS studies show that highly hydrophilic polymers are effective in protecting liposomes from oxidation and ITC measurements indicate that the degree of hydrophobicity dictates the polymer-membrane interactions: hydrophilic triblock copolymers only weakly adsorb at the membrane surface, whereas hydrophobic triblock copolymers insert into the lipid bilayer. The combination of DLS and ITC results thus indicates that the protecting effect of the triblock copolymers comes from polymer adsorption, but not from polymer insertion, as the latter disrupts lipid packing and enhances membrane permeability. Our findings help identify a key structural feature of triblock copolymers necessary for their roles as cell membrane preservers. The new insights of polymer-membrane interactions afforded by the current study should aid in the design of novel polymeric cell membrane sealants and/or permeation enhancers.
Supplementary Material
Acknowledgments
We very kindly thank Songi Han and Chi-Yuan Cheng for their fruitful discussions and valuable comments to this work. This work was supported in part by the National Institutes of Health (1R01 NS056313-01A1 to JM). J.-Y.W. and K.Y.C.L. acknowledge for the support of the March of Dimes (No.6-FY07-357), the NSF (MCB-0920316) and the University of Chicago NSF-MRSEC program (DMR-0820054). We thank the Biophysical Core Facility at the University of Chicago for access to the ITC instrument and Dr. E. Solomaha for help with ITC measurements.
Footnotes
Supporting Information: DLS analysis of POPC-PLPC-POPG liposome as a function of concentrations of P1107 and PEG-8K without incubation before and after oxidation; ITC of P188 and P181 with POPC-PLPC-POPG liposomes at 25°C; ITC of release of POPC LUVs pre-incorporated with P181 and P333 into water and the fitting parameters obtained for both uptake and release experiments at 25 °C; 300 MHz 1H NMR spectrum of P188 (a) before oxidation (b) after oxidation in D2O at 25°C. This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Jia-Yu Wang, Email: jiayuwang@uchicago.edu.
Jeremy Marks, Email: jmarks@uchicago.edu.
References
- 1.Cheng CY, Wang JY, Kausik R, Lee KYC, Han S. Manuscript is under review [Google Scholar]
- 2.Alexandridis P. Curr Opin Colloid Interface Sci. 1997;2:478. [Google Scholar]
- 3.Munshi N, Rapoport N, Pitt WG. Cancer Lett. 1997;118:13. doi: 10.1016/s0304-3835(97)00218-8. [DOI] [PubMed] [Google Scholar]
- 4.Lee RC, River LP, Pan FS, Ji L, Wollmann RL. Proc Nat Acad Sci USA. 1992;89:4524. doi: 10.1073/pnas.89.10.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kabanova AV, Batrakovaa EV, Alakhovb VY. Adv Drug Deliv Rev. 2002;54:759. doi: 10.1016/s0169-409x(02)00047-9. [DOI] [PubMed] [Google Scholar]
- 6.Armstrong JK, Meiselman HJ, Fisher TC. Thromb Res. 1995;437:1995. doi: 10.1016/0049-3848(95)00134-d. [DOI] [PubMed] [Google Scholar]
- 7.Merchant A, Holmes WH, Capelli-Schellpfeffer M, Lee RC, Toner M. J Surg Res. 1998;74:131. doi: 10.1006/jsre.1997.5252. [DOI] [PubMed] [Google Scholar]
- 8.Greenbaum BBK, Hannig J, Carrillo CS, Beckett MA, Weichselbaum RR, Lee RC. Burns. 2004;30:539. doi: 10.1016/j.burns.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 9.Serbest G, Horwitz J, Barbee K. Journal of Neurotrauma. 2005;22:119. doi: 10.1089/neu.2005.22.119. [DOI] [PubMed] [Google Scholar]
- 10.Krylova OO, Pohl P. Biochemistry. 2004;43:3696. doi: 10.1021/bi035768l. [DOI] [PubMed] [Google Scholar]
- 11.Erukova VY, Krylova OO, Antonenko YN, Melik-Nubarov NS. Biochim Biophys Acta. 2000;1468:73. doi: 10.1016/s0005-2736(00)00244-3. [DOI] [PubMed] [Google Scholar]
- 12.Miller DW, Batrakova EV, Kabanov AV. Pharm Res. 1999;16:396. doi: 10.1023/a:1018873702411. [DOI] [PubMed] [Google Scholar]
- 13.Wang JY, Chin J, Marks JD, Lee KYC. Langmuir. 2010;26:12953. doi: 10.1021/la101841a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Halliwell B, Gutteridge JMC. Methods in Enzymology. Vol. 186. Academic Press; San Diago: 1990. p. 1. [DOI] [PubMed] [Google Scholar]
- 15.Zweier JL, P K, Lutty GA. Proc Nat Acad Sci USA. 1988;85:4046. doi: 10.1073/pnas.85.11.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borst JW, Visser NV, Kouptsova O, Visser AJ. Biochim Biophys Acta. 2000;1487:61. doi: 10.1016/s1388-1981(00)00084-6. [DOI] [PubMed] [Google Scholar]
- 17.Ayuyan AG, Cohen FS. Biophys J. 2006;91:2172. doi: 10.1529/biophysj.106.087387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heuvingh J, Bonneau S. Biophys J. 2009;97:2904. doi: 10.1016/j.bpj.2009.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hannig J, Zhang D, Canaday DJ, Beckett MA, Astumian RD, Weichselbaum RR, Lee RC. Radiat Res. 2000;154:171. doi: 10.1667/0033-7587(2000)154[0171:ssompb]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 20.Frim DM, Wright DA, Curry DJ, Cromie W, Lee R, Kang UJ. Cognitive Neuroscience and Neuropsychology. 2004;15:171. doi: 10.1097/00001756-200401190-00033. [DOI] [PubMed] [Google Scholar]
- 21.Fusco S, Borzacchiello A, Netti PA. J Bioact Compat Pol. 2006;21:149. [Google Scholar]
- 22.MacDonald RC, MacDonald RI, Menco BPM, Takeshita K, Subbarao NK, Hu L. Biochim Biophys Acta. 1991;1061:297. doi: 10.1016/0005-2736(91)90295-j. [DOI] [PubMed] [Google Scholar]
- 23.Hope MJ, Bally MB, Webb G, Cullis PR. Biochim Biophys Acta. 1985;812:55. doi: 10.1016/0005-2736(85)90521-8. [DOI] [PubMed] [Google Scholar]
- 24.Niki E. Methods Enzymol. 1990;186:100. doi: 10.1016/0076-6879(90)86095-d. [DOI] [PubMed] [Google Scholar]
- 25.Krasowska A, Rosiak D, Szkapiak K, Oswiecimska M, Witek S, Lukaszewicz M. Cell Mol Biol Lett. 2001;6:71. [PubMed] [Google Scholar]
- 26.Heerklotz HH, Binder H, Epand RM. Biophys J. 1999;76:2606. doi: 10.1016/S0006-3495(99)77413-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu L, Davis TA, Porter NA. J Am Chem Soc. 2009;131:13037. doi: 10.1021/ja9029076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schnitzer E, Pinchuk I, Bor A, Leikin-Frenkel A, Lichtenberg D. Chem Phys Lipids. 2007;126:95. doi: 10.1016/j.chemphyslip.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 29.Meyer F, Smit B. Proc Natl Acad Sci USA. 2009;106:3654. doi: 10.1073/pnas.0809959106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noguchi N, Takahashi M, Tsuchiya J, Yamashita H, Komuro E, Niki E. Biochem Pharmacol. 1998;55:785. doi: 10.1016/s0006-2952(97)00533-9. [DOI] [PubMed] [Google Scholar]
- 31.Liu Z, Yu W, Liu Z. Chem Phys Lipids. 1999;103:125. doi: 10.1016/s0009-3084(99)00101-2. [DOI] [PubMed] [Google Scholar]
- 32.Liégeois C, Lermusieau G, Collin S. J Agric Food Chem. 2000;48:1129. doi: 10.1021/jf9911242. [DOI] [PubMed] [Google Scholar]
- 33.Olsher M, Yoon SI, Chong PLG. Biochemistry. 2005;44:2080–2087. doi: 10.1021/bi047710s. [DOI] [PubMed] [Google Scholar]
- 34.Faria A, Calhau C, Freitas VD, Mateus N. J Agrc Food Chem. 2006;54:2392. doi: 10.1021/jf0526487. [DOI] [PubMed] [Google Scholar]
- 35.Riske KA, Sudbrack TP, Archilha NL, Uchoa AF, Schroder AP, Marques CM, Baptista MS, Itri R. Biophys J. 2009;97:1362. doi: 10.1016/j.bpj.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.This is not surprising as our earlier study13 pertaining to osmotic stress has suggested that the interaction between P188 and phospholipid membranes is fairly weak and it requires at least 3 h for enough P188 to accumulate on the lipid vesicles to be effective.
- 37.Pananilam JT, Bischof JC, Lee RC, Cravalho EG, Tompkins RG, Yarmush ML, Toner M. Ann N Y Acad Sci. 1994;720:111. doi: 10.1111/j.1749-6632.1994.tb30439.x. [DOI] [PubMed] [Google Scholar]
- 38.Borgens RB, Shi R, Bohnert D. The Journal of experimental biology. 2002;205:1. doi: 10.1242/jeb.205.1.1. [DOI] [PubMed] [Google Scholar]
- 39.Palmer JS, Cromie WJ, Lee RC. The Journal of urology. 1998;159:2136. doi: 10.1016/S0022-5347(01)63295-6. [DOI] [PubMed] [Google Scholar]
- 40.Marks JD, Pan CY, Bushell T, Cromie W, Lee RC. FAEBS J. 2001:1107. doi: 10.1096/fj.00-0547fje. [DOI] [PubMed] [Google Scholar]
- 41.Hannig J, Yu J, Beckett M, Weichselbaum R, Lee RC. Int J Rad Biol. 1999;75:379. doi: 10.1080/095530099140555. [DOI] [PubMed] [Google Scholar]
- 42.Liu-Snyder P, Logan MP, Shi R, Smith DT, Borgens RB. The Journal of Experimental Biology. 2007;210:1455. doi: 10.1242/jeb.02756. [DOI] [PubMed] [Google Scholar]
- 43.Yin H, Xu L, N Porter A. Chem Rev. 2011;111:5944. doi: 10.1021/cr200084z. [DOI] [PubMed] [Google Scholar]
- 44.Heerklotz H, Seelig J. Biochim Biophys Acta. 2000;1508:69. doi: 10.1016/s0304-4157(00)00009-5. [DOI] [PubMed] [Google Scholar]
- 45.Gabriel GJ, Pool JG, Som A, Dabkowski JM, Coughlin EB, Muthukumar M, Tew GN. Langmuir. 2008;24:12480. doi: 10.1021/la802232p. [DOI] [PubMed] [Google Scholar]
- 46.Wu G, Lee KYC. J Phys Chem B. 2009;113:15522. doi: 10.1021/jp906331m. [DOI] [PubMed] [Google Scholar]
- 47.Arnold K, Zschoernig O, Barthel D, Herold W. Biochim Biophys Acta. 1990;1022:303. doi: 10.1016/0005-2736(90)90278-v. [DOI] [PubMed] [Google Scholar]
- 48.Evans E, Deedham D. Macromolecules. 1988;21:1822. [Google Scholar]
- 49.Lehtonen JYA, Kinnumen PK. BIophys J. 1995;68:525. doi: 10.1016/S0006-3495(95)80214-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McIntyre TM, Zimmermant GA, Prescott SM. J Biol Chem. 1999;274:25189. doi: 10.1074/jbc.274.36.25189. [DOI] [PubMed] [Google Scholar]
- 51.Pamplona R. Biochim Biophys Acta. 2008;1777:1249. doi: 10.1016/j.bbabio.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 52.Barclay LRC, U IK. J Am Chem Soc. 1981;103:6478. [Google Scholar]
- 53.Tirosh O, Kohen R, Katzhendler J, Gorodetsky R, Barenholz Y. J Chem Soc, Perkin Trans. 1997;2:383. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.