

Abstract
Monozygotic (identical) twins have been widely used in genetic studies to determine the relative contributions of heredity and the environment in human diseases. Discordance in disease manifestation between affected monozygotic twins has been attributed to either environmental factors or different patterns of X chromosome inactivation (XCI). However, recent studies have identified genetic and epigenetic differences between monozygotic twins, thereby challenging the accepted experimental model for distinguishing the effects of nature and nurture. Here, we report the genomic and epigenomic sequences in skin fibroblasts of a discordant monozygotic twin pair with Rett syndrome, an X-linked neurodevelopmental disorder characterized by autistic features, epileptic seizures, gait ataxia and stereotypical hand movements. The twins shared the same de novo mutation in exon 4 of the MECP2 gene (G269AfsX288), which was paternal in origin and occurred during spermatogenesis. The XCI patterns in the twins did not differ in lymphocytes, skin fibroblasts, and hair cells (which originate from ectoderm as does neuronal tissue). No reproducible differences were detected between the twins in single nucleotide polymorphisms (SNPs), insertion-deletion polymorphisms (indels), or copy number variations. Differences in DNA methylation between the twins were detected in fibroblasts in the upstream regions of genes involved in brain function and skeletal tissues such as Mohawk Homeobox (MKX), Brain-type Creatine Kinase (CKB), and FYN Tyrosine Kinase Protooncogene (FYN). The level of methylation in these upstream regions was inversely correlated with the level of gene expression. Thus, differences in DNA methylation patterns likely underlie the discordance in Rett phenotypes between the twins.
Introduction
Rett syndrome (RTT) is an X-linked dominant neurodevelopmental disorder that predominantly affects females with an incidence of one in 10,000–15,000 female births [1], [2]. Affected patients manifest various neuro-psychiatric features, including autistic features, epileptic seizures, gait ataxia, and stereotypical hand movements [1]. Mutation of the MECP2 gene (Xq28) accounts for approximately 90% of cases with classic RTT [3].
Twins with clinically well-characterized RTT have been identified [4]–[10]. To date, MECP2 mutations have been confirmed in only two cases, a point mutation (R294X) and a deletion in exon 3, respectively [9], [10]. Phenotypic differences between the twins has been attributed to differences in their X chromosome inactivation (XCI) patterns, with skewing in favor of the paternal allele (the presumptive mutated allele) in the twin with the more severe clinical phenotype [9].
If the concordance rate between monozygotic (MZ) twins approaches 100%, then the trait under study is likely to be under genetic control with high penetrance. For some traits, however, MZ concordance rates are much lower than 100%, indicating a role for environmental and/or stochastic epigenetic factors in modifying the phenotype associated with the underlying genotype. Epigenetic mechanisms are widely believed to mediate the effect of environmental factors on the genome and, as a result, the use of twins in epigenetic research is becoming increasingly popular [11], [12]. Comparative analysis of the epigenome has recently been conducted in twins showing discordances in mental disease [13], [14], multiple sclerosis [15], or without any specific disease [16]. Some of these studies examined DNA methylation at specific imprinted loci [17]–[20], whereas, genome-wide DNA methylation was analyzed in others [14]–[16], [21]. In this report, we compared whole genome sequences and DNA methylation patterns in discordant RTT twins with a MECP2 mutation in order to determine the genetic or epigenetic factor(s) responsible for the inter-twin differences in clinical severity. To our knowledge, this is the first study to make use of this approach in discordant twins with a disease with a disease (Rett syndrome) involving failure of an epigenetic mechanism.
Materials and Methods
Patients’ Samples
Peripheral blood leukocytes and skin fibroblasts were obtained from the twin patients. Written informed consent was obtained from parents of the twins before collection of the patient samples and prior to publishing details of the patient cases. The study protocol was reviewed and approved by the research ethics committees of University of Yamanashi (Nos. 523, 699), National Institute of Genetics (Nos. 22-1, 23-19), and Keio University (No. 20080016). This study was conducted in accordance with the principles expressed in the Declaration of Helsinki.
DNA Sequencing of the MECP2 Gene
DNA was extracted from peripheral blood leukocytes and skin fibroblasts according to standard procedures. PCR was performed using seven primers designed to cover the entire MECP2 coding region as described previously (Amir et al. 1999). The PCR product was purified using a commercial PCR purification kit (Qiagen, Valencia, CA, USA) and bidirectional sequenced using an ABI Prism 310 sequencer (Applied Biosystems, Foster City, CA, USA). Sequence data of the subjects were compared to the published reference genomic sequences (RefSeqGene on chromosome X NCBI Reference Sequence: NG_007107.1).
Generating Mouse Hybrid Cells Containing a Single Patient-derived X-chromosome
The construction of mouse A9 cells containing a single human X chromosome was carried out as reported previously [22]. Briefly, mouse A9 cells were fused with recipient cells for the introduction of a single human chromosome X. The parental origin of a single patient-derived human chromosome X in each hybrid clone was determined by polymorphic analysis.
Bisulfite Treatment
Genomic DNA was extracted using a DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany), and was subjected to sodium bisulfite modification with an EZ DNA Methylation-Gold kit (Zymo Research, CA, USA) as described previously [23].
X-chromosome Inactivation Analyses
The human androgen receptor gene locus (HUMARA) assay using methylation-specific PCR was performed as described previously [24]. Briefly, the assay uses a bisulfite-treatment followed by PCR; the inactive X pattern is based on the methylated allele at the HUMARA locus and the active X pattern is based on the unmethylated allele at the same locus. Both patterns were used to calculate the XCI ratio.
Whole-genome Sequencing
High-quality genomic DNA from the twins were fragmented using a Covaris Acoustic Solubilizer S220. A paired-end library from each sheared DNA was prepared using the Illumina Paired End DNA Sample Prep kit according to the manufacturer’s protocol. The concentration and size distribution of the libraries were quantified using an Agilent 2100 Bioanalyzer. The libraries were run on the Illumina HiSeq2000 sequencers and read length was 100 bp.
Mapping and Variant Calling
Paired-end reads were aligned to the human reference sequence (NCBI Build 36.1) using the Burrows-Wheeler Aligner program [25], and duplicated reads were removed using the Picard program. Variant calling from the pileup file obtained by the SAMtools program [26] was performed using the following filter: (i) paired-end reads were aligned uniquely for at least two reads of each orientation, (ii) mapped read depth for variant calling was 20x or more coverage, (iii) mismatches with a base quality score of less than 13 were discarded. Variants with a read ratio ≥0.3, which is defined as the ratio of variant read depth to total read depth at each position, were genotyped as heterozygous variants and those with a ratio ≥0.8 were genotyped as homozygous variants. The BreakdancerMax-1.1 program [27] was used to detect the structural variants for 10x or more coverage. To detect copy number variants (CNVs), short reads were mapped to the human reference genome, which was masked by RepeatMasker and TRF programs, using mrFAST (v2.0.0.5) and allowing up to 5% mismatches. Then, copy number over 5 kb was predicted by mrCaNaVaR (v0.3).
BeadChip Array Analysis
SNP genotyping and CNV detection were performed using the Illumina Infinium HD Assay system (HumanOmni2.5-Quad). Genomic DNA from the twins were tested on duplicated arrays. Data analysis for SNPs and CNV probes was performed using the Illumina’s GenomeStudio software.
Digital Droplet PCR Assay
The digital droplet PCR assay was performed as described previously [28], [29]. Briefly, target primer pairs were designed using ProbeFinder software from Roche Applied Science. The Droplet Digital PCR workflow begins by partitioning the universal probe library (UPL) probe (Roche, Tokyo, Japan) reaction mix containing DNA into aqueous droplets in oil via the QX100 Droplet Generator; after transfer of droplets to a 96-well PCR plate, a 2-step thermocycling protocol (95°C × 10 min; 40 cycles of [94°C × 30 s, 60°C × 60 s (ramp rate set to 2°C/s)], 98°C × 10 min) is carried out in a conventional thermal cycler, and the PCR plate is then transferred to the QX100 Droplet Reader (a droplet flow cytometer) for automatic reading of samples in all wells. Bio-Rad QX100 reagents and consumables were used for the experiments including droplet generator oil (186–3005), DG8™ cartridges and gaskets (186–3006), droplet reader oil (186–3004), and ddPCR supermix for probes (186–3010). UPL probe assays were used at a final concentration of 1× (1 µM of each forward and reverse target primer; 2 µM of each RNaseP forward and reverse primer; 250 nM of each FAM UPL probe) in all ddPCR reactions. All reactions for each primer set were run in triplicate.
Bisulfite Sequencing
Bisulfite modified DNA was amplified by PCR with the primers MECP2-BF1 (5′-GTTAGGTTTTAGGGTGGGTAATTTT-3′) and MECP2-BR1 (5′-CCCCTCCAACTATTAATTAACTACTTTC-3′), MECP2-BF2 (5′- GAGGTTTTGGTATGTATTTTTTTT-3′) and MECP2-BR2 (5′- ATTACCCACCCTAAAACCTAAC- 3′) specific to the MECP2- promoter region, MKX-BF (5′-TTAGGGTTGTAGGTGTAAAA-3′) and MKX-BR (5′-TACTATTAACCCCTAACAAAAAAAC-3′) specific to the MKX- upstream region, CKB-BF (5′-GATTTGTTTAAGGTTAGGGTAT-3′) and CKBBR (5′-CCTCAAATCCTTAAATATCTAAATCCC-3′) specific to the CKB-upstream region and, FYN-BF (5′-AATTTTTAAAAATATATAGATTTTTTTT- 3′) and FYN-BR (5′-TATCAAAAAACCCCTTAAAAACTAC-3′) specific to the FYN-upstream region using one cycle of 95°C for 10 min, 35 cycles of 95°C for 30 s, 55°C for 30 s, 72°C for 30 s, with a final cycle of 72°C for 7 min. Each PCR product was cloned into a pTAC-2 vector using a TA PCR Cloning Kit (BioDynamics, Tokyo, Japan) and sequenced.
Quantitative Reverse Transcription PCR Assay (qRT-PCR)
Total RNA was reverse-transcribed with random primers and reverse transcriptase (TOYOBO, Osaka, Japan) according to the manufacturer’s instructions. One tenth of the reaction was used in the PCR amplification. Gene expression was measured by qRT-PCR on an ABI Prism 7500 with a THUNDERBIRD SYBR Green PCR mix (TOYOBO) using primers MECP2 (QT00065933, Qiagen), MKX-F (5′-ATCGCACAGACACTCTGGAA-3′) and MKX-R (5′-CCATAGCTGCGTTGATCTCCT-3′), CKB-F (5′-GCCTCACCCAGATTGAAACTCTCT-3′) and CKB-R (5′-GGTTGGGCAGCTTGATATGCAC-3′), FYN-F (5′-TCAAGTCTGACGTGTGGTCTT-3′) and FYN-R (5′-CTCCCGGTTGTTCATGCCT-3′). The expression level of each gene was normalized against that of human GAPDH. All qRT-PCRs were performed three times using duplicated samples. The Mann-Whitney U test was performed to compare the relative expression levels between twins.
Array-based Genome-wide DNA Methylation Analysis
Array-based genome-wide DNA methylation analysis was performed using the Infinium HumanMethylation450 BeadChip (Illumina) according to the manufacturer’s instructions. GenomeStudio normalizes data using different internal controls that are present on the HumanMethylation450 BeadChip. It also normalizes data depending on internal background probes. The methylation status of specific cytosines is indicated by an average (AVG) beta value where 1 corresponds to complete methylation and 0 to no methylation. Signals of probes with P≥0.05 were excluded from the analysis. The comparison of methylation patterns between the twins was based on the difference in (AVG) beta value of each CpG site. DiffScore, a value obtained by an Illumina software (GenomeStudio Methylation module, ver.1.9), is used for illustration of the difference of two groups of data. For a locus covered by multiple probes, the DiffScores across probes were averaged (DiffScore <|374.344|; control = 0). An absolute value >80 was considered to indicate differentially methylated CpG sites between the twins. Methylation microarray data were deposited in the GEO database (accession number GSE43141).
Results
Probands
The twins were delivered spontaneously at 37 weeks of gestation to healthy, non-consanguineous parents, when both parents were 25 years old. Their 16-year-old brother was healthy. Twin 1 (RS1), who has a milder phenotype, developed apparently normally until 2.5 years of age; she was able to use a spoon, to run and jump, and to climb stairs. At age 2, she communicated using two-word sentences. At 2.5 years, she started to lose learned words and the ability to communicate. At 3 years and 4 months, her EEG was abnormal, but she did not have seizures. At 3 years and 5 months, she lost purposeful hand skills and started to exhibit stereotypical hand movements. At 3.5 years, she weighed 12 kg (−1.6 SD), measured 91 cm (−1.1 SD), and had an occipito-frontal circumference (OFC) of 47.5 cm (−0.7 SD). At age 12 years, she had generalized convulsions and her EEG showed epileptic discharges. Since then, an antiepileptic drug has been administered and her seizures are currently well-controlled. At age 13 years, she could run and jump, was rather hyperactive and thus slimmer than twin 2, was able to reach for and grasp desired objects, liked to swim and to watch children’s TV programs.
Twin 2 (RS2), who has a more severe phenotype, had an OFC at birth of 32.3 cm (−0.6 SD). Her development during the first 6 months appeared normal but she soon started to lag. She was able to hold her head steady at 6 months, could roll over at 9 months, and could sit up by herself at 9 months. She had marked hypotonia and never walked. At age 12 months, she spoke using simple words, such as “momma” and “dada,” and could grasp a toy, but she lost these abilities later and started to exhibit stereotypical hand movements. At age 3.5 years, her weight was 11.34 kg (−2.0 SD), her length, 94.9 cm (−2.1 SD), and her OFC, 45.3 cm (−2.1 SD). At 2 years and one month, she had afebrile seizures, and her EEG showed epileptiform spike discharges. At 6 years of age, an antiepileptic drug was administered. At age 7 years, she was unable to stand, walk, speak or communicate with others. At age 13 years, she could not stand and required a wheelchair.
The clinical score used to grade the severity of RTT [30] was 7 for RS1 (milder) and 27 for RS2 (more severe). Based on more recent diagnostic criteria for RTT, RS2 showed the most important criterion (a period of regression) and fulfilled all other main criteria (loss of acquired purposeful hand skills, loss of acquired spoken language, gait abnormalities and stereotypical hand movements) indicating that she has “typical RTT”; by contrast, although RS1 showed the most important criterion (a period of regression), she only fulfilled two of the four main criteria (loss of acquired spoken language and stereotypical hand movements) indicating that she has “atypical RTT” [31]. Twenty-one informative microsatellite markers [32] were genotyped in the twins, and identical alleles were observed at all markers confirming their monozygosity.
Mutation Detection of MECP2 Gene
DNA sequencing analyses demonstrated that both twins had the same frame-shift mutation resulting from a 1 bp deletion (c.806delG, p.Gly269AlafsX288) in exon 4 of MECP2 in the middle of the transcriptional repression domain (Fig. 1A). This mutation has previously been identified in two other RTT patients [33]. The mutation was not found in either the father or the mother (data not shown), indicating that it is de novo.
Figure 1. Mutation analyses of the MECP2 gene in the RTT twins.

(A) DNA sequencing demonstrated that both twin 1 (RS1) and twin 2 (RS2) had the same mutation (1 bp “G” deletion in exon 4). (B) Determination of the parental origin of the mutation. (Upper) Sequencing of MECP2 in a somatic hybrid cell clone carrying an RS1 derived maternal X chromosome. (Lower) Sequencing of MECP2 in a somatic hybrid cell clone carrying an RS1 derived paternal X chromosome. The parent of origin of the X chromosome was determined by the HUMARA X chromosome inactivation assay.
Parent-of-origin of the MECP2 Mutation
To determine whether the de novo mutation occurred in the germ cells of the father or the mother, mono-chromosomal hybrid cell lines were established from the twins. We performed quinacrine plus Hoechst 33258 staining and identified several clones from each twin carrying a single paternal or maternal chromosome X (data not shown). The human androgen receptor (HUMARA) assay (Fig. 1B, left), followed by DNA sequencing analyses (Fig. 1B, right), demonstrated that the MECP2 mutation was paternal in origin. This finding is consistent with previous reports of paternally-derived MECP2 mutations in Rett syndrome patients [34]–[36].
XCI Patterns in Various Tissues of the Twins
Rett syndrome is an X-linked dominant disease caused by a MECP2 mutation. Thus, a favorable XCI pattern is one in which the predominant cell population has the active X with the normal MECP2 allele. This was the pattern expected of twin 1 (RS1) who had the milder phenotype. The opposite pattern, that is, a predominant cell population with the active X carrying the mutant allele, was expected in the second twin (RS2) who showed a more severe phenotype.
In this context, we examined the patterns using the HUMARA assay based on methylation-specific PCR [24]. As a result, the patterns in peripheral blood lymphocytes were 53:47 and 47:53 in RS1 and RS2, respectively, for the active normal allele to active mutant allele (Fig. 2A), and the patterns in skin fibroblasts were 55:45 and 52:48 in RS1 and RS2, respectively (Fig. 2B).
Figure 2. X chromosome inactivation analyses.
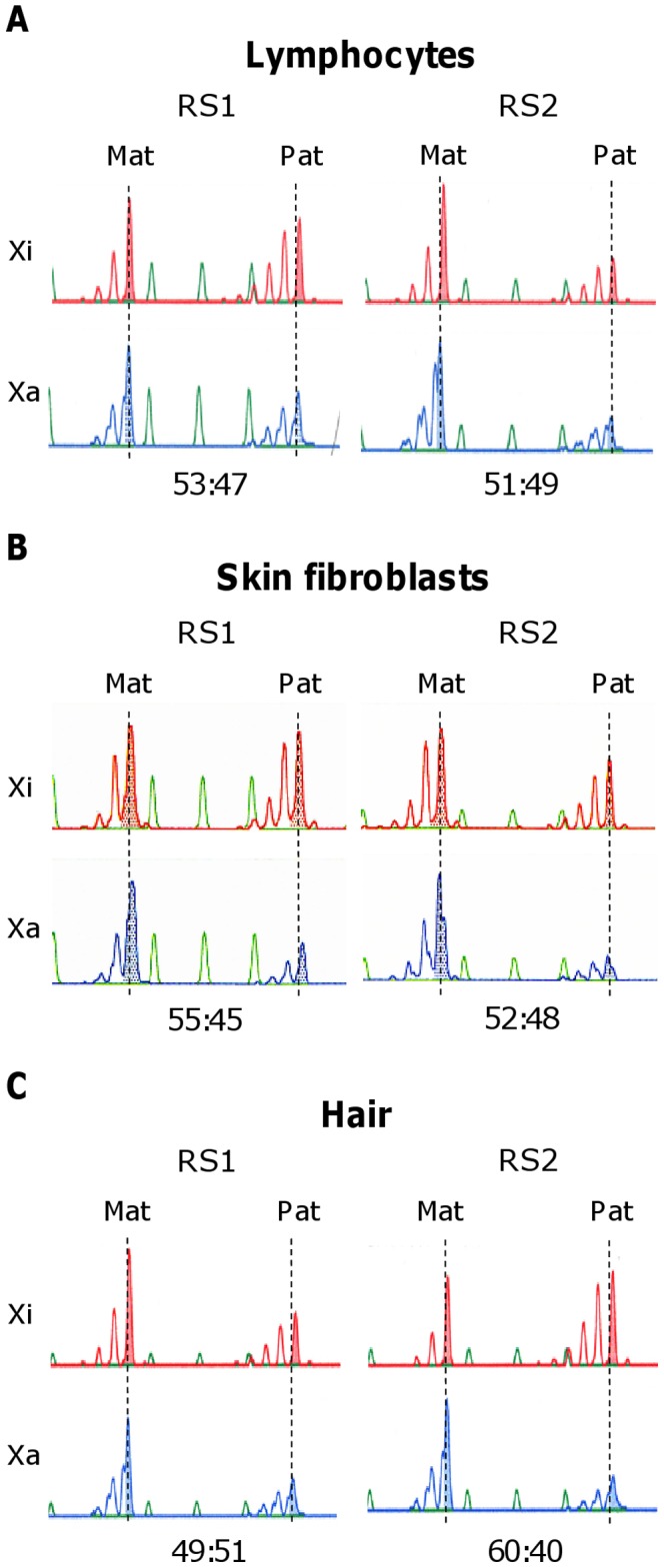
Xi: X inactivation pattern based on the inactive X chromosome, Xa: X-inactivation pattern based on the active X chromosome. Mat: X chromosome inherited from the mother, Pat: X chromosome inherited from the father. Xi and Xa are differentiated by their methylation status at the androgen receptor gene locus. The maternal X and the paternal X are also differentiated by CAG repeat polymorphism at this locus. The X chromosome inactivation patterns showed no differences between the twins in lymphoblasts (A), skin fibroblasts (B), or hair cells (C).
Lymphocytes and skin fibroblasts originate from the embryonic mesoderm, whereas the origin of neuronal tissue (the target tissue of this syndrome) is ectodermal. Since XCI patterns vary among tissues of different embryonic origin but are similar within those of the same embryonic origin [37], we analyzed XCI patterns in hair cells, which are of ectodermal origin. This analysis yielded ratios of 49:51 and 60:40 for RS1 and RS2, respectively (Fig. 2C). Overall, the analyses of the various tissues indicated that there was no difference in XCI patterns between the twins.
SNP Genotyping in the Twins
A total of 24,500,000 chromosomal SNPs were genotyped in the twins using the Illumina Human 610-Quad BeadChips. The overall rate of success for SNP genotyping was 99.99% (24,474,618). In the 24,474,618 SNPs that were successfully genotyped in both twins, there were 19 SNP differences. Each SNP with a difference was either heterozygous in one of the twins and homozygous in the other twin, or homozygous in both twins but with different nucleotides. These putative 19 SNP differences were validated using Solexa whole genome sequencing by HiSeq2000 (Illumina). However, the base sequences of all 19 SNPs were identical in the twins, suggesting typing errors as the source of the 19 SNPs in the Illumina Human 610-Quad BeadChips.
In the Illumina sequencing, the average insert size of each paired-end library was 322 bp (RS1) and 329 bp (RS2) with 100 bp paired-end reads, respectively. After alignment to the human reference genome and removal of PCR duplications, we obtained high-quality nucleotide sequences of 114.9 Gb (40.2× coverage) from RS1 and 103.0 Gb (36.1× coverage) from RS2. The read coverage was 99.9% of the human reference genome (both RS1 and RS2). Comparison of their genomes revealed no specific variants in positions covered with at least 20× depth (at least two reads of each orientation).
Indel Genotyping in the Twins
Using Solexa whole genome sequencing by HiSeq2000 (Illumina), we also investigated the twins for differences with respect to deletions, insertions, inversions, intra-chromosome translocations and inter-chromosome translocations. We detected one difference, a deletion, between the twins. We attempted to validate this putative deletion by PCR using a primer pair encompassing the affected region. However, the PCR failed to demonstrate the deletion, suggesting that the supposed deletion was a detection error in the Solexa whole genome sequencing (Illumina).
CNV Detection in the Twins
In addition to the SNP typing probes, 2,419,193 CNV probes in the Illumina Human 610-Quad BeadChips were used to detect CNVs in the twins. With respect to these CNVs, cnvPartition 1.0.2. demonstrated that 2,410,998 were matched between the twins, whereas 8,195 were not matched. Solexa whole genome sequencing was used to validate these putative 8,195 CNV differences; copy number differences were found for 29 CNVs, with RS1 having three copies and RS2 two copies.
A digital PCR assay was used for further validation of these 29 different CNVs; this analysis found no copy number differences between the twins. Overall, therefore, we could not find any genotypic differences between these monozygotic twins.
DNA Methylation and Expression Differences between the Twins within the MECP2 Region
It is generally accepted that an unfavorable XCI pattern is associated with a more severe RTT phenotype because of the reduced expression of normal MECP2 [38]. As genome-wide differences in DNA methylation have also been reported between MZ twins [39], we postulated that MECP2 expression might also be reduced by hypermethylation of the MECP2 promoter region in the twin that shows the more severe phenotype. However, bisulfite sequencing did not show any DNA methylation difference in the promoter region of fibroblasts from the twins (RS1, 50.3%; RS2, 50.0%) (Fig. 3A). In agreement with the methylation results, a qRT-PCR assay did not find a significant difference in MECP2 expression in the fibroblasts of the twins (Fig. 3B).
Figure 3. DNA methylation and expression analyses for the MECP2 gene.
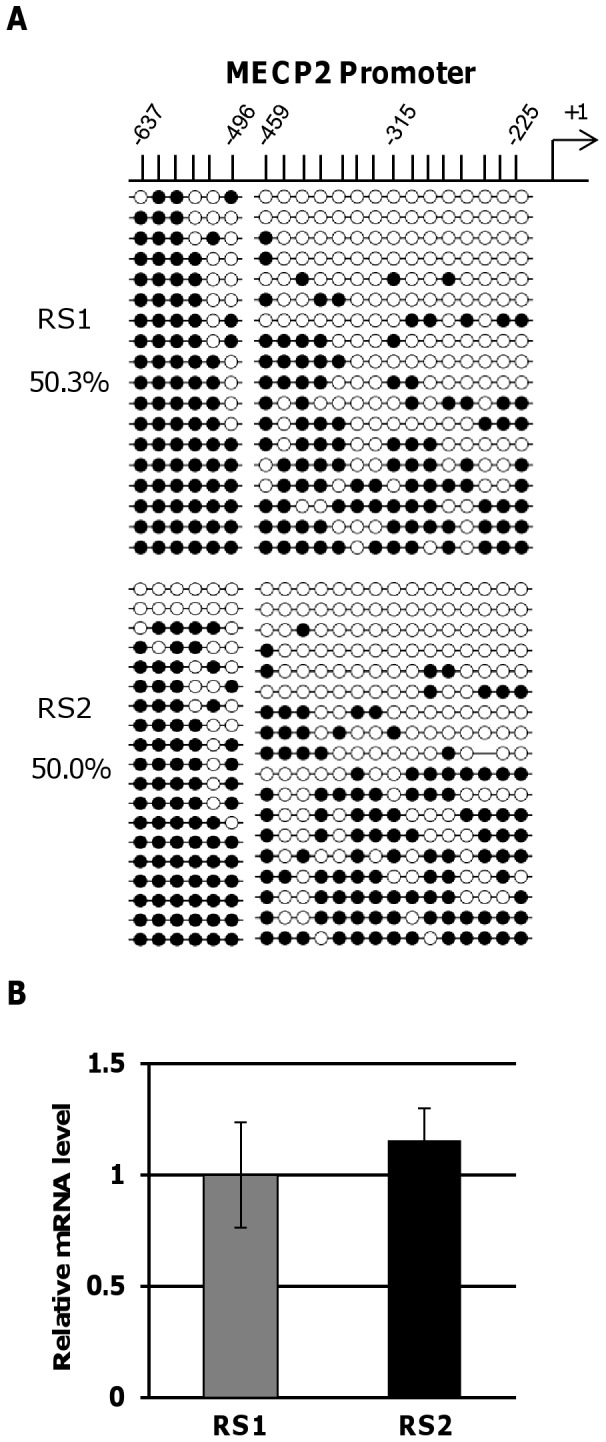
(A) DNA methylation profile in the upstream region of MECP2 by bisulfite sequencing. The upper number indicates the position of the CpG from the transcription start site of MECP2. (B) Results of a qRT-PCR assay for MECP2 gene.
Genome-wide Survey of DNA Methylation Difference between the Twins
Although we did not find any DNA methylation differences within the MECP2 region, it was still possible that such differences were present elsewhere in the genomes of the twins [39], and these differences might alter gene expression patterns to produce the different clinical phenotypes. Therefore, we searched for differentially methylated regions between the twins using an array method that allowed us to determine the DNA methylation status of 450,000 CpG dinucleotide sites in the human genome (mostly located in gene promoter regions) [40].
After normalization and data processing of the array, the full dataset contained 485,577 loci. We found that the methylation levels of these loci mostly matched in the fibroblast samples from RS1 and RS2 (R2 = 0.9612) (Fig. 4A). However, 252 loci showed a distinct DNA methylation difference (DiffScore >|80|) between the twins: RS1 (with the milder phenotype) showed higher DNA methylation than RS2 (with the more severe phenotype) at 100 loci; whereas, RS2 showed higher DNA methylation than RS1 at 152 loci (Fig. 4B).
Figure 4. Results of genome-wide analysis of DNA methylation in the twins.
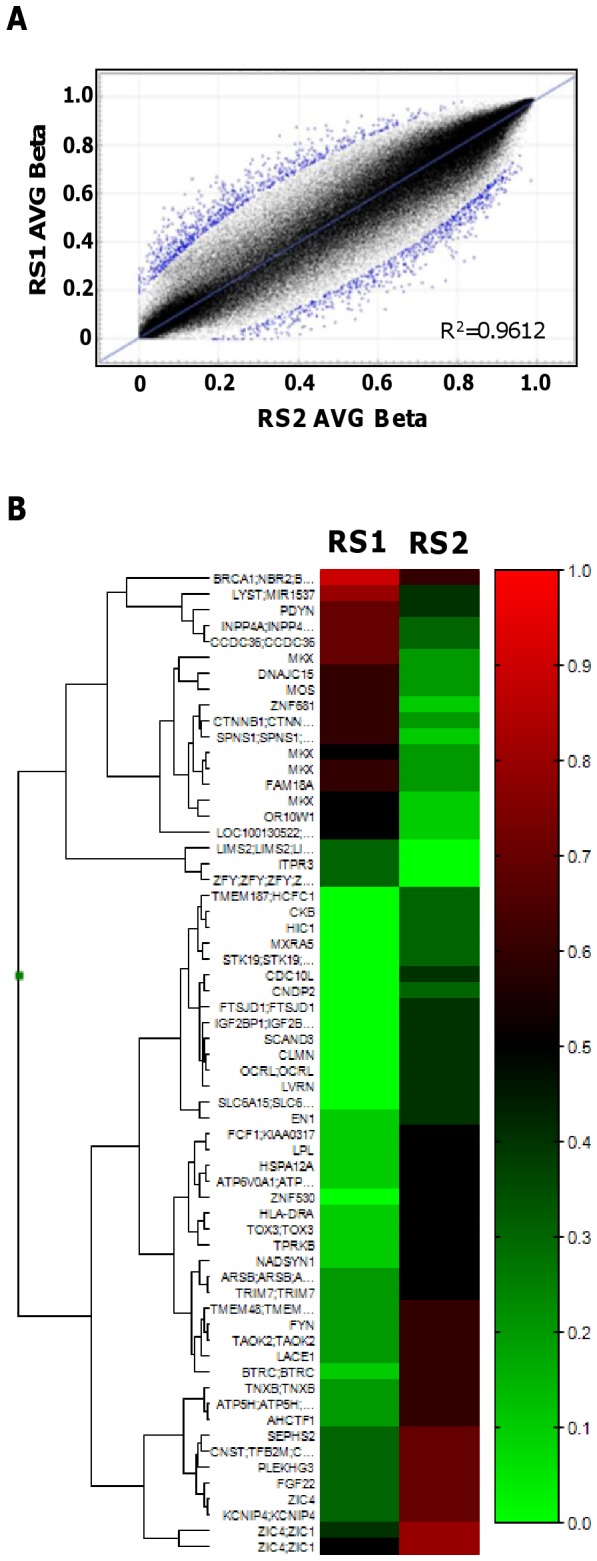
(A) Correlation of DNA methylation at the CpG sites analyzed between the twins based on an average (AVG) beta value. (B) Differentially methylated CpG sites between the twins. Red indicates hypermethylation and green indicates hypomethylation.
Validation of DNA methylation and expression status at selected sites
Among the 252 loci identified in the above analysis, we selected 14 that were located upstream of the transcription start site. We performed a bisulfite sequencing analysis to confirm the methylation status at these 14 loci. Significant differences in methylation status between the twins were confirmed for 3 of the 14 loci, namely Mohawk Homeobox (MKX), Brain-type Creatine Kinase (CKB), and FYN Tyrosine Kinase Protooncogene (FYN). The differentially methylated sites are located upstream of the transcription start site of these genes, which might contribute to the clinical differences between the twins. For the upstream region of MKX, RS1 showed higher DNA methylation than RS2; however, for the upstream regions of CKB and FYN, RS2 showed higher DNA methylation levels (Fig. 5A). These results were consistent with those from the genome-wide methylation analysis. As expected, MKX expression was lower in RS1 than RS2, and CKB and FYN expression was higher in RS1 than RS2 (Fig. 5B). These expression patterns were consistent with the methylation status of each gene obtained from the genome-wide and bisulfite sequencing methylation analyses.
Figure 5. Validation of DNA methylation status and expression difference between the twins.

(A) DNA methylation difference between the twins by bisulfite sequencing in the upstream regions of Mohawk Homeobox (MKX), Brain-type Creatine Kinase (CKB), and FYN Tyrosine Kinase Protooncogene (FYN) genes. The proportion (%) of methylated CpG sites is shown. (B) Expression difference between the twins. *p<0.05.
Discussion
In the present study, we examined the genome, epigenome and expression patterns of MZ twins discordant for RTT. We found that (1) the twins shared the same de novo MECP2 mutation; (2) the de novo mutation was of paternal origin (occurred in spermatogenesis); (3) XCI patterns did not differ in various peripheral tissues between the twins; (4) no inter-twin difference was found in whole genome sequences; (5) there were no differences in DNA methylation of the MECP2 promoter region, nor did MECP2 expression differ between the twins; (6) the DNA methylation status of a number of loci varied between the twins; (7) this DNA methylation difference was confirmed by the effect on expression of three genes, which may contribute to clinical differences between the twins. These results indicate that epigenetic differences, but not genetic differences, appear to be associated with the discordance between these twins.
Thanks to the advances in next generation sequencing technology, several whole genome sequencing studies have now been conducted to identify differences between MZ twins. Whereas SNP differences have not been reported between MZ twins, CNV differences between MZ twins have been reported [40]–[43]. A possible mechanism for this phenomenon can be suggested from the observation that aphidicolin (an inhibitor of eukaryotic nuclear DNA replication) can induce replication stress in normal human cells leading to a high frequency of copy number changes across the human genome that closely resemble CNVs [44]. Thus, CNV differences between twins might be induced by environmental factors. However, there are also lines of evidence that suggest CNVs do not differ between MZ twins [15], [45], [46]. In our study, candidate CNV loci identified by a combination of next-generation sequencing and a CNV/SNP chip assay could not be validated by a digital droplet-based PCR quantitative assay. Our analysis indicates that the DNA sequences in primary skin fibroblasts are stable post-zygotically. This conclusion is supported by a recent study of induced pluripotent stem cells (iPSCs) using a digital droplet PCR assay; this study reported that somatic reprogramming did not lead to de novo CNVs in iPSCs derived from the fibroblasts [29]. Our results also caution that CNV data obtained by next-generation sequencing or a CNV/SNP chip assay should be validated by a quantitative assay.
Initially, DNA methylation analysis was performed at the single gene level by a method based on Southern blotting; subsequently, a methylation-specific PCR assay in combination with sodium-bisulfite treatment of DNA was used [23], [47]. Recently, it has become possible to investigate genome-wide DNA methylation patterns using a method based on a CpG island microarray with sodium-bisulfite treated DNA [48] or with methylated DNA immunoprecipitation (MeDIP) [49]. In this study, we used an assay based on a CpG island microarray with sodium-bisulfite treated DNA. The assay encompassed 450,000 CpG dinucleotides in the human genome, and identified a number of loci with different DNA methylation patterns between the twins. However, the target of this method was mainly gene promoter regions and the coverage was only ∼2% of the human genome. Therefore, it is still possible that there are other regions where DNA methylation differences are more distinct between the twins. Next generation sequencing methods with MeDIP or a new sequencer which allows direct reading of 5-methylcytosine may be able to determine whether this possibility actually occurs. These methods will be useful for ongoing twin cohort studies [50].
In the comprehensive DNA methylation assay, we identified loci with differential methylation in the upstream regions of some genes. We were able to show that the twins showed gene expression differences that were in accord with the methylation patterns of three genes, namely MKX, CKB and FYN.
MKX encodes a transcription repressor that is mainly expressed in the embryonic progenitor cell populations of skeletal muscle, tendon, bone and cartilage, and which is an essential factor for musculoskeletal and tendon differentiation [51], [52]. Knockout mice experiments demonstrated that deficiency of this molecule leads to abnormal tendon sheaths and morphological abnormalities in muscle satellite cells. In our study, over-expression due to promoter hypomethylation was observed in the twin with the more severe phenotype (RS2). Although this analysis was confined to fibroblasts and the significance of its over-expression in these cells is not well understood, it is possible that the expression difference was associated with the clinical differences in bones and muscles between the twins, because the more severely affected individual (RS2) had greater muscle weakness than her sibling (RS1).
CKB encodes a brain isoform of creatine kinase that plays a crucial role in brain energy homeostasis and an important role in GABA neurons by activating the neuron-specific K+-Cl- co-transporter KCC2 [53]. Furthermore, CKB is decreased in the brains of patients with neurodegenerative disorders including Alzheimer disease, Huntington disease, and Pick disease [54]–[56]. CKB also has an important role in the bone-reabsorption function of osteoclasts [57]. Although down-regulation due to DNA methylation was only confirmed in peripheral tissues, it is intriguing to speculate that the decreased CKB expression found in RS2 might contribute to her more severe neurodevelopmental features.
FYN, a member of the Src family of kinases associated with neuronal signaling, regulates phosphorylation and trafficking of N-methyl-D-aspartate receptors (NMDARs) [58], molecules that are important to synaptic plasticity and learning [59]. FYN is also one of the dephosphorylation-target substrates of striatal-enriched protein tyrosine phosphatase (STEP). STEP dysregulation is involved in neuropsychiatric disorders, such as Alzheimer’s disease, schizophrenia, and fragile X syndrome [60]. Therefore, down-regulation of this molecule due to hypermethylation may be associated with the greater mental retardation observed in the more severely affected twin (RS2), although it was obviously not possible to confirm the methylation and expression status in neuronal cells of the twins.
In this study, we mainly targeted upstream promoter regions of genes, because the methylation status of these regions is known to be associated with differences in gene expression patterns. However, a recent study suggested that the methylation status of intragenic (alternative promoter) regions may also be important to the control of gene expression [61]. Therefore, a whole-genome DNA methylation analysis by MeDIP-sequencing will be necessary to determine the influence of possible methylation differences in intragenic regions in the etiology of the discordance between the twins.
DNA methylation studies have been performed in patients with mental diseases, and disease-specific aberrant methylation has been found in neuronal gene regions in these patients [13]. Moreover, it has been reported that there is a correlation between peripheral DNA methylation status and brain function as assessed by positron emission tomography [62]. However, since DNA methylation status varies among tissues, it will definitely be important to determine the specific DNA methylation status of the brains of patients.
A recent development in clinical studies of neurological diseases, including RTT, is to exploit iPSCs in order to understand the consequences of genetic changes in target tissues, such as neuronal cells [63]. Although iPSCs are believed to faithfully recapture the developmental course of the disease of interest, iPSCs cannot maintain stochastic or environmentally-induced alterations in epigenetic status, including DNA methylation, because the epigenetic status is erased during the establishment of iPSC cell lines. Thus, twin-derived-iPSCs will not recapture the original DNA methylation status. It will require further technical breakthroughs, such as development of real-time imaging systems to monitor DNA methylation status in patient brains, to advance our understanding of the role of changes in methylation in the development of neurological diseases.
Acknowledgments
We thank the patient and their parents for their cooperation in this study. We also thank Mr. Hidenori Sato, Mr. Kazushi Endo, Dr. Kazuya Iwamoto, Dr. Tadafumi Kato, Dr. Yusuke Miyanari and Prof. Hiroyuki Sasaki for their valuable advice.
Funding Statement
This work was supported by the Grants-in-Aid for Scientific Research (K.M. and T.K.), the Grant-in-Aid for Exploratory Research (A.T. and T.K.), and the Grant-in-Aid for Scientific Research on Innovative Areas “Genome Science” (A.T. and T.K.) from the Ministry of Education, Culture Sports, Science and Technology, Japan, and the Grant from the Kawano foundation for medical research, Japan (T.K.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Hagberg B, Aicardi J, Dias K, Ramos O (1983) A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases. Ann Neurol 14: 471–479. [DOI] [PubMed] [Google Scholar]
- 2. Bienvenu T, Philippe C, De Roux N, Raynaud M, Bonnefond JP, et al. (2006) The incidence of Rett syndrome in France. Pediatr Neurol 34: 372–375. [DOI] [PubMed] [Google Scholar]
- 3. Zoghbi HY (2005) MeCP2 dysfunction in humans and mice. J Child Neurol 20: 736–740. [DOI] [PubMed] [Google Scholar]
- 4. Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, et al. (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23: 185–188. [DOI] [PubMed] [Google Scholar]
- 5. Tariverdian G, Kantner G, Vogel F (1987) A monozygotic twin pair with Rett syndrome. Hum Genet 75: 88–90. [DOI] [PubMed] [Google Scholar]
- 6. Partington MW (1988) Rett syndrome in monozygotic twins. Am J Med Genet 29: 633–637. [DOI] [PubMed] [Google Scholar]
- 7. Bruck I, Philippart M, Giraldi D, Antoniuk S (1991) Difference in early development of presumed monozygotic twins with Rett syndrome. Am J Med Genet 39: 415–417. [DOI] [PubMed] [Google Scholar]
- 8. Ogawa A, Mitsudome A, Yasumoto S, Matsumoto T (1997) Japanese monozygotic twins with Rett syndrome. Brain Dev 19: 568–570. [DOI] [PubMed] [Google Scholar]
- 9. Ishii T, Makita Y, Ogawa A, Amamiya S, Yamamoto M, et al. (2001) The role of different X-inactivation pattern on the variable clinical phenotype with Rett syndrome. Brain Dev 23 (Suppl 1)S161–164. [DOI] [PubMed] [Google Scholar]
- 10. Mittal K, Kabra M, Juyal R, BK T (2011) De novo deletion in MECP2 in a monozygotic twin pair: a case report. BMC Med Genet 12: 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coolen MW, Statham AL, Qu W, Campbell MJ, Henders AK, et al. (2011) Impact of the genome on the epigenome is manifested in DNA methylation patterns of imprinted regions in monozygotic and dizygotic twins. PLoS One 6: e25590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bell JT, Saffery R (2012) The value of twins in epigenetic epidemiology. Int J Epidemiol 41: 140–150. [DOI] [PubMed] [Google Scholar]
- 13. Kuratomi G, Iwamoto K, Bundo M, Kusumi I, Kato N, et al. (2008) Aberrant DNA methylation associated with bipolar disorder identified from discordant monozygotic twins. Mol Psychiatry13: 429–441. [DOI] [PubMed] [Google Scholar]
- 14. Sugawara H, Iwamoto K, Bundo M, Ueda J, Miyauchi T, et al. (2011) Hypermethylation of serotonin transporter gene in bipolar disorder detected by epigenome analysis of discordant monozygotic twins. Transl Psychiatry 1: e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baranzini SE, Mudge J, van Velkinburgh JC, Khankhanian P, Khrebtukova I, et al. (2010) Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature 464: 1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yu CC, Furukawa M, Kobayashi K, Shikishima C, Cha PC, et al. (2012) Genome-wide DNA methylation and gene expression analyses of monozygotic twins discordant for intelligence levels. PLoS One 7: e47081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Laborie LB, Mackay DJ, Temple IK, Molven A, Søvik O, et al. (2010) DNA hypomethylation, transient neonatal diabetes, and prune belly sequence in one of two identical twins. Eur J Pediatr 169: 207–213. [DOI] [PubMed] [Google Scholar]
- 18. Tierling S, Souren NY, Reither S, Zang KD, Meng-Hentschel J, et al. (2011) DNA methylation studies on imprinted loci in a male monozygotic twin pair discordant for Beckwith-Wiedemann syndrome. Clin Genet 79: 546–553. [DOI] [PubMed] [Google Scholar]
- 19. Ollikainen M, Craig JM (2011) Epigenetic discordance at imprinting control regions in twins. Epigenomics 3: 295–306. [DOI] [PubMed] [Google Scholar]
- 20. Niederhoffer KY, Peñaherrera M, Pugash D, Rupps R, Arbour L, et al. (2012) Beckwith-Wiedemann syndrome in sibs discordant for IC2 methylation. Am J Med Genet A 158A: 1662–1669. [DOI] [PubMed] [Google Scholar]
- 21.Furukawa H, Oka S, Matsui T, Hashimoto A, Arinuma Y, et al.. (2012) Genome, Epigenome and Transcriptome Analyses of a Pair of Monozygotic Twins Discordant for Systemic Lupus Erythematosus. Hum Immunol Nov 28. doi:pii: S0198–8859(12)00614–3. 10.1016/j.humimm.2012.11.007. [Epub ahead of print]. [DOI] [PubMed]
- 22. Kugoh H, Mitsuya K, Meguro M, Shigenami K, Schulz TC, et al. (1999) Mouse A9 cells containing single human chromosomes for analysis of genomic imprinting. DNA Res 6: 165–172. [DOI] [PubMed] [Google Scholar]
- 23. Kubota T, Das S, Christian SL, Baylin SB, Herman JG, et al. (1997) Methylation-specific PCR simplifies imprinting analysis. Nat Genet 16: 16–17. [DOI] [PubMed] [Google Scholar]
- 24. Kubota T, Nonoyama S, Tonoki H, Masuno M, Imaizumi K, et al. (1999) A new assay for the analysis of X-chromosome inactivation based on methylation-specific PCR. Hum Genet 104: 49–55. [DOI] [PubMed] [Google Scholar]
- 25. Li H, Durbin R (2009a) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, et al., 1000 Genome Project Data Processing Subgroup (2009b) The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, et al. (2009) BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods 6: 677–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heredia NJ, Belgrader P, Wang S, Koehler R, Regan J, et al.. (2012) Droplet Digital™ PCR quantitation of HER2 expression in FFPE breast cancer samples. Methods pii: S1046–2023(12)00252–6. doi: 10.1016/j.ymeth.2012.09.012. [Epub ahead of print]. [DOI] [PubMed]
- 29. Abyzov A, Mariani J, Palejev D, Zhang Y, Haney MS, et al. (2012) Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature 492: 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kerr AM, Nomura Y, Armstrong D, Anvret M, Belichenko PV, et al. (2001) Guidelines for reporting clinical features in cases with MECP2 mutations. Brain Dev 23: 208–211. [DOI] [PubMed] [Google Scholar]
- 31. Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, et al. (2010) Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol 68(6): 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sakai J, Hashiyada M, Kanetake J, Takahashi S, Kanawaku Y, et al. (2006) Simulation of kinship tests using the Monte Carlo method. Res Pract Forens Med 49: 213–218. [Google Scholar]
- 33. Auranen M, Vanhala R, Vosman M, Levander M, Varilo T, et al. (2001) MECP2 gene analysis in classical Rett syndrome and in patients with Rett-like features. Neurology 56: 611–617. [DOI] [PubMed] [Google Scholar]
- 34. Girard M, Couvert P, Carrié A, Tardieu M, Chelly J, et al. (2001) Parental origin of de novo MECP2 mutations in Rett syndrome. Eur J Hum Genet. 9: 231–236. [DOI] [PubMed] [Google Scholar]
- 35. Trappe R, Laccone F, Cobilanschi J, Meins M, Huppke P, et al. (2001) MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin. Am J Hum Genet 68: 1093–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhu X, Li M, Pan H, Bao X, Zhang J, et al. (2010) Analysis of the parental origin of de novo MECP2 mutations and X chromosome inactivation in 24 sporadic patients with Rett syndrome in China. J Child Neurol 25: 842–848. [DOI] [PubMed] [Google Scholar]
- 37. Azofeifa J, Waldherr R, Cremer M (1996) X-chromosome methylation ratios as indicators of chromosomal activity: evidence of intraindividual divergencies among tissues of different embryonal origin. Hum Genet 97: 330–333. [DOI] [PubMed] [Google Scholar]
- 38. Knudsen GP, Neilson TC, Pedersen J, Kerr A, Schwartz M, et al. (2006) Eur J Hum Genet. 14: 1189–1194. [DOI] [PubMed] [Google Scholar]
- 39. Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, et al. (2005) Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA 102: 10604–10609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bruder CE, Piotrowski A, Gijsbers AA, Andersson R, Erickson S, et al. (2008) Phenotypically concordant and discordant monozygotic twins display different DNA copy-number-variation profiles. Am J Hum Genet 82: 763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sasaki H, Emi M, Iijima H, Ito N, Sato H, et al. (2011) Copy number loss of (src homology 2 domain containing)-transforming protein 2 (SHC2) gene: discordant loss in monozygotic twins and frequent loss in patients with multiple system atrophy. Mol Brain 4: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Breckpot J, Thienpont B, Gewillig M, Allegaert K, Vermeesch JR, et al. (2012) Differences in Copy Number Variation between Discordant Monozygotic Twins as a Model for Exploring Chromosomal Mosaicism in Congenital Heart Defects. Mol Syndromol 2: 81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ehli EA, Abdellaoui A, Hu Y, Hottenga JJ, Kattenberg M, et al. (2012) De novo and inherited CNVs in MZ twin pairs selected for discordance and concordance on Attention Problems. Eur J Hum Genet 20: 1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Arlt MF, Mulle JG, Schaibley VM, Ragland RL, Durkin SG, et al. (2009) Replication stress induces genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am J Hum Genet 84: 339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ono S, Imamura A, Tasaki S, Kurotaki N, Ozawa H, et al. (2010) Failure to confirm CNVs as of aetiological significance in twin pairs discordant for schizophrenia. Twin Res Hum Genet 13: 455–460. [DOI] [PubMed] [Google Scholar]
- 46. Veenma D, Brosens E, de Jong E, van de Ven C, Meeussen C, et al. (2012) Copy number detection in discordant monozygotic twins of Congenital Diaphragmatic Hernia (CDH) and Esophageal Atresia (EA) cohorts. Eur J Hum Genet 20: 298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kubota T, Sutcliffe JS, Aradhya S, Gillessen-Kaesbach G, Christian SL, et al. (1996) Validation studies of SNRPN methylation as a diagnostic test for Prader- Willi syndrome. Am J Med Genet 66: 77–80. [DOI] [PubMed] [Google Scholar]
- 48. Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H (2011) Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet 88: 450–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sakazume S, Ohashi H, Sasaki Y, Harada N, Nakanishi K, et al. (2012) Spread of X-chromosome inactivation into chromosome 15 is associated with Prader-Willi syndrome phenotype in a boy with a t(X;15)(p21.1;q11.2) translocation. Hum Genet 131: 121–130. [DOI] [PubMed] [Google Scholar]
- 50.Loke YJ, Novakovic B, Ollikainen M, Wallace EM, Umstad MP, et al. (2012 ) The Peri/Postnatal Epigenetic Twins Study (PETS). Twin Res Hum Genet Nov 22: 1–8. [Epub ahead of print]. [DOI] [PubMed]
- 51. Ito Y, Toriuchi N, Yoshitaka T, Ueno-Kudoh H, Sato T, et al. (2010) The Mohawk homeobox gene is a critical regulator of tendon differentiation. Proc Natl Acad Sci U S A 107: 10538–10542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liu W, Watson SS, Lan Y, Keene DR, Ovitt CE, et al. (2010) The atypical homeodomain transcription factor Mohawk controls tendon morphogenesis. Mol Cell Biol 30: 4797–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Inoue K, Yamada J, Ueno S, Fukuda A (2006) Brain-type creatine kinase activates neuron-specific K+-Cl- co-transporter KCC2. J Neurochem 96: 598–608. [DOI] [PubMed] [Google Scholar]
- 54. Aksenov M, Aksenova M, Butterfield DA, Markesbery WR (2000) Oxidative modification of creatine kinase BB in Alzheimer’s disease brain. J. Neurochem 74: 2520–2527. [DOI] [PubMed] [Google Scholar]
- 55. Burbaeva GS Savushkina OK, Boksha IS (2003) Creatine kinase BB in brain in schizophrenia. World J Biol Psychiatry 4: 177–183. [DOI] [PubMed] [Google Scholar]
- 56. Lin YS, Wang CH, Chern Y (2011) Besides Huntington’s disease, does brain-type creatine kinase play a role in other forms of hearing impairment resulting from a common pathological cause? Aging 3: 657–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chang EJ, Ha J, Oerlemans F, Lee YJ, Lee SW, et al. (2008) Brain-type creatine kinase has a crucial role in osteoclast-mediated bone resorption. Nat Med 14: 966–972. [DOI] [PubMed] [Google Scholar]
- 58. Trepanier CH, Jackson MF, MacDonald JF (2012) Regulation of NMDA receptors by the tyrosine kinase Fyn. FEBS J 279: 12–19. [DOI] [PubMed] [Google Scholar]
- 59. Goebel-Goody SM, Baum M, Paspalas CD, Fernandez SM, Carty NC, et al. (2012a) Therapeutic implications for striatal-enriched protein tyrosine phosphatase (STEP) in neuropsychiatric disorders. Pharmacol Rev 64: 65–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Goebel-Goody SM, Wilson-Wallis ED, Royston S, Tagliatela SM, Naegele JR, et al. (2012b) Genetic manipulation of STEP reverses behavioral abnormalities in a fragile X syndrome mouse model. Genes Brain Behav 11: 586–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, et al. (2010) Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466: 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shumay E, Logan J, Volkow ND, Fowler JS (2012) Evidence that the methylation state of the monoamine oxidase A (MAOA) gene predicts brain activity of MAO A enzyme in healthy men. Epigenetics 7: 1151–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, et al. (2010) A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 143: 527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]