

Abstract
Background
Previous molecular genetic studies of physiology and pigmentation of sheep skin have focused primarily on a limited number of genes and proteins. To identify additional genes that may play important roles in coat color regulation, Illumina sequencing technology was used to catalog global gene expression profiles in skin of sheep with white versus black coat color.
Results
There were 90,006 and 74,533 unigenes assembled from the reads obtained from white and black sheep skin, respectively. Genes encoding for the ribosomal proteins and keratin associated proteins were most highly expressed. A total of 2,235 known genes were differentially expressed in black versus white sheep skin, with 479 genes up-regulated and 1,756 genes down-regulated. A total of 845 novel genes were differentially expressed in black versus white sheep skin, consisting of 107 genes which were up-regulated (including 2 highly expressed genes exclusively expressed in black sheep skin) and 738 genes that were down-regulated. There was also a total of 49 known coat color genes expressed in sheep skin, from which 13 genes showed higher expression in black sheep skin. Many of these up-regulated genes, such as DCT, MATP, TYR and TYRP1, are members of the components of melanosomes and their precursor ontology category.
Conclusion
The white and black sheep skin transcriptome profiles obtained provide a valuable resource for future research to understand the network of gene expression controlling skin physiology and melanogenesis in sheep.
Keywords: Sheep skin, Transcriptome, Gene expression, Pigmentation, Melanogenesis
Background
Sheep are the most important fiber producing animals worldwide. Fiber diameter, length and color are key traits contributing to the economic value of sheep and are determined by both genetics [1,2] and environment [3]. Factors that determine coat color in sheep are becoming of increasing interest. White fleece holds greatest economic value due to its ability to be dyed to virtually any color, whereas interest in natural colors is increasing due to the green revolution and consumer preference for natural products.
Coat color genes are good candidates for facilitation of traceability of animal breeds [4]. Coat color is determined by amounts and types of melanin produced and released by melanocytes resident in the skin [5,6]. The genetic basis for coat color is well understood in rodents [7,8], with many common genes also implicated in regulation of coat color in other species, including sheep. For example, MC1R and ASIP are known to be major regulators of coat color in mice and MC1R[9] and ASIP[10] loci are functionally linked to undesirable coat color phenotypes in sheep. In addition, tyrosinase-related protein 1 (TYRP1) is a strong positional candidate gene for color variation in Soay sheep [11]. Recent studies have combined SNP analysis and gene expression profiling to dissect the basis for the piebald pigmentation phenotype in Merino sheep [12]. Despite considerable knowledge of the genetic regulation of coat color in mice and identification of loci involved in coat color regulation in fiber producing species, the molecular mechanisms, at the level of gene expression, associated with differences in coat color phenotype are not well understood. This information is critical not only to enhanced basic understanding of regulation of melanogenesis, but also to the identification of novel pharmacological and molecular genetics approaches to regulate or select for coat color in fiber producing species.
Transcriptional profiling is a powerful approach for identification of genes globally and functionally expressed in various tissues including skin [13]. Limited information is currently available regarding differences in transcriptome profiles of skin associated with coat color in fiber producing species. To investigate genes that may play important roles in coat color regulation in sheep skin and gain insight into molecular mechanisms responsible for biochemistry of skin and fibers (including pigmentation) in animals producing hair such as sheep and alpaca, we investigated the transcriptome profiles in skin of sheep with black versus white coat color using high throughput RNA deep sequencing. Results provided novel insight into differences in gene expression associated with coat color, including key genes implicated in the melanogenesis pathway.
Results
Assembly of unigenes
After the raw reads were filtered, 51,297,002 clean reads with 52.2% GC percentage and 51,655,390 clean reads with 53.7% GC percentage were obtained from white and black sheep skin, respectively. These clean reads were assembled into unigenes, yielding 90,006 and 74,533 unigenes from white and black sheep skin, respectively. There were 2,892 and 2,884 unigenes with sequence size greater than 3,000 nucleotides in white and black sheep skin, respectively. The longest unigene sequenced was more than 9,000 nt in length and the average size of the majority of coding sequence (CDS) identified was 300 nt. There were 1,367 unigenes with more than 3,000 nt of CDS (Figure 1).
Figure 1.

Length distribution and abundance of all unigenes identified in sheep skin.
Functional classification of the unigenes
BLAST analysis (e-value < 0.00001) of the sheep skin unigenes against the protein and nucleotide databases revealed 37,768 known genes, of which, 36,438 were annotated through COG classification analysis. These genes were grouped into 25 classes based on their putative functions and the largest group of genes was classified into general function only (15%; Figure 2). The known genes were also annotated through GO classification analysis and grouped into 3 categories (biological process, 46.1%; cellular component, 36.1%; molecular function, 16%) based on their putative functions (Figure 3).
Figure 2.

COG functional classification of all unigenes in sheep skin.
Figure 3.
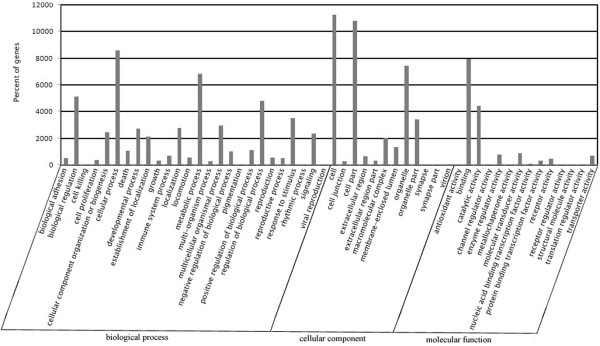
GO functional classification of all unigenes in sheep skin.
Genes highly expressed in sheep skin
The top 30 genes most highly expressed in sheep skin included genes of the keratin family and ribosomal proteins (Table 1). The most highly expressed gene in sheep skin was 40S ribosomal protein S29. The other highly expressed genes in sheep skin included, NADH dehydrogenase subunit 5 (NADH5), cytochrome c oxidase subunit I (COX1), NADH-ubiquinone oxidoreductase chain 4 (ND4), keratin associated protein 9.2 (KAP9.2), keratin 27 (Krt27), high-sulfur keratin BIIIB4 protein, hair keratin cysteine rich protein (Dmpk), keratin-associated protein 1.4 (KRTAP1.4) and keratin-associated protein 3–2 (KAP3.2). The list of most highly expressed genes also included 4 unknown genes.
Table 1.
Top 30 highly expressed genes in sheep skin
| Gene name | FPKM (White) | FPKM (Black) |
|---|---|---|
| Keratin associated protein 9.2 |
2584.8 |
821.2 |
| Keratin 27 |
2607.2 |
1067.3 |
| Complete mitochondrial genome |
2674.7 |
3041.1 |
| Unknown (unigene10779_all) |
2748.0 |
3383.1 |
| PREDICTED: ferritin, heavy polypeptide 1 |
2888.1 |
2553.0 |
| Ribosomal protein P2-like |
2895.4 |
2725.2 |
| SJCHGC01393 protein |
2901.1 |
3225.6 |
| NADH dehydrogenase subunit 5 |
2932.5 |
2388.7 |
| Cystatin-M precursor |
2986.9 |
782. 3 |
| Unknown |
3057.6 |
2405.2 |
| 40S ribosomal protein S15 |
3076.5 |
2540.7 |
| High-sulfur keratin BIIIB4 protein |
3091.8 |
1541.3 |
| 60S ribosomal protein l18a-like |
3167.0 |
3583.3 |
| 40S ribosomal protein S8 |
3400.9 |
3428.3 |
| 40S ribosomal protein S17 |
3456.8 |
3098.2 |
| 40S ribosomal protein S2 |
3617.6 |
3951.0 |
| PREDICTED: 40S ribosomal protein s15a |
3621.0 |
3604.1 |
| High-sulfur keratin BIIIB4 protein |
3717.6 |
2178.6 |
| Hair keratin cysteine rich protein |
3729.8 |
2563.5 |
| Keratin-associated protein 9.2 |
3753.7 |
1379.9 |
| Bos taurus ubiquitin C (UBC) |
3912.4 |
5532.3 |
| Keratin-associated protein 1.4 |
3945.3 |
2057.2 |
| 40S ribosomal protein S11 |
4019.3 |
3719.5 |
| PREDICTED: rrna promoter binding protein-like |
4062.3 |
4910.0 |
| Keratin associated protein 9.2 |
4628.2 |
1689.3 |
| Cytochrome c oxidase subunit I |
5279.6 |
3998.8 |
| Keratin-associated protein 3-2 |
6143.7 |
3911.5 |
| NADH-ubiquinone oxidoreductase chain 4 |
6684.7 |
5468.1 |
| Unknown (unigene19263_all) |
7622.1 |
12646.3 |
| PREDICTED: 40S ribosomal protein S29-like | 19925.8 | 9994.6 |
Genes encoding transcription factors expressed in sheep skin
In the transcriptome of sheep skin, there was at least 527 genes identified encoding for transcription factors (Additional file 1: Table S1). The highly expressed transcription factor genes in both white and black sheep skin include transcription factor jun-B-like, nascent polypeptide-associated complex subunit alpha isoform b, endothelial differentiation-related factor 1 isoform alpha, homeobox protein DLX-3 (DLX3), transcription factor AP-1 (JUN), cyclic AMP-dependent transcription factor ATF-4 (ATF4), and transcription factor GATA3 (GATA3) (Table 2). Most of these transcription factors do not show differential expression between white and black skin. However, some less abundantly expressed transcription factors such as Pbx, Tcf4, Nr2f1, Sox11 and Sox4, were differentially expressed in white and black sheep skin. Transcription factors that are known to regulate mRNA expression of coat color genes, such as MITF and CREB/ATF bZIP transcription factor were also found to be expressed in sheep skin. Interestingly, both genes were expressed approximately 2 times higher in white sheep skin than black sheep skin.
Table 2.
Highly expressed transcription factors in sheep skin
| Gene name | FPKM (White) | FPKM (Black) |
|---|---|---|
| Transcription factor jun-B-like |
206.6 |
368.3 |
| Cyclic AMP-dependent transcription factor ATF-4 |
301.0 |
340.3 |
| Transcription factor AP-1 |
125.5 |
205.5 |
| Endothelial differentiation-related factor 1 isoform alpha |
94.0 |
140.7 |
| homeobox protein DLX-3 |
132.6 |
90.1 |
| transcription factor GATA3 | 61.7 | 63.2 |
Differentially expressed genes in white versus black sheep skin
Using an algorithm based on a previously described method [14], genes differentially expressed between white and black sheep skin were identified. There were a total of 2,235 known genes identified as differentially expressed in white versus black sheep skin, of which 1,756 were down-regulated (≤ 2 fold) and 479 were up-regulated (≥ 2 fold) in skin from black sheep compared with skin from white sheep (see Additional file 2: Table S2). For the GO analysis, 1,904, 1,784 and 1,787 differentially expressed genes were grouped in cellular component, molecular function and biological process categories, respectively. Most of the differentially expressed genes were classified into two GO categories (cellular process and cell and cell part; Table 3 and Figure 4). The majority of the GO terms including pigmentation do not appear to be significantly enriched in the differentially expressed genes.
Table 3.
Total number of genes and genes with difference in each GO term
| GO terms | Total number of genes | Total number of genes with difference | Corrected P-value |
|---|---|---|---|
| Biological adhesion |
673 |
89 |
1 |
| Biological regulation |
8650 |
1141 |
1 |
| Cell killing |
32 |
5 |
1 |
| Cell proliferation |
470 |
76 |
1 |
| Cellular component organization or biogenesis |
3698 |
515 |
1 |
| Cellular process |
11404 |
1441 |
1 |
| Death |
678 |
92 |
1 |
| Developmental process |
3927 |
549 |
1 |
| Establishment of localization |
3191 |
416 |
1 |
| Growth |
309 |
34 |
1 |
| Immune system process |
1278 |
175 |
1 |
| Localization |
3783 |
92 |
1 |
| Locomotion |
882 |
126 |
1 |
| Metabolic process |
7062 |
944 |
1 |
| Multi-organism process |
963 |
132 |
1 |
| Multicellular organismal process |
5437 |
642 |
1 |
| Negative regulation of biological process |
2792 |
398 |
1 |
| Pigmentation |
69 |
21 |
0.40 |
| Positive regulation of biological process |
3228 |
447 |
1 |
| Regulation of biological process |
8093 |
1078 |
1 |
| Reproduction |
1347 |
147 |
1 |
| Reproductive process |
1342 |
147 |
1 |
| Response to stimulus |
6174 |
753 |
1 |
| Rhythmic process |
184 |
21 |
1 |
| Signaling |
3997 |
469 |
1 |
| Viral reproduction |
534 |
43 |
1 |
| Cell |
13120 |
1724 |
7.08 × 10-8 |
| Cell junction |
679 |
86 |
1 |
| Cell part |
13110 |
1724 |
4.13 × 10-8 |
| Extracellular region |
1685 |
256 |
0.15 |
| Extracellular region part |
960 |
157 |
0.09 |
| Macromolecular complex |
3942 |
495 |
1 |
| Membrane-enclosed lumen |
3111 |
420 |
1 |
| Organelle |
9471 |
1284 |
6.93 × 10-5 |
| Organelle part |
6280 |
845 |
0.72 |
| Synapse |
477 |
61 |
1 |
| Synapse part |
353 |
44 |
1 |
| Antioxidant activity |
55 |
4 |
|
| Binding |
10938 |
1519 |
1.11 × 10-15 |
| Catalytic activity |
5291 |
725 |
1 |
| Channel regulator activity |
76 |
13 |
1 |
| Enzyme regulator activity |
903 |
123 |
1 |
| Molecular transducer activity |
1898 |
136 |
1 |
| Nucleic acid binding transcription factor activity |
817 |
116 |
1 |
| Protein binding transcription factor activity |
495 |
66 |
1 |
| Receptor activity |
2057 |
157 |
1 |
| Receptor regulator activity |
35 |
5 |
1 |
| Structural molecule activity |
792 |
76 |
1 |
| Translation regulator activity |
27 |
2 |
1 |
| Transporter activity | 1175 | 153 | 1 |
Figure 4.
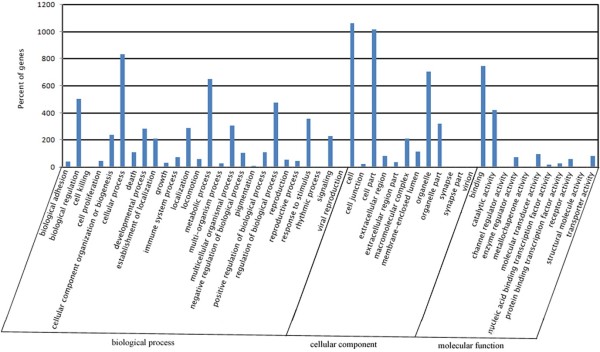
GO functional classification of differentially expressed unigenes in black versus white sheep skin.
A total of 845 novel genes were identified as differentially expressed, of which 738 were down-regulated (≤ 2 fold) and 107 were up-regulated (≥ 2fold) in skin from black sheep compared with skin from white sheep. Of the 107 up-regulated genes, 16 genes were exclusively expressed in black sheep skin (including 2 genes highly expressed), and 26 genes were expressed 8 to 256 times higher in black versus white sheep skin (see Additional file 3: Table S3).
In order to validate the transcriptome sequencing results, we selected 10 genes at random for real time PCR to determine their relative expression in black and white sheep skin. These genes, identified as differentially expressed in black versus white sheep skin based on transcriptome sequencing analysis, included known genes related to coat color in mammals. The results of real time PCR showed that 8 of the 10 selected genes had significantly higher expression in black sheep skin compared with white sheep skin, which was consistent with the transcriptome sequencing data. Among the differentially expressed genes, TYR showed the greatest differential expression between black and white sheep skin (Figure 5).
Figure 5.
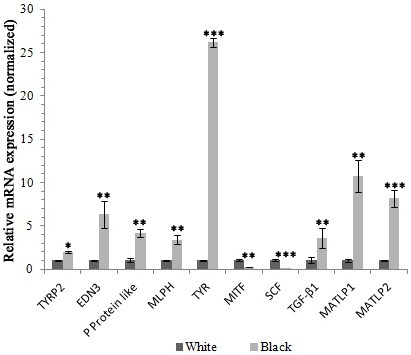
Real time PCR validation of differentially expressed genes in black versus white sheep skin. Abundance of target genes was normalized relative to abundance of â-actin gene. Bars in each panel represent the mean ± standard error (n = 3), * P < 0.05; ** P < 0.01; *** P < 0.001.
KEGG pathway analysis
Of the 2,235 known genes differentially expressed in white versus black sheep skin, 1,903 had a specific KEGG pathway annotation. Of these KEGG pathway annotated genes, 324 were down-regulated in black sheep skin. These down-regulated genes are mainly involved in oxidative phosphorylation, and glycolysis and/or gluconeogenesis. Remaining KEGG pathway annotated genes were associated with 241 pathways including those functionally related to coat color in skin such as melanogenesis, tyrosine metabolism and Wnt signaling. For example, there were 20, 53 and 27 differentially expressed genes involved in tyrosine metabolism, Wnt signaling and melanogenesis pathways, respectively. The enriched GO terms for genes identified in sheep skin transcriptome related to pigmentation and melanogenesis and their relative expression in black versus white skin are shown in Table 4 and Figure 6.
Table 4.
Differentially expressed genes and their GO terms related to pigmentation and melanogenesis in black versus white sheep skin
| GO terms | Genes | Relative expression in black vs white sheep skin | Fold change | FDR p-value | Corrected p-value of GO term |
|---|---|---|---|---|---|
| Developmental pigmentation |
Tyrosinase |
Up-regulation |
96.88 |
1.89 × 10-137 |
0.00273 |
| G-protein coupled receptor 143 |
Up-regulation |
35.63 |
1.32 × 10-28 |
||
| Pallidin-like protein |
Down-regulation |
0.16 |
1.47 × 10-17 |
||
| P protein-like |
Up-regulation |
17.65 |
4.78 × 10-5 |
||
| Melanophilin |
Up-regulation |
27.75 |
1.94 × 10-158 |
||
| Pigment cell differentiation |
Pallidin-like protein |
Down-regulation |
0.16 |
1.47 × 10-17 |
1 |
| P protein-like |
Up-regulation |
17.65 |
4.78 × 10-5 |
||
| Melanophilin |
Up-regulation |
27.75 |
1.94 × 10-158 |
||
| Cellular pigmentation and pigment granule localization |
G-protein coupled receptor 143 |
Up-regulation |
35.63 |
1.32 × 10-28 |
1 |
| Melanophilin |
Up-regulation |
27.75 |
1.94 × 10-158 |
||
| Pigment metabolic process involved in developmental pigmentation and pigmentation |
Tyrosinase |
Up-regulation |
96.88 |
1.89 × 10-137 |
1 |
| G-protein coupled receptor 143 |
Up-regulation |
35.63 |
1.32 × 10-28 |
||
| P protein-like |
Up-regulation |
17.65 |
4.78 × 10-5 |
||
| Melanin metabolic process | Aralkylamine N-acetyltransferase (AANAT) |
Up-regulation |
9.80 |
7.9 × 10-5 |
1 |
| Chain E, Crystal Structure Of The 14-3-3 Zeta:serotonin N- Acetyltransferase |
Up-regulation |
7.84 |
1.44 × 10-6 |
||
| Tyrosinase |
Up-regulation |
96.88 |
1.89 × 10-137 |
||
| P protein-like |
Up-regulation |
17.65 |
4.78 × 10-5 |
||
| Membrane-associated transporter protein-like isoform 1 |
Up-regulation |
32.75 |
1.03 × 10-42 |
||
| Membrane-associated transporter protein-like isoform 2 | Up-regulation | 4099.12 | 3.46 × 10-13 |
Figure 6.

Differentially expressed coat color genes in sheep skin and their involvement in the melanogenesis pathway. Genes with red frame are up-regulated and genes with green frame are down-regulated in black versus white sheep skin.
Differential expression of known coat color genes
Approximately 127 genes involved in different pathways controlling coat color formation have been identified in the mouse [15]. Those known coat color genes are routinely classified into six general functions including: Melanocyte development, Components of melanosomes and their precursors, Melanosome construction/protein routing, Melanosome transport, Eumelanin and pheomelanin and Systemic effects [15]. Expression of a total of 49 of aforementioned coat color genes was detected in sheep skin in present studies, and 13 genes showed higher expression in black sheep skin and 8 genes showed higher expression in white sheep skin. Interestingly, all genes encoding for the components of melanosomes and their precursors had higher expression in black sheep skin (see Additional file 4: Table S4). The coat color genes in the ‘Eumelanin and pheomelanin’ functional category showed higher expression in black sheep skin, while genes in the ‘Melanosome construction/protein routing (HPS-related)’ category displayed lower expression in black sheep skin. Among the coat color genes showing higher expression in black sheep skin, TYRP1 showed the highest expression in black sheep skin versus white sheep skin, followed by TYR, MLPH, MATP and Si (Table 5). The genes associated with oculocutaneous albinism (OCA) such as HPS1, HPS3, HPS4, HPS5 and HPS6 were expressed in sheep skin but most of them did not show differential expression associated with coat color.
Table 5.
Highly up-regulated coat color genes in black sheep skin
| Gene name | Fold change | FDR P-value | Classification | Function |
|---|---|---|---|---|
| Membrane-associated transporter protein (Matp) |
32.75 |
1.03 × 10-42 |
Components of melanosomes and their precursors |
Apparent transporter |
| Silver (Si) |
25.2 |
0 |
Components of melanosomes and their precursors |
Melanosome matrix |
| Tyrosinase (Tyr) |
96.88 |
1.89 × 10-137 |
Components of melanosomes and their precursors |
Melanosomal enzyme |
| Tyrosinase-related protein 1 (Tyrp1) |
284.17 |
0 |
Components of melanosomes and their precursors |
Melanosomal protein |
| Melanophilin (Mlph) | 27.75 | 1.94 × 10-158 | Melanosome transport | Melanosome transport |
Discussion
Mammalian coat color exhibits a wide range of shades and is dictated by melanin production in melanocytes (melanogenesis). Melanogenesis involves a complex molecular regulation [7]. In order to understand the molecular mechanisms of coat color formation, previous studies have reported the generation of ESTs from both sheep and alpaca skin through traditional Sanger sequencing [16,17]. A previous study examined differences in gene expression associated with black spots in fleece of Corriedale sheep using microarray technology [18]. To further investigate genes that may play important roles in sheep skin, particularly in fiber/coat pigmentation, over 100 million transcriptome sequence reads were generated from white and black sheep skin using the Illumina technology. From these reads there were 37,768 known unigenes identified as expressed in sheep skin, among which 2,235 were differentially expressed in black versus white sheep skin. It is acknowledged that study design was not optimal due to limited biological replication because single pooled samples (n = 3 per coat color) were used in transcriptome sequencing analysis and the same three samples from white sheep skin and from black sheep skin were used individually for quantitative real time PCR validation of the sequencing results. Despite such limitations, results have significantly enhanced understanding of sheep skin transcriptome composition and potential differences in gene expression associated with coat color that are foundational to further study in the future.
Genes encoding for ribosomal proteins, keratin family members and keratin associated proteins were among the most highly expressed genes detected in sheep skin. The ribosome is a central player in the translation system and its function is to decode the nucleotide sequence carried by the mRNA and convert it into an amino acid primary structure [19]. Abundant presence of ribosomal proteins in sheep skin suggests the importance of high rates of protein translation in sheep skin. In channel catfish skin, the expression of ribosomal proteins was high presumably due to higher levels of translational activities [20,21]. Of the top 30 highly expressed genes in sheep skin, all 9 keratin family members and keratin associated proteins displayed down regulation in black sheep skin, which was the same as observed in piebald Merino sheep skin [12]. Collectively, results support Garcia’s view that no single keratin gene alone appears to be responsible for the coat color trait [12]. Hair keratins contain a much higher content of cysteine residues in their non-helical domains and thus form tougher and more durable structures via intermolecular disulfide bond formation [22]. Therefore, high expression of keratins is likely crucial for fleece strength. Genes encoding for important oxidative and dehydrolytic enzymes such as NADH5 and COX1 were also highly expressed in sheep skin. The coenzyme NAD (nicotinamide adenine dinucleotide) is a key electron-carrier which mediates hundreds of reactions. The redox state of the NAD–NADH couple plays a central role in energy metabolism [23], signal transduction [24], and transcriptional regulation [25], which is consistent with the need for mitochondrial biogenesis, energy and other proteins during the strong metabolism characteristic of adult sheep skin development [26].
The human hair follicle (HF) has a variable response to potent androgens, such as testosterone (T) and dihydrotestosterone (DHT). The pilosebaceous unit (including HF and sebaceous gland) enzymatically converts weak androgens, such as dehydroepiandrosterone (DHEA) and androstenedione (AD), to more potent androgens, such as T and DHT. In HF of scalp, androgens shorten the anagen growth phase of the hair cycle, causing the HF to regress and recede. The conversion of androgens is dependent on oxidized-reduced pyridine cofactors, NAD, NADH, and NADPH [27]. So, the high level of expression of NADH likely improves the conversion of androgens in certain body regions, influencing terminal hair growth.
Transcription factors (TFs) perform important regulatory functions by controlling a variety of cellular processes [28]. In the mouse genome, 1,445 genes were identified to encode for TFs and 983 were expressed in the brain [29]. In the current studies expression of 527 TF genes was detected in sheep skin, including general TFs such as endothelial differentiation-related factor 1 isoform alpha, DLX3, JUN, ATF4 and GATA3. The high level of expression of these genes detected in skin reflects their importance in regulation of general transcriptional pathways in sheep skin.
Several novel genes were also identified in sheep skin, and a portion of such genes were differentially expressed. Two of the novel genes detected lacked ORF in sequence reads detected, and were highly abundant and exclusively expressed in black sheep skin. BLAST analysis of these 2 novel genes did not find any similar sequences in NCBI database (including EST), suggesting that they could be specific to sheep skin. The differentiated phenotype of melanocytes must be due, at least in part, to differential transcription of melanocyte-specific genes [30]. Thus, these two novel genes may play an important role in promoting pigmentation and dark coat colors.
The GO and KEGG pathway analyses of differentially expressed genes revealed that most were associated with the function of cell and cell part ontology categories. Of particular interest in our dataset were pathways related to pigmentation and melanogenesis. Of the differentially expressed genes, the genes in the category related to ‘the components of melanosomes and their precursor’ and ‘Eumelanin and pheomelanin’ were up-regulated in skin from sheep with black coat color. The function of genes in “the components of melanosomes and their precursor” and ‘Eumelanin and pheomelanin’ are melanin synthesis and the switch between eumelanin and pheomelanin [15]. The darker pigmentation of skin, and possibly of hair, is associated with a higher numbers of melanosomes, although the number of melanocytes remains constant [7,31]. Melanocytes in black hair follicles contain the greatest number of melanosomes (which are eumelanosomes), while the melanosomes in brown hair bulbs are smaller and those in blonde hair are very poorly melanised. The relationship of less melanin with lighter skin/hair phenotype has been reported in several species, including humans [32], alpaca [17], llama [33] and horse [34]. In both domestic sheep and Soay sheep, light coat color is known to be due to a decrease in the ratio of eumelanin to pheomelanin, relative to black coat color [35].
Genes in the ‘Melanosome construction/protein routing (HPS-related)’ ontology category, such as HPS5, Lysosomal trafficking regulator (Lyst) and Pallidin, were all down-regulated in skin of sheep with black coat color. The functions of genes in the ‘Melanosome construction/protein routing (HPS-related)’ categories are related to organelle biogenesis [15]. The key to melanin production is the organelle that is the site of melanogenesis, the melanosome, whose architecture, intracellular distribution and enzyme catalog are critical [30]. HPS5 protein is a component of the biogenesis of lysosome-related organelles complex-2 (BLOC-2) and its deficiency can result in Hermansky–Pudlak syndrome (HPS-5) [36]. HPS is a disorder of lysosome-related organelle (melanosome) biogenesis, resulting in oculocutaneous albinism [37,38]. It has been reported that HPS5 melanocytes have an approximately normal contingent of the melanogenic protein, TYR[36]. Elucidation of the relationship between lower level of expression of HPS5 and other genes in this ontology category with black coat color phenotype will require further investigation.
Among the differentially expressed coat color genes, TYRP1 showed the greatest level of differential expression in black versus white sheep skin. TYRP1, one of the members of the tyrosinase family, is a I type membrane bound protein that is expressed in both melanocytes and the retinal epithelium. TYRP1 is involved in the distal eumelanic pathway and plays a role in stabilizing TYR, which is the critical and rate-determining enzyme in melanogenesis [39]. There existed a significant association between coat color and TYRP1 in Soay sheep [11]. In the free-living Soay sheep, coat color is either dark brown or light tawny color. The light phenotype is determined by homozygosity of a single recessive amino acid change (G → T transversion) at coding position 869 in the TYRP1 gene [11]. This is consistent with studies in domestic sheep, where light coat color is known to be due to a decrease in the ratio of eumelanin to pheomelanin, relative to black coat color [35].
Conclusions
In summary, to our knowledge this is the first report of transcriptome analysis of sheep skin from animals with white and black coat color. The present studies have described and revealed a set differentially expressed known and novel genes in sheep skin potentially related to coat color and other physiological functions. The 16 novel genes exclusively expressed in skin of sheep with black coat color are of particular interest for further studies to elucidate their functional roles in coat color regulation. Results are foundational for future studies to potentially manipulate coat color via pharmacological and genetic approaches.
Methods
Sheep skin sampling and total RNA extraction
Housing and care of sheep and collection of skin samples for use in the described experiments were conducted in accordance with the International Guiding Principles for Biomedical Research Involving Animals (http://www.cioms.ch/frame 1985 texts of guidelines.htm). The animals were locally anaesthetized with hydrochloridum (1.5 ml of 3%, i.h.), following the approval (reference number 2010[088]) provided by the Animal Hospital of Shanxi Agricultural University to decrease the animal suffering. Six healthy 2-year-old white and black female Sunite sheep 3 sheep per color) were selected for sample collection from the sheep farm in Sunite, Inner Mongolia, China. A piece of skin (8 mm in diameter) from the neck was collected via punch skin biopsy under local anesthesia and immediately placed in liquid nitrogen. Total RNA from the sample was extracted using Trizol reagent (Invitrogen, USA) according to the manufacturer’s instructions. The RNA integrity was evaluated by gel electrophoresis and the RNA purity was checked by the ratio of OD260/OD280 and RIN value. RNA samples with RIN value greater than 7.5 and OD260/OD280 ratio greater than 1.7 were selected for deep sequencing.
Library generation and sequencing
Three RNA samples from black or white sheep skin were pooled before mRNA isolation. Beads with Oligo (dT) were used to isolate poly (A) mRNA from sheep skin total RNA. The isolated mRNA was fragmented followed by first-strand cDNA synthesis using random hexamer-primers. The second-strand cDNA was synthesized using buffer, dNTPs, RNaseH and DNA polymerase I. The short cDNA fragments were purified using QiaQuick PCR extraction kit (Qiagen, USA). The fragment ends were repaired and A tailed followed by ligation to sequencing adaptors. Suitable size fragments were selected following agarose gel electrophoresis and used as templates for PCR amplification. Sequencing of the library was performed using Illumina HiSeq™ 2000.
Unigene assembly and functional annotation
Raw reads were cleaned by removing adaptors and low quality reads before assembly. Unigene assembly was carried out using the short reads assembly program, Trinity (http://www.genomics.cn). Blastx alignment (e-value < 0.00001) between the unigenes and protein databases (nr, Swiss-Prot, KEGG and COG) was performed, and the best aligned results were used to decide sequence direction of the unigenes. If results of different databases conflicted with each other, a priority order of nr > Swiss-Prot,>KEGG > COG was followed when deciding sequence direction of the unigenes. If a unigene was not aligned to one of the above databases, ESTScan software was used to determine its sequence direction. Unigene sequences were first aligned by blastx to protein databases and then aligned by blastn to nucleotide database nt (e-value < 0.00001), retrieving proteins with the highest sequence similarity with the given unigenes along with their protein functional annotations. Proteins with the highest ranks in the blast results were taken to determine the coding region sequences of unigenes. Coding sequences were translated into amino acid sequences with the standard codon usage. Gene Ontology (GO) functional annotation was based on nr annotation [40]. Blast2GO program (http://www.blast2go.com) was used to assign GO annotations, and WEGO software (http://wego.genomics.org.cn/cgi-bin/wego/index.pl) was used to perform GO functional classification for all unigenes.
Identification of differentially expressed genes and pathway analysis
A rigorous algorithm based a previously described method [14] was used to identify differentially expressed genes between white and black skin. The FDR (false discovery rate) value of ≤ 0.001 and RPKM ratio of > 2 were used in the analysis [41]. Differentially expressed genes (DEG) were mapped to each term of GO database (http://www.geneontology.org/) and the gene numbers for each GO term were calculated. A list of genes and gene numbers for every GO term was obtained. Hypergeometric test was used to find significantly enriched GO terms in DEG against the genome background. The calculated p-values went through Bonferroni correction, using corrected p-value ≤ 0.05 as a threshold. GO terms fulfilling this condition were defined as significantly enriched GO terms in DEG. With the help of KEGG [42] pathway database (http://www.genome.jp/kegg/pathway.html), the biological complex behaviors of the DEG were further studied.
Validation of mRNA expressed differentially in skin of sheep with white versus black coat color
Ten genes were selected at random from the differentially expressed genes for validation by quantitative real time PCR analysis. Total RNA was isolated from the same 6 sheep skin samples used for RNA sequencing. One μg of DNase-treated RNA was converted to cDNA using oligo dT primer and MMLV cDNA kit mix (TaKaRa, Dalian, China). The cDNA was then used for real time PCR quantification of mRNAs using mRNA specific primers (Additional file 5: Table S5). β-actin was used as an endogenous control. Quantitative real-time PCR was performed in triplicate on the Stratagene Mx3005P system. The 10 μL PCR reaction included 5 μL SYBR Premix Ex TaqTM II (TaKaRa, Dalian, China), 0.5 μL specific forward primer, 0.5 μL reverse primer, 0.5 μL ROX reference dye, 2 μL diluted (4 times) cDNA and 1.5 μL water. Cycling parameters were 95°C for 30 sec, followed by 40 cycles of 95°C for 5 sec, 56°C or 58°C for 20 sec and 72°C for 15 sec. Melting curve analyses were performed following amplifications. Quantification of selected mRNA transcript abundance was performed using the comparative threshold cycle (CT) method [43]. The difference in abundance of mRNA for the genes was determined by analysis of variance.
Abbreviations
MITF: Microphthalmia-associated transcription factor; TYRP1: Tyrosinase-related protein 1; TYR: Tyrosinase; DCT: Tyrosinase-related protein2; MLPH: Melanophilin; MATP: Membrane-associated transporter protein; Si: Silver; MC1R: Melanocortin 1 receptor; ASIP: Agouti/agouti signaling protein.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RWF designed the study, performed the analysis and wrote the paper. JBY and GWS participated in the design of the study, analyzed the data and critically revised the manuscript. JMB, JSX and XYJ performed real time PCR. LY and YFS took part in sample collection and RNA extraction. RB, HDW, WJG, XYH, MH and XT took part in the data analysis and discussion of the manuscript. CSD conceived the study, analyzed the data and participated in writing. All authors read and approved the final manuscript.
Supplementary Material
Transcription factors expressed in sheep skin. List of transcription factors and their expression levels in sheep skin.
Differentially expressed known genes in black versus white sheep. List of known genes expressed differentially with fold change and P value.
Differentially expressed novel genes in black versus white sheep skin. List of novel genes expressed differentially with fold change and P value.
Differentially expressed known coat color genes in black versus white sheep skin. List of coat color genes expressed differentially with fold change and P value.
Primers used for quantitative real time PCR. List of primers for 10 genes used in quantitative real time PCR analysis to verify differential expression of genes identified by RNA-Seq analysis.
Contributor Information
Ruiwen Fan, Email: ruiwenfan@163.com.
Jianshan Xie, Email: xiejianshan008@yahoo.com.cn.
Junming Bai, Email: baibai84aa@163.com.
Haidong Wang, Email: whd1232123@163.com.
Xue Tian, Email: tianxue_81@126.com.
Rui Bai, Email: chinabairui@yahoo.com.cn.
Xiaoyun Jia, Email: gssjxy@hotmail.com.
Lei Yang, Email: yl1984wy1988@163.com.
Yunfei Song, Email: sxjcsyf@126.com.
Muren Herrid, Email: mherrid3@une.edu.au.
Wenjun Gao, Email: wenjungao437@126.com.
Xiaoyan He, Email: hexiaoyan630805@yahoo.com.cn.
Jianbo Yao, Email: jianbo.yao@mail.wvu.edu.
George W Smith, Email: smithge7@msu.edu.
Changsheng Dong, Email: cs_dong@sxau.edu.cn.
Acknowledgements
The study was sponsored by the National Natural Science Foundation of China (30571070, 31201868) and China Postdoctoral Science Foundation funded project (20100481306).
References
- Lamoreux M, Wakamatsu K, Ito S. Interaction of major coat color gene functions in mice as studied by chemical analysis of eumelanin and pheomelanin. Pigm Cell Res. 2002;14(1):23–31. doi: 10.1034/j.1600-0749.2001.140105.x. [DOI] [PubMed] [Google Scholar]
- Bunge R, Thomas DL, Nash TG, Lupton CJ. Performance of hair breeds of sheep in Southern Illinois: wool production and fleece quality. J Anim Sci. 1996;74:25–30. doi: 10.2527/1996.74125x. [DOI] [PubMed] [Google Scholar]
- Kidson S, Fabian B. The effect of temperature on tyrosinase activity in Himalayan mouse skin. J Exp Zool. 2005;215(1):91–97. doi: 10.1002/jez.1402150111. [DOI] [PubMed] [Google Scholar]
- Crepaldi P, Nicoloso L. SNPs in coat colour genes in goats. Ital J Anim Sci. 2010;6(1s):91–93. [Google Scholar]
- Ito S, Wakamatsu K, OZEKI H. Chemical analysis of melanins and its application to the study of the regulation of melanogenesis. Pigm Cell Res. 2000;13:103–109. doi: 10.1034/j.1600-0749.13.s8.19.x. [DOI] [PubMed] [Google Scholar]
- Ito S, Wakamatsu K. Chemistry of Mixed Melanogenesis-Pivotal Roles of Dopaquinone. Photochem Photobiol. 2008;84(3):582–592. doi: 10.1111/j.1751-1097.2007.00238.x. [DOI] [PubMed] [Google Scholar]
- Slominski A, Tobin DJ, Shibahara S, Wortsman J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol Rev. 2004;84(4):1155–1228. doi: 10.1152/physrev.00044.2003. [DOI] [PubMed] [Google Scholar]
- Steingrímsson E, Copeland NG, Jenkins NA. Mouse coat color mutations: from fancy mice to functional genomics. Dev Dynam. 2006;235(9):2401–2411. doi: 10.1002/dvdy.20840. [DOI] [PubMed] [Google Scholar]
- Våge DI, Klungland H, Lu D, Cone RD. Molecular and pharmacological characterization of dominant black coat color in sheep. Mamm Genome. 1999;10(1):39–43. doi: 10.1007/s003359900939. [DOI] [PubMed] [Google Scholar]
- Norris BJ, Whan VA. A gene duplication affecting expression of the ovine ASIP gene is responsible for white and black sheep. Genome Res. 2008;18(8):1282–1293. doi: 10.1101/gr.072090.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratten J, Beraldi D, Lowder B, McRae A, Visscher P, Pemberton J, Slate J. Compelling evidence that a single nucleotide substitution in TYRP1 is responsible for coat-colour polymorphism in a free-living population of Soay sheep. P Roy Soc Lond B Bio. 2007;274(1610):619–626. doi: 10.1098/rspb.2006.3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Gámez E, Reverter A, Whan V, McWilliam SM, Arranz JJ, Kijas J. Using regulatory and epistatic networks to extend the findings of a genome scan: identifying the gene drivers of pigmentation in Merino sheep. PLoS One. 2011;6(6):e21158. doi: 10.1371/journal.pone.0021158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tae-Hun K, Nam-Soon K, Dajeong L, Kyung-Tai L, Jung-Hwa O, Hye-Sook P, Gil-Won J, Hyung-Yong K, Mina J, Bong-Hwan C. Generation and analysis of large-scale expressed sequence tags (ESTs) from a full-length enriched cDNA library of porcine backfat tissue. BMC Genomics. 2006;7:36–44. doi: 10.1186/1471-2164-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res. 1997;7(10):986–996. doi: 10.1101/gr.7.10.986. [DOI] [PubMed] [Google Scholar]
- Bennett DC, Lamoreux ML. The color loci of mice–a genetic century. Pigm Cell Res. 2003;16(4):333–444. doi: 10.1034/j.1600-0749.2003.00067.x. [DOI] [PubMed] [Google Scholar]
- Hu X, Gao Y, Feng C, Liu Q, Wang X, Du Z, Wang Q, Li N. Advanced technologies for genomic analysis in farm animals and its application for QTL mapping. Genetica. 2009;136(2):371–386. doi: 10.1007/s10709-008-9338-7. [DOI] [PubMed] [Google Scholar]
- Fan R, Dong Y, Cao J, Bai R, Zhu Z, Li P, Zhang J, He X, Lü L, Yao J. Gene expression profile in white alpaca (Vicugna pacos) skin. Animal. 2011;5(8):1157–1161. doi: 10.1017/S1751731111000280. [DOI] [PubMed] [Google Scholar]
- Peñagaricano F, Zorrilla P, Naya H, Robello C, Urioste JI. Gene expression analysis identifies new candidate genes associated with the development of black skin spots in Corriedale sheep. J Appl Genet. 2012;53(1):99–106. doi: 10.1007/s13353-011-0066-9. [DOI] [PubMed] [Google Scholar]
- Marshall RA, Aitken CE, Dorywalska M, Puglisi JD. Translation at the single-molecule level. Annu Rev Biochem. 2008;77:177–203. doi: 10.1146/annurev.biochem.77.070606.101431. [DOI] [PubMed] [Google Scholar]
- Patterson A, Karsi A, Feng J, Liu Z. Translational machinery of channel catfish: II. Complementary DNA and expression of the complete set of 47 60S ribosomal proteins. Gene. 2003;305(2):151–160. doi: 10.1016/S0378-1119(02)01183-6. [DOI] [PubMed] [Google Scholar]
- Karsi A, Cao D, Li P, Patterson A, Kocabas A, Feng J, Ju Z, Mickett KD, Liu Z. Transcriptome analysis of channel catfish (Ictalurus punctatus): initial analysis of gene expression and microsatellite-containing cDNAs in the skin. Gene. 2002;285(1):157–168. doi: 10.1016/s0378-1119(02)00414-6. [DOI] [PubMed] [Google Scholar]
- Rouse JG, Van Dyke ME. A review of keratin-based biomaterials for biomedical applications. Materials. 2010;3(2):999–1014. doi: 10.3390/ma3020999. [DOI] [Google Scholar]
- Bakker BM, Overkamp KM, Maris AJA, Kötter P, Luttik MAH, Dijken JP, Pronk JT. Stoichiometry and compartmentation of NADH metabolism in Saccharomyces cerevisiae. FEMS Microbiol Rev. 2006;25(1):15–37. doi: 10.1111/j.1574-6976.2001.tb00570.x. [DOI] [PubMed] [Google Scholar]
- Berger F, Ramýìrez-Hernández MH, Ziegler M. The new life of a centenarian: signalling functions of NAD (P) Trends Biochem Sci. 2004;29(3):111–118. doi: 10.1016/j.tibs.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Piston DW, Goodman RH. Regulation of corepressor function by nuclear NADH. Science Signalling. 2002;295(5561):1895–1897. doi: 10.1126/science.1069300. [DOI] [PubMed] [Google Scholar]
- Harle-Bachor C, Boukamp P. Telomerase activity in the regenerative basal layer of the epidermis in human skin and in immortal and carcinoma-derived skin keratinocytes. PNAS. 1996;93(13):6476–6481. doi: 10.1073/pnas.93.13.6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya ME. Steroid chemistry and hormone controls during the hair follicle cycle. Annny Acad Sci. 1991;642(1):376–383. doi: 10.1111/j.1749-6632.1991.tb24402.x. [DOI] [PubMed] [Google Scholar]
- Han X, Wu X, Chung WY, Li T, Nekrutenko A, Altman NS, Chen G, Ma H. Transcriptome of embryonic and neonatal mouse cortex by high-throughput RNA sequencing. PNAS. 2009;106(31):12741–12746. doi: 10.1073/pnas.0902417106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray PA, Fu H, Luo P, Zhao Q, Yu J, Ferrari A, Tenzen T, Yuk D, Tsung EF, Cai Z. Mouse brain organization revealed through direct genome-scale TF expression analysis. Sci STKE. 2004;306(5705):2255–2257. doi: 10.1126/science.1104935. [DOI] [PubMed] [Google Scholar]
- Jackson IJ. Molecular and developmental genetics of mouse coat color. Annu Rev Genet. 1994;28(1):189–217. doi: 10.1146/annurev.ge.28.120194.001201. [DOI] [PubMed] [Google Scholar]
- Rees JL. Genetics of hair and skin color. Annu Rev Genet. 2003;37(1):67–90. doi: 10.1146/annurev.genet.37.110801.143233. [DOI] [PubMed] [Google Scholar]
- Commo S, Gaillard O, Bernard B. Human hair greying is linked to a specific depletion of hair follicle melanocytes affecting both the bulb and the outer root sheath. Brit J Dermatol. 2004;150(3):435–443. doi: 10.1046/j.1365-2133.2004.05787.x. [DOI] [PubMed] [Google Scholar]
- Cecchi T, Valbonesi A, Passamonti P, Frank E, Renieri C. Quantitative variation of melanins in llama (Lama glama L.) Small Ruminant Res. 2007;71(1–3):52–58. [Google Scholar]
- Sponenberg DP, Ito S, Eng LA, Schwink K. Pigment types of various color genotypes of horses. Pigm Cell Res. 1988;1(6):410–413. doi: 10.1111/j.1600-0749.1988.tb00144.x. [DOI] [PubMed] [Google Scholar]
- Aliev G, Rachkovsky M, Ito S, Wakamatsu K, Ivanov A. Pigment types in selected color genotypes of Asiatic sheep. Pigm Cell Res. 1990;3(4):177–180. doi: 10.1111/j.1600-0749.1990.tb00286.x. [DOI] [PubMed] [Google Scholar]
- Helip-Wooley A, Westbroek W, Dorward HM, Koshoffer A, Huizing M, Boissy RE, Gahl WA. Improper trafficking of melanocyte-specific proteins in Hermansky–Pudlak syndrome type-5. J Invest Dermatol. 2007;127(6):1471–1478. doi: 10.1038/sj.jid.5700737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huizing M, Boissy RE, Gahl WA. Hermansky–Pudlak syndrome: vesicle formation from yeast to man. Pigm Cell Res. 2002;15(6):405–419. doi: 10.1034/j.1600-0749.2002.02074.x. [DOI] [PubMed] [Google Scholar]
- Wei ML. Hermansky–Pudlak syndrome: a disease of protein trafficking and organelle function. Pigm Cell Res. 2006;19(1):19–42. doi: 10.1111/j.1600-0749.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Imokawa G, Bennett DC, Hearing VJ. Tyrosinase stabilization by Tyrp1 (the brown locus protein) JBC. 1998;273(48):31801–31805. doi: 10.1074/jbc.273.48.31801. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann Appl Stat. 2001;29(4):1165–1188. doi: 10.1214/aos/1013699998. [DOI] [Google Scholar]
- Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999;27(1):29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-[Delta][Delta] CT method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transcription factors expressed in sheep skin. List of transcription factors and their expression levels in sheep skin.
Differentially expressed known genes in black versus white sheep. List of known genes expressed differentially with fold change and P value.
Differentially expressed novel genes in black versus white sheep skin. List of novel genes expressed differentially with fold change and P value.
Differentially expressed known coat color genes in black versus white sheep skin. List of coat color genes expressed differentially with fold change and P value.
Primers used for quantitative real time PCR. List of primers for 10 genes used in quantitative real time PCR analysis to verify differential expression of genes identified by RNA-Seq analysis.