

Abstract
Genetic alterations in cancer tissues may reflect the mutational fingerprint of environmental carcinogens. Here we review the evidence that support the role of aristolochic acid (AA) in inducing a mutational fingerprint in the tumor suppressor gene TP53 in urothelial carcinomas of the upper urinary tract (UUT). Exposure to AA, a nitrophenathrene carboxylic acid present in certain herbal remedies and in flour prepared from wheat grain contaminated with seeds of Aristolochia clematitis, has been linked to chronic nephropathy and UUT. TP53 mutations in UUT of individuals exposed to AA reveal a unique pattern of mutations characterised by A to T transversions on the non-transcribed strand, which cluster at hotspots rarely mutated in other cancers. This unusual pattern, originally discovered in UUTs from two different populations, one in Taiwan, and one in the Balkans, has been reproduced experimentally by treating mouse cells that harbour human TP53 sequences with AA. The convergence of molecular epidemiological and experimental data establishes a clear causal association between exposure to the human carcinogen AA and UUT cancer. Despite bans on the sale of herbs containing AA, their use continues, raising global public health concern and an urgent need to identify populations at risk.
Keywords: Aristolochic acid(s), TP53 mutation, nephropathy, herbal remedies, hotspot mutations, urothelial cancer
1. Introduction
Exposure to certain environmental mutagens produces specific mutational patterns, so-called “fingerprints”, in tumor cell DNA. These patterns, defined by the locations, frequencies, types of mutations and strand preference, are influenced by the reactivity of the mutagenic agent, DNA sequence context, epigenetic modifications, DNA repair, replication processes (e.g., translesion DNA synthesis), transcriptional activity, and biological selection [1]. Mutations in the tumor suppressor gene TP53 are among the most frequent mutational events observed in human cancers and, in some cases, reflect the mutational fingerprint of environmental carcinogens [2].
The best documented examples of TP53 mutational fingerprints in human cancers are a) exposure to sunlight and the presence of tandem CC to TT transitions in nonmelanoma skin cancers [3]; b) exposure to polycyclic aromatic hydrocarbon (PAH) through tobacco smoking or smoky coal exposure and G to T transversions in lung cancers, with the mutated guanines located at specific codons on the non-transcribed strand of DNA [4–6]; and c) dietary exposure to aflatoxin B1 and the presence of G to T transversions at the third base of codon 249 (AGG to AGT) in HBV-related hepatocellular carcinoma [7]. Recent genome-wide sequencing analyses have shown that, in tumors of patients exposed to UV or tobacco, the global mutation patterns observed are consistent with fingerprints derived from collating TP53 mutation data from multiple patients exposed to these agents [8;9]. Taken together, these data illustrate the usefulness of mutational analyses of the TP53 gene or of the whole genome in providing clues to prior exposure and in demonstrating causal links between exposure to environmental mutagens and human cancer.
The International Agency for Research on Cancer (IARC) TP53 Database (http://p53.iarc.fr/) includes data on the prevalence and patterns of TP53 mutations in human cancers, annotations of tumor phenotype, patient characteristics, and the structural and functional impact of the mutations [10]. This information can be compared with data generated in cells or in organisms exposed to environmental agents believed to contribute to the mutational burden in humans. Additionally, using cells obtained from mice genetically engineered to harbor human TP53 sequences, iconic TP53 mutation patterns found in human tumors have been reproduced with carcinogens known to initiate the cancers in question [11–13]. Recently, resources such as the International Cancer Genome Consortium (ICGC) data repository (http://www.icgc.org/), containing genome wide data on tumor somatic mutations, have become available, providing valuable tools for studies of factors that influence mutation load and patterns in human cancers.
The disparity in the incidence of various cancers worldwide suggests that environmental and life-style factors play a critical role in human cancers. Data on upper urinary tract tumors (UUT) provide a striking example of this phenomenon. Thus, pockets of high incidence of relatively rare UUT occur in rural villages in the Balkans [14], while, in Taiwan, the incidence of UUT is the highest of any country in the world [15;16]. Among the several environmental agents that have been investigated, ingestion of aristolochic acid (AA) found in Aristolochia plants proved to be the causal agent of UUT in both of these high-risk populations. Mutational analysis of tumors in these two cohorts, coupled with DNA adduct studies [17–21], has provided definitive molecular epidemiologic evidence for incriminating AA in the etiology of UUT [17;19–26]. No such strong molecular, clinical or epidemiologic evidence exists for the myotoxin ochratoxin A (OTA), one of many environmental agents hypothesized as playing a role in the etiology of BEN [27].
The purpose of this article is to describe how patterns of TP53 mutations in UUT patients from Taiwan and the Balkans differ from those observed in other cancers, and how these mutations are recapitulated in experimental models. We will then examine why these observations support the conclusion that AA induces fingerprint TP53 mutations in UUT.
2. Human exposure to Aristolochia
Plants of the genus Aristolochia have been used for medicinal purposes for centuries [24]. All Aristolochia sp contain nitrophenanthrene carboxylic acids, including AAI and AAII (Figure 1; in this article, we refer to either compound or to both as “aristolochic acid”). Importantly, AA-I and AA-II do not have identical biological effects; for example in certain strains of mice, AA-I is nephrotoxic while AA-II is not [28]. However, although Aristolochia clematitis contains significant amounts of both analogs, AL-DNA adducts recovered from the renal cortex of patients with endemic nephropathy contain much less AL-II than AA-I [19]; thus, mutagenicity and carcinogenicity in humans are likely to be attributed to AA-I.
Figure 1.
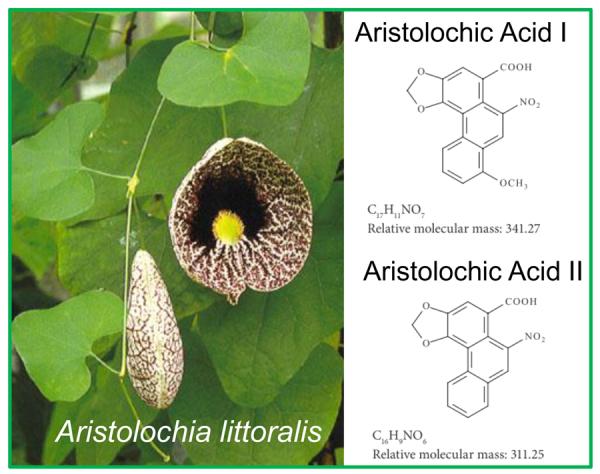
Aristolochia littoralis and structures of aristolochic acids.
A diverse palette of benefits has been sought from Aristolochia herbal remedies, including relief from snakebites, arthritis, and gout, and, prominently, for facilitation of childbirth [22]. In some regions of the world, particularly the Far East, and possibly on the Indian subcontinent, its use continues to be widespread, although challenging to document, particularly where self-administration is commonplace. However, in Taiwan, national prescription databases reveal that millions of Taiwanese have ingested herbal remedies containing Aristolochia [29].
Two decades ago, the inadvertent use of Aristolochia herbs in a weight reduction clinic in Belgium resulted in iatrogenic renal disease, resulting from exposure to AA [30]. The episode drew widespread attention to the nephrotoxic and carcinogenic effects of AA, a substance known since the early 1980s to be tumorigenic in rodents [31;32]. As such, importation of Aristolochia herbs is officially banned in many but not all countries. In the Belgian clinic, an herbal preparation containing Aristolochia fangchi, instead of the intended herb diuretic Stephania tetranda, had been administered as part of the slimming regimen. Within 10 years following this exposure, 100 cases of nephrotoxicity, many accompanied by UUT, occurred in the women who consumed the herbal preparation [30]. In this group of patients, the connection between exposure to AA and development of UUT was well documented. The reactive metabolic intermediate of AA binds covalently to purine bases in DNA, and the adducted base persists in target tissues due to the inability of the global genomic repair system to recognize and excise the lesion [33]. Aristolactam-DNA (AL-DNA) adducts in tissue of patients were detectable years later [30].
Both the cluster of 1800 Belgian women and JD Wang's elegant studies of the general population of Taiwan lend themselves to a quantitative analysis of the dose response relationship. Thus, 1800 Belgian women received 0.025mg/kg of AA a day over a period averaging 13 months with 5% of this cohort developing end stage renal failure up to 85 months after last ingesting Aristolochia fangchi [34]. And, in large-scale population studies in Taiwan, the amount of AA contained in various herbal products has been associated, in a dose-dependent manner with the risk of developing urothelial cancer or chronic renal disease [15;35].
Life-long dietary exposure to AA occurs in rural communities of the Danube river basin. This unusual circumstance provides additional data independently linking ingestion of AA with renal toxicity and UUT [36–38]. Epidemiological evidence, accumulating since the 1950's, suggested that clusters of chronic nephropathy cases in the Balkans (frequently referred to in the literature as Balkan Endemic Nephropathy, BEN) are associated with UUT. Recently, BEN was attributed to the ingestion of bread prepared from wheat grain harvested in fields where Aristolochia clematitis also grows [37]. Detection of AL-DNA adducts in the renal cortex of UUT patients residing in rural areas where nephropathy and UUT are endemic, but not in UUT patients from Zagreb and Belgrade [19;20], firmly established the association between AA exposure, endemic nephropathy and UUT in the Balkans [19–22].
The most recent information linking AA exposure to AL-DNA adducts in tissues of patients with UUT comes from a study of 151 cases in Taiwan [17]. This study presents compelling evidence for a causal link between AA and TP53 mutation patterns in tumor DNA. As detailed in the following section, the TP53 mutation spectrum in AA-UUT display an unusual pattern, readily distinguishable from all the other mutation spectra that have emerged to date from among the >27,000 human tumor mutations in the IARC TP53 database.
3. Mutation Analyses
3.1 In human cancers, somatic A:T>T:A mutations are rare compared with other types of base changes, yet they dominate the mutation pattern in UUT tumors from patients exposed to aristolochic acid
A:T>T:A mutations represent about 5% of somatic TP53 mutations in all types of cancers combined (IARC TP53 Database, R15). The proportion of A:T>T:A mutations among all reported single base substitutions varies among cancer types and, in most cases, the proportion observed in TP53 are similar to that observed in whole genome data extracted from the ICGC data portal (Table 1). Hepatocellular cancers and chronic lymphocytic leukaemia (CLL) show somewhat higher proportions of A:T>T:A mutations than other cancer types, but A:T>T:A transversions account consistently for less than 15% of the total single base substitutions observed (Table 1). Remarkably, A to T transversions constitute 58% (101/173) of TP53 sequence changes found in UUT linked to AA exposure (abbreviated “AA-UUT”), but represent less than 2% (1/68) in UUT patients with no suspected exposure to AA (abbreviated “non-AA-UUT”)(p=4.3E-19 for AA-UUT vs nonAA-UUT), or 5.9% (818/13854) in all other types of cancers combined (p=1.1E-74 for AA-UUT vs other cancers), in accordance with the general rarity of this type of substitution in other types of cancer (Suppl. Table 1). All mutation data in AA-UUT patients, obtained in recent molecular epidemiology studies of the two high risk populations (Taiwan and Balkan rural communities), were concordant despite significant differences in the dose, frequency and timing of exposure [17;19–21]. The AA-UUT data used in the present analyses also include one case of documented exposure in the UK to an Aristolochia-containing herbal preparation [39].
Table 1.
Frequency of human cancer A:T>T:A mutations in the TP53 gene (IARC TP53 Database, R15) and in the whole genome (ICGC data 2012)
| A:T>T:A (% of point 1 mutations) | ||
|---|---|---|
| TP53 | Whole genome | |
| AA-UUT | 58.91 | no data |
| nonAA-UUT | 1.47 | no data |
| Breast | 6.11 | 4.62 |
| CLL * | 8.8 | 13.7 |
| CRC | 3.63 | 5.01 |
| Brain, GBM | 2.2 | 2.63 |
| Liver | 9.65 | 12.46 |
| Lung, ADC | 3.65 | 8.48 |
| Myeloma * | 12.5 | 5.38 |
| Pancreas | 7.98 | 6.85 |
| Stomach | 5.77 | 5.38 |
less than 60 total point mutations
Abbreviations: CLL, chronic lymphocytic leukaemia; CRC, colorectal cancer; GBM, glioblastoma; ADC, adenocarcinoma.
The proportion of A:T>T:A transversions is thus a highly distinguishing feature of the AA-UUT mutation spectrum of TP53. Additionally, TP53 A:T>T:A mutations in AA-UUTs are almost exclusively oriented such that the pre-mutated adenine is positioned on the non-transcribed strand (Suppl. Table 1), suggesting the impact of transcription-coupled repair of AA-derived DNA adducts on the mutation spectrum [40]. A:T>T:A transversions observed in other human cancers do not show this marked strand bias (Suppl. Table 1). Adenine is the principal DNA base subjected to electrophilic attack by metabolically activated aristolochic acid, and A to T transversions are predicted by the known miscoding properties of AL-dA adducts in mammalian cells [41].
3.2 A:T>T:A mutations in upper urinary tract tumors from aristolochic acid-exposed patients occur at hotspots that differ from common sites of A to T base changes in other types of cancers
The set of codons affected by A:T>T:A mutations in AA-UUT is unusual (Figure 2A), compared to those of other types of human tumors (including nonAA-UUT tumors) (Figure 2B). Several reiterated A:T>T:A mutations in the TP53 coding sequences of AA-UUT are conspicuously rare or entirely absent in other tumor types. Of the 101 AA-UUT A:T>T:A mutations, 12 distinct coding mutations are present in two or more patients, yet these reiterated sites are found very rarely in other cancers, and are comparatively underrepresented amongst the 1,167 A:T>T:A mutations in all other tumor types combined (Table 2). Several of these unusual A:T>T:A mutations were, found in two independent studies [17;21], and have been induced in cell cultures treated with AA (mutations in bold in Table 2) as discussed below. Among these, p.N131Y (c.391A>T) is of special note. Eight AA-UUTs harbor this mutation (8/173; 4.6%), yet among >27,000 TP53 mutations reported in the IARC DB (all types of cancers combined worldwide excluding AA-related cancers), only five p.N131Y mutations have ever been described (<0.002%). p.N131Y is thus a promising specific biomarker of AA exposure. Based on experimental assays and prediction models, this mutation is deleterious, causing a complete loss of function (LOF) in p53 transactivation activity [10;42].
Figure 2.
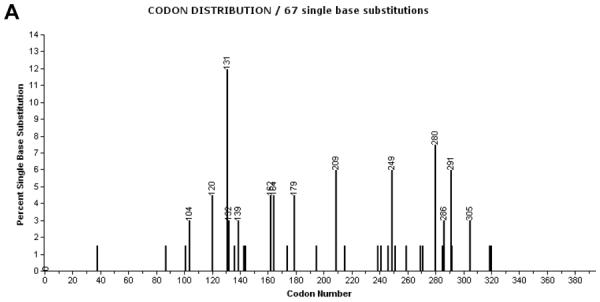
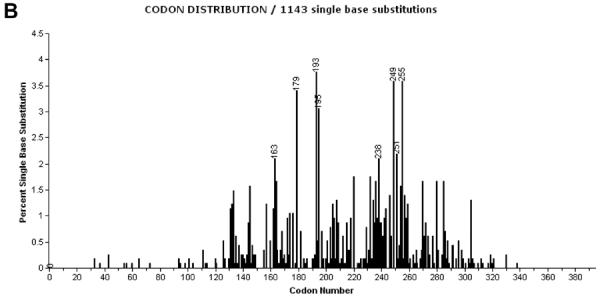
Codon distribution of A:T>T:A substitutions in TP53 coding sequence in (A) AA-UUT cancers [17;19;21;39], and in (B) other cancer types (IARC TP53 Database, R15, excluding AA-related cancers and cell-lines). Codon labels are shown for mutations representing more than 2% of all coding mutations. Note that intronic mutations located at splice sites (first and last nucleotides of introns) are not depicted in this chart.
Table 2.
Recurrent A:T>T:A coding TP53 mutations in AA-UUT
| A:T>T:A mutation | Number of records (%) in AA-UUT | Number of records (%) in Other tumor types (IARC DB) |
|---|---|---|
| p.Q104L * | 2 (1.9%) | 1 (<0.005%) |
| p.K120M | 3 (2.9%) | 1 (<0.005%) |
| p.N131Y | 8 (7.9%) | 5 (<0.005%) |
| p.K139X | 2 (1.9%) | 3 (<0.005%) |
| p.I162F | 3 (2.9%) | 10 (0.008%) |
| p.K164X | 3 (2.9%) | 13 (0.01%) |
| p.H179L | 3 (2.9%) | 32 (0.03%) |
| p.R209X | 4 (3.9%) | 8 (0.007%) |
| p.R249W | 4 (3.9%) | 37 (0.03%) |
| P.R280S | 4 (3.9%) | 11 (0.009%) |
| p.K291X | 4 (3.9%) | 4 (<0.005%) |
| p.K305X | 2 (1.9%) | 15 (0.01%) |
| AUA:T>T:A | 101 | 1167 |
In contrast to A:T>T:A transversions, other types of base substitutions in AA-UUT revealed no unusual features. For example, A:T>G:C transitions have been found in UUT at codons 163, 205 and 220, as in other cancers (Suppl. Figure 1).
Remarkably, more than 30% (34/101) of A:T>T:A mutations reported in AA-UUT occur at splice sites. Taking into account all possible A:T>T:A substitutions in the TP53 sequence, only 3.5% of A:T>T:A mutations would be expected to occur at splice sites, if A:T>T:A transversions occurred at random with no predilection for specific sequence contexts (Table 3). In nonAA-UUT cancers, splice site mutations have not been reported and, in other types of cancer, the overall percentage has been found to be 5%. Thus, splice site mutations in AA-UUT tumors are almost tenfold more frequent than expected, and about sixfold more common than in other cancers.
Table 3.
Expected versus observed A:T>T:A mutations (TP53 exons 2–10) in relation to functional impact of single base substitutions
| Type | Effect | Expected1 | Observed in AA-UUT2 | Observed in Other tumor types (IARC DB)3 |
|---|---|---|---|---|
| A:T>T:A | missense LOF | 18.43 | 34.65 | 64.59 |
| missense others | 54.66 | 9.90 | 18.18 | |
| nonsense | 6.21 | 20.79 | 11.48 | |
| silent | 17.18 | 0.99 | 0.96 | |
| Splice4 | 3.52 | 33.66 | 4.78 | |
| Others | missense LOF | 15.20 | 48.57 | 66.25 |
| missense others | 54.80 | 24.29 | 15.06 | |
| nonsense | 3.06 | 18.57 | 12.03 | |
| silent | 23.84 | 7.14 | 2.55 | |
| splice | 3.13 | 1.43 | 4.11 |
Proportion of mutations (percent, of 483 AT>TA and 2907 other mutations) taking into account all possible single base substitutions in the TP53 sequence (including only exons and first and last two nucleotides of introns)
Proportion of mutations (percent, of 101 AT>TA and 70 other mutations) observed in AA-UUT cancers
Proportion of mutations (percent, of 209 AT>TA and 3141 other mutations) observed in cancers other than UUT (IARC DB)
Splice mutations are defined here as substitutions occurring at the first two and last two nucleotides of introns
The mechanism underlying this striking feature of the AA-UUT mutation spectrum remains unclear. The canonical splice acceptor sequence -ag- and its canonical flanking bases [(Py)-ag-G] may constitute a preferred context for DNA adduction by AA. Indeed, the data imply that the PyAPu motif is a preferred site for AA mutagenesis as analysis of the 101 A:T>T:A transversions in AA-UUT cancers shows that mutation-prone adenines are flanked by a 5'pyrimidine and a 3'purine in most instances [17] (Figure 3A). When A:T>T:A leading to missense and nonsense mutations are considered exclusively, the consensus sequence would be (C/G)APu (Figure 3B).
Figure 3.

Sequence context of A>T mutations in AA-UUT cancers. Motif surrounding the A nucleotide involved in the A>T mutations associated with AA exposure in UUT, taking into account (A) all A>T mutations, or (B) only A>T resulting in missense or nonsense mutations.
Exome and whole genome sequencing of AA-UUT tumors and studies of AA-immortalised cell clones should help to establish whether the PyAPu consensus sequence is inherently vulnerable to mutation by AA. Thus, with whole genome analysis, it should be possible to determine whether A:T base pairs at intronic branch sites, which are in themselves PyAPu motifs, are selectively mutation-prone in genes other than TP53. Curiously, of the 34 TP53 A:T>T:A splice site mutations in AA-UUT, ten were located in the acceptor site of intron-6, and 9 were in intron-8. These sites have no obvious feature that would explain this high incidence of mutations. Moreover, when all other cancer types are combined, base substitutions at TP53 splice sites are only slightly more common at introns 5 and 6 than at other introns (Table 4). Flanking sequences of the acceptor sites of these two introns are very similar (cttagGT and cctagGT). Although prediction tools suggest that altered splicing results from intronic base substitutions (IARC TP53 Database), outcomes of these mutations in human cells are unknown. Therefore, the reason for the high prevalence of AA-UUT splice site mutations remains to be clarified. It is conceivable that splice site mutations in TP53 from other types of tumors are more prevalent than reported in public databases because of a bias towards mutation analyses/experimental designs based on exons or cDNA sequencing [43].
Table 4.
TP53 A:T>T:A mutations among splice site mutations observed in nonAA, non-UUT cancers (IARC DB)
| Intron number | Splice site point mutations (N=326) | A:T>T:A (%) |
|---|---|---|
| 3 | 10 (3.1%) | 1 (3.5%) |
| 4 | 51 (15.6%) | 5 (17.2%) |
| 5 | 74 (22.7%) | 5 (17.2%) |
| 6 | 63 (19.3%) | 6 (20.7%) |
| 7 | 46 (14.1%) | 4 (13.8%) |
| 8 | 48 (14.7%) | 3 (10.3%) |
| 9 | 34 (10.4%) | 5 (17.2%) |
Nonsense mutations resulting from A:T>T:A transversions are three times more numerous than expected in AA-UUTs, and nonsense mutations resulting from other types of base substitutions are six times more numerous in AA-UUT than expected (Table 3). As nonsense and most splice mutations are expected to confer a complete loss of function, these data suggest a strong selection for total loss of the p53 wild-type functions in UUT. Although most tumorigenic mutations show complete loss of function (some also acquire simultaneously a novel oncogenic gain of function), some missense mutations retain partial wild-type p53 ability to transactivate p53-responsive genes [42]. For A:T>T:A and other types of mutations, loss of function due to missense mutations are more frequent than expected in AA-UUT (Table 3), again suggesting a strong requirement for total loss of p53 function.
3.3 AA-induced mutations in experimental systems
Laboratory studies have demonstrated that AL-dA is the predominant DNA lesion in AA-exposed cells, and that the adducted base can miscode during translesion synthesis, thereby generating a T:A base pair at the site of the original A:T pair in subsequent rounds of DNA synthesis [41]. Mutagenesis assays using microorganisms, mammalian cells and genetically engineered laboratory rodents have been used to study the induction of base substitution mutations by environmental agents. Such tests have shown that AA is a potent mutagen (IARC monograph volume 82, 2002). Tumor analysis in an early rodent cancer study of AA demonstrated the activation of ras genes by A:T>T:A transversion at the critical codons. ACB (allele-specific competitive blocker)-PCR, a DNA amplification method that permits detection of mutations present in low abundance in a cell population, recorded the induction by AA of A >T substitutions in codon 61 (CAA to CTA) of the Hras oncogene [44]. Recently, oncogene-activating A>T mutations at the second nucleotide of HRAS codon 61 were observed in a subset of patients with AA-UUT [17].
The frequencies of specific types of base alterations resulting from mutagen exposure in various sequence contexts can be assessed in certain mutagenesis protocols, for example, by sequencing a reporter gene that provides a selection strategy for enumeration of mutants. Transgenic mouse and BigBlue™ rat studies verify that exposure to AA results in a high frequency of A>T transversions [45;46]. Recapitulation of the exposure-linked human tumor TP53 mutation spectrum in laboratory experiments would strengthen arguments for a causal link. However, the strand bias or preferred sequence contexts observed in human cancer genes cannot be recapitulated when certain reporter gene assays are employed (eg, those using nonexpressed transgenes). As no efficient strategies were available for recovering TP53 mutations in cells exposed to a mutagen, one us (MH) constructed the Hupki (human p53 knock-in) mouse, a strain genetically altered to express human TP53 instead of mouse Trp53, and then exploited the ability of primary mouse embryonic fibroblasts to form immortalised clones when p53 is disabled by a mutation [12;47;48]. In this experimental system, Hupki embryonic fibroblasts (HUFs) are exposed in vitro to a mutagen, and the TP53 gene of immortalised clones, which arise after several weeks, is sequenced [48]. The biological process of immortalization serves as a selection strategy for recovering TP53 mutants, analogous to the use of drugs to recover mutants in classical mutation assays. In experiments with several human carcinogens, the HUF assay generated mutation data that were consistent with human tumor TP53 mutation spectra [11;49].
A striking recapitulation of the human TP53 mutational spectrum in HUFs was observed in experiments in which the cells were exposed to AA. Of 37 mutations recovered in four separate experiments, 21 (57%) were A:T>T:A transversions, and 20/21 (95%) involved mutation on the non-transcribed strand. This result is consistent with AA-UUT data (57% vs 58% and 95% vs 98%, respectively). However, the most persuasive data with respect to the specificity of the AA-UUT spectrum is evident from a comparison of A:T>T:A transversion sites in the TP53 sequence. Figure 4 depicts the extent of overlap in A:T>T:A substitution locations in the two datasets. There are 46 observed A:T>T:A distinct mutations in AA-UUT, and 18 in HUF, l1 of which (23.9% and 61.1% respectively) are common to the two datasets (left panel). Of the 11 mutations in common, most were observed in more than two UUT patients (Figure 4, right panel). Notably, of the 12 A:T>T:A transversions found in more than one AA-UUT and thus, potential mutational hotspots (Table 2), ten have been reproduced in the AA-HUF experiments despite the small HUF experimental data set. The concordant transversions include the prominent p.N131Y in AA-UUT discussed above. The selection of TP53 p.N131Y in immortalized HUFs (i.e. fibroblasts) argues against the possibility that p.N131Y is an AA-UUT signature mutation due to special p.N131Y gain-of-function properties uniquely active in urothelial cells. The preferred sequence context for A:T>T:A derived from nonsense and missense mutations in the tumors (Figure 3B) also matches the HUF data. In contrast, among 163 HUF mutant TP53 clones recovered from experiments with other human carcinogens, no A:T>T:A mutations identical to those found in AA-UUT were found [48–52], and unpublished data), although other mutagens specifically targeting A:T base pairs have yet to be tested in this assay.
Figure 4.
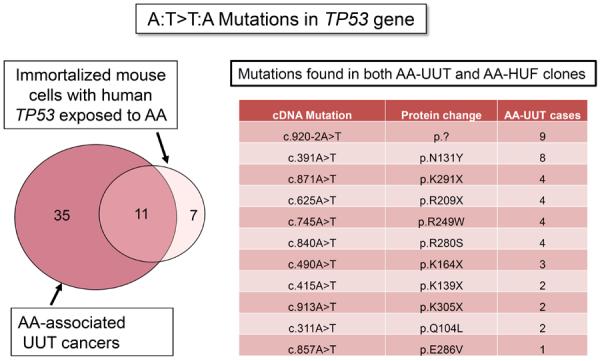
Overlap in identity of specific A to T mutations in AA-UUT and in AA HUFs. Left panel: Overlap between the specific A:T>T:A AA-associated mutations in Hupki (N=18 unique mutations) and in AA-UUT (N=46 unique mutations). Right panel: details on the specific mutations overlapping between Hupki and human data. Mutations are described using NCBI reference sequence NM_000546 for cDNA description and NP_000537 for protein description.
As was found in AA-UUT patients, the proportion of mutations in AA-HUFs generating a stop codon is high (6/37, 16.2%). Curiously, only one intron splice mutation (intron 8 splice acceptor) was detected, a clear departure from the mutation pattern observed in AA-exposed patients. A tentative explanation for this difference may lie in the methods used to screen HUFs and tumors for mutations. Patient DNA was sequenced using the Roche AmpliChip p53 microarray which was designed to detect splice site mutations with high efficiency. Genome sequencing of AA-HUF clones and HUF clones induced by other carcinogens will provide data for comparisons with next-generation sequencing studies in human tumors.
4. Discussion and Future Directions
The most plausible explanation for the unique UUT mutation pattern in the Balkan and Taiwanese populations, reinforced by the strong concordance between tumor mutations in patients and AA-induced TP53 mutations in HUFs, is that AA exposure plays a major role in causing genetic damage to TP53 and in the subsequent development of UUT.
TP53 A:T>T:A mutations have been linked to another environmental carcinogen, vinyl chloride, in angiosarcomas of vinyl chloride-exposed factory workers [53;54]. These data remain very limited (only 14 mutations described) and show different hotspot mutations than the ones observed in AA-UUT. Thus, the pattern of AA-related TP53 mutations appears to be highly specific for AA.
Molecular epidemiological and experimental data supporting the deduction that AA induces A:T>T:A mutations in UUT has parallels in the extensive research performed on aflatoxin B1 exposure and the remarkable hotspot codon 249 TP53 mutation (c.747G>T; p.R249S) found in liver tumors. This single mutation is considered a signature for aflatoxin exposure and a risk marker for hepatocellular carcinoma (HCC) (Table 5). Although A:T>T:A mutations in AA-associated UUT are more dispersed in the TP53 gene than in the aflatoxin B1 (AFB1)-linked hotspot mutation in HCC, distinctive hotspot signature mutations in UUT-AA are evident (Table 2). Thus, the case of AA-UUT represents another compelling example, with strong mechanistic support, of the concept that exposure to environmental carcinogens may generate “signature” mutations in genes that drive tumor development in target tissues. With the advent of nanopore/semiconductor DNA sequencing methods [55] and other next generation sequencing technologies, the analysis of AA-associated mutations in circulating tumor cells, or in exfoliated cells in urine could be explored in strategies for noninvasive measurements and prospective study design. It would also be important to investigate the role of genetic factors in DNA repair and of enzymes involved in forming DNA adducts, thereby shaping mutation patterns and influencing cancer risk [56;57].
Table 5.
Hypothesis: dietary ingestion of AA may result in A:T>T:A mutations in human cancers of the upper urinary tract*
| Strength of association: | Biological plausibility |
|---|---|
|
| |
| Consistency | • AA is a potent mutagen and carcinogen in laboratory studies (IARC monograph, volume 82, 2002) |
| • Dose-response correlation between AA exposure and incidence of UUT [15;34] | |
| • AL-DNA adducts detected in individuals exposed to AA, but not in unexposed cohorts [17–22;30] | • AA is metabolized to an electrophilic intermediate that binds covalently to purines in DNA [57] |
| Specificity | |
| • Frequent A:T>T:A (non-transcribed strand), especially at codons 131, 209, 249, and splice sites in tumors of AA exposed patients, but uncommon in other cancers and in UUT of patients with no AA exposure history (This review; [18]) | • AL-dA DNA adducts are miscoding lesions that produce A>T transversions in vitro [18;22;41] |
| • Treatment of cells with AA induces A:T>T:A mutations with a strong strand bias in TP53 that occur at locations identical to those found in tumors of humans exposed to AA (this review and [48;50]) | |
| • A:T>T:A mutations (“genome scarring”) as a biomarker of UUT cancer risk | |
| Temporality | |
| • A:T>T:A mutations in disease-free individuals predict higher UUT risk (prospective) | |
| • AL-dA adducts predominate, and can persist in tissues for decades because they are poorly repaired [22;40] | • AL-DNA adducts are found in the target organ of exposed patients and experimental animals [20;30;45;46] |
The original table (Table 1 of Hussain et al. [7] presented evidence in support of the hypothesis that aflatoxin B1 exposure can cause codon 249 G>T mutations in the development of hepatocellular carcinoma (HCC). It has been adapted here to the evidence for the role of AA in causing deleterious TP53 mutations in the development of UUT cancer. Italics indicate associations that are lacking which future studies/new technologies might examine with respect to AA exposure and UUT cancer.
Recent investigations in Taiwan raise the possibility that AA-associated UUT cancer is a disease of global proportions, with the number of persons at risk being orders of magnitude higher than hitherto appreciated [17]. In Taiwan alone, one in three persons has used some form of Aristolochia herbal preparations [15]. Moreover, the incidence of UUT in Taiwan is the highest recorded world-wide [15;16]. Case-control studies in Taiwan, with prescription documentation, would be required to investigate a direct relationship between cancer risk and the estimated exposure to Aristolochia species. Despite official bans on the sale and use of Aristolochia herbs for medicinal purposes, self-administration of this widely used herbal remedy continues, adding to the millions of people already exposed.
Supplementary Material
Acknowledgements
MH gratefully acknowledges support from the Deutsches Krebsforschungszentrum (DKFZ) and the Yorkshire Cancer Research charity, UK. MO acknowledges support from IARC. APG and MM are supported by grants from the National Institutes of Environmental Health (ES-04068) and from Henry and Marsha Laufer.
Abbreviations
- AA
Aristolochic acid(s)
- AL-DNA adducts
aristolactam-DNA adducts
- AL-dA
7-(deoxyadenosin-N6-yl)aristolactam
- BEN
Balkan endemic nephropathy
- COSMIC
Catalogue of Somatic Mutations in Cancer
- IARC
International Agency for Research on Cancer
- IARC DB
IARC TP53 Database
- ICGC
International Cancer Genome Consortium
- TP53
the human p53 tumor suppressor gene
- LOF
loss of wild-type function
- Trp53
the mouse p53 tumor suppressor gene
- UUT
upper urinary tract tumors
- AA-UUT
UUT tumors from patient cohorts with documented or suspected exposure to AA
- nonAA-UUT
UUT tumors from patient cohorts with no known or suspected exposure to AA
- HUF
Hupki embryonic fibroblasts
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Vogelstein B, Kinzler KW. Carcinogens leave fingerprints. Nature. 1992;355:209–210. doi: 10.1038/355209a0. [DOI] [PubMed] [Google Scholar]
- [2].Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- [3].Giglia-Mari G, Sarasin A. TP53 mutations in human skin cancers. Hum. Mutat. 2003;21:217–228. doi: 10.1002/humu.10179. [DOI] [PubMed] [Google Scholar]
- [4].Pfeifer GP, Denissenko MF, Olivier M, Tretyakova N, Hecht SS, Hainaut P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking- associated cancers. Oncogene. 2002;21:7435–7451. doi: 10.1038/sj.onc.1205803. [DOI] [PubMed] [Google Scholar]
- [5].Pfeifer GP, Hainaut P. On the origin of G --> T transversions in lung cancer. Mutat. Res. 2003;526:39–43. doi: 10.1016/s0027-5107(03)00013-7. [DOI] [PubMed] [Google Scholar]
- [6].DeMarini DM, Landi S, Tian D, Hanley NM, Li X, Hu F, Roop BC, Mass MJ, Keohavong P, Gao W, Olivier M, Hainaut P, Mumford JL. Lung tumor KRAS and TP53 mutations in nonsmokers reflect exposure to PAH-rich coal combustion emissions. Cancer Res. 2001;61:6679–6681. [PubMed] [Google Scholar]
- [7].Hussain SP, Schwank J, Staib F, Wang XW, Harris CC. TP53 mutations and hepatocellular carcinoma: insights into the etiology and pathogenesis of liver cancer. Oncogene. 2007;26:2166–2176. doi: 10.1038/sj.onc.1210279. [DOI] [PubMed] [Google Scholar]
- [8].Pleasance ED, Stephens PJ, O'Meara S, McBride DJ, Meynert A, Jones D, Lin ML, Beare D, Lau KW, Greenman C, Varela I, Nik-Zainal S, Davies HR, Ordonez GR, Mudie LJ, Latimer C, Edkins S, Stebbings L, Chen L, Jia M, Leroy C, Marshall J, Menzies A, Butler A, Teague JW, Mangion J, Sun YA, McLaughlin SF, Peckham HE, Tsung EF, Costa GL, Lee CC, Minna JD, Gazdar A, Birney E, Rhodes MD, McKernan KJ, Stratton MR, Futreal PA, Campbell PJ. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordonez GR, Bignell GR, Ye K, Alipaz J, Bauer MJ, Beare D, Butler A, Carter RJ, Chen L, Cox AJ, Edkins S, Kokko-Gonzales PI, Gormley NA, Grocock RJ, Haudenschild CD, Hims MM, James T, Jia M, Kingsbury Z, Leroy C, Marshall J, Menzies A, Mudie LJ, Ning Z, Royce T, Schulz-Trieglaff OB, Spiridou A, Stebbings LA, Szajkowski L, Teague J, Williamson D, Chin L, Ross MT, Campbell PJ, Bentley DR, Futreal PA, Stratton MR. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, Olivier M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–629. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- [11].Besaratinia A, Pfeifer GP. Applications of the human p53 knock-in (Hupki) mouse model for human carcinogen testing. FASEB J. 2010;24:2612–2619. doi: 10.1096/fj.10-157263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Luo JL, Yang Q, Tong WM, Hergenhahn M, Wang ZQ, Hollstein M. Knock-in mice with a chimeric human/murine p53 gene develop normally and show wild-type p53 responses to DNA damaging agents: a new biomedical research tool. Oncogene. 2001;20:320–328. doi: 10.1038/sj.onc.1204080. [DOI] [PubMed] [Google Scholar]
- [13].Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use, Cold Spring Harb. Perspect. Biol. 2010;2:a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Petronic V. Tumors of the upper urothelium and endemic nephropathy. In: Radovanovic Z, Sindic M, Polenakovic M, Djukanovic L, Petronic V, editors. Endemic Nephropathy, Institute for Textbook Publishing. Belgrade, Serbia: 2000. pp. 350–439. [Google Scholar]
- [15].Lai MN, Wang SM, Chen PC, Chen YY, Wang JD. Population-based case-control study of Chinese herbal products containing aristolochic acid and urinary tract cancer risk. J Natl. Cancer Inst. 2010;102:179–186. doi: 10.1093/jnci/djp467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yang MH, Chen KK, Yen CC, Wang WS, Chang YH, Huang WJ, Fan FS, Chiou TJ, Liu JH, Chen PM. Unusually high incidence of upper urinary tract urothelial carcinoma in Taiwan. Urology. 2002;59:681–687. doi: 10.1016/s0090-4295(02)01529-7. [DOI] [PubMed] [Google Scholar]
- [17].Chen CH, Dickman KG, Moriya M, Zavadil J, Sidorenko VS, Edwards KL, Gnatenko DV, Wu L, Turesky RJ, Wu XR, Pu YS, Grollman AP. Aristolochic acid-associated urothelial cancer in Taiwan. Proc. Natl. Acad. Sci. U. S. A. 2012;109:8241–8246. doi: 10.1073/pnas.1119920109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Grollman AP. Aristolochic acid nephropathy: Harbinger of a global iatrogenic disease. Environ Mol Mutagen. 2013;54:1–7. doi: 10.1002/em.21756. [DOI] [PubMed] [Google Scholar]
- [19].Grollman AP, Shibutani S, Moriya M, Miller F, Wu L, Moll U, Suzuki N, Fernandes A, Rosenquist T, Medverec Z, Jakovina K, Brdar B, Slade N, Turesky RJ, Goodenough AK, Rieger R, Vukelic M, Jelakovic B. Aristolochic acid and the etiology of endemic (Balkan) nephropathy. Proc. Natl. Acad. Sci. U. S. A. 2007;104:12129–12134. doi: 10.1073/pnas.0701248104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jelakovic B, Karanovic S, Vukovic-Lela I, Miller F, Edwards KL, Nikolic J, Tomic K, Slade N, Brdar B, Turesky RJ, Stipancic Z, Dittrich D, Grollman AP, Dickman KG. Aristolactam-DNA adducts are a biomarker of environmental exposure to aristolochic acid. Kidney Int. 2012;81:559–567. doi: 10.1038/ki.2011.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Moriya M, Slade N, Brdar B, Medverec Z, Tomic K, Jelakovic B, Wu L, Truong S, Fernandes A, Grollman AP. TP53 Mutational signature for aristolochic acid: an environmental carcinogen. Int. J Cancer. 2011;129:1532–1536. doi: 10.1002/ijc.26077. [DOI] [PubMed] [Google Scholar]
- [22].Arlt VM, Stiborova M, vom BJ, Simoes ML, Lord GM, Nortier JL, Hollstein M, Phillips DH, Schmeiser HH. Aristolochic acid mutagenesis: molecular clues to the aetiology of Balkan endemic nephropathy-associated urothelial cancer. Carcinogenesis. 2007;28:2253–2261. doi: 10.1093/carcin/bgm082. [DOI] [PubMed] [Google Scholar]
- [23].De Broe ME. Chinese herbs nephropathy and Balkan endemic nephropathy: toward a single entity, aristolochic acid nephropathy. Kidney Int. 2012;81:513–515. doi: 10.1038/ki.2011.428. [DOI] [PubMed] [Google Scholar]
- [24].Grollman AP, Scarborough J, Jelakovic B. Aristolochic acid nephropathy: an environmental and iatrogenic disease. Adv. Mol. Tox. 2009;3:211–227. [Google Scholar]
- [25].Olivier M, Hollstein M, Schmeiser HH, Straif K, Wild CP. Upper urinary tract urothelial cancers: where it is A:T. Nat. Rev. Cancer. 2012;12:503–504. doi: 10.1038/nrc3311. [DOI] [PubMed] [Google Scholar]
- [26].Schetter AJ, Harris CC. Tumor suppressor p53 (TP53) at the crossroads of the exposome and the cancer genome. Proc. Natl. Acad. Sci. U. S. A. 2012;109:7955–7956. doi: 10.1073/pnas.1205457109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Djukanovic L, Radovanovic Z. Balkan endemic nephropathy. In: De Broe M, Porter G, Bennett W, Verppoten G, editors. Clinical Nephrotoxins. Kluwer, Dordrecht: 2003. pp. 588–601. [Google Scholar]
- [28].Shibutani S, Dong H, Suzuki N, Ueda S, Miller F, Grollman AP. Selective toxicity of aristolochic acids I and II. Drug Metab Dispos. 2007;35:1217–1222. doi: 10.1124/dmd.107.014688. [DOI] [PubMed] [Google Scholar]
- [29].Hsieh SC, Lin IH, Tseng WL, Lee CH, Wang JD. Prescription profile of potentially aristolochic acid containing Chinese herbal products: an analysis of National Health Insurance data in Taiwan between 1997 and 2003. Chin Med. 2008;3:13. doi: 10.1186/1749-8546-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nortier JL, Martinez MC, Schmeiser HH, Arlt VM, Bieler CA, Petein M, Depierreux MF, De PL, Abramowicz D, Vereerstraeten P, Vanherweghem JL. Urothelial carcinoma associated with the use of a Chinese herb (Aristolochia fangchi) N. Engl. J Med. 2000;342:1686–1692. doi: 10.1056/NEJM200006083422301. [DOI] [PubMed] [Google Scholar]
- [31].Mengs U. On the histopathogenesis of rat forestomach carcinoma caused by aristolochic acid. Arch. Toxicol. 1983;52:209–220. doi: 10.1007/BF00333900. [DOI] [PubMed] [Google Scholar]
- [32].Mengs U. Tumour induction in mice following exposure to aristolochic acid. Arch. Toxicol. 1988;61:504–505. doi: 10.1007/BF00293699. [DOI] [PubMed] [Google Scholar]
- [33].Sidorenko VS, Yeo JE, Bonala RR, Johnson F, Scharer OD, Grollman AP. Lack of recognition by global-genome nucleotide excision repair accounts for the high mutagenicity and persistence of aristolactam-DNA adducts. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Martinez MC, Nortier J, Vereerstraeten P, Vanherweghem JL. Progression rate of Chinese herb nephropathy: impact of Aristolochia fangchi ingested dose. Nephrol. Dial. Transplant. 2002;17:408–412. doi: 10.1093/ndt/17.3.408. [DOI] [PubMed] [Google Scholar]
- [35].Lai MN, Lai JN, Chen PC, Hsieh SC, Hu FC, Wang JD. Risks of kidney failure associated with consumption of herbal products containing Mu Tong or Fangchi: a population-based case-control study. Am. J Kidney Dis. 2010;55:507–518. doi: 10.1053/j.ajkd.2009.10.055. [DOI] [PubMed] [Google Scholar]
- [36].Grollman AP, Jelakovic B. Role of environmental toxins in endemic (Balkan) nephropathy. October 2006, Zagreb, Croatia. J Am. Soc. Nephrol. 2007;18:2817–2823. doi: 10.1681/ASN.2007050537. [DOI] [PubMed] [Google Scholar]
- [37].Hranjec T, Kovac A, Kos J, Mao W, Chen JJ, Grollman AP, Jelakovic B. Endemic nephropathy: the case for chronic poisoning by aristolochia. Croat. Med. J. 2005;46:116–125. [PubMed] [Google Scholar]
- [38].Ivic M. Etiology of endemic nephropathy. Lijec Vjesn. 1969;91:1273–1281. [PubMed] [Google Scholar]
- [39].Lord GM, Hollstein M, Arlt VM, Roufosse C, Pusey CD, Cook T, Schmeiser HH. DNA adducts and p53 mutations in a patient with aristolochic acid-associated nephropathy. Am. J Kidney Dis. 2004;43:e11–e17. doi: 10.1053/j.ajkd.2003.11.024. [DOI] [PubMed] [Google Scholar]
- [40].Sidorenko VS, Yeo JE, Bonala RR, Johnson F, Scharer OD, Grollman AP. Lack of recognition by global-genome nucleotide excision repair accounts for the high mutagenicity and persistence of aristolactam-DNA adducts. Nucleic Acids Res. 2012;40:2494–2505. doi: 10.1093/nar/gkr1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Attaluri S, Bonala RR, Yang IY, Lukin MA, Wen Y, Grollman AP, Moriya M, Iden CR, Johnson F. DNA adducts of aristolochic acid II: total synthesis and site-specific mutagenesis studies in mammalian cells. Nucleic Acids Res. 2010;38:339–352. doi: 10.1093/nar/gkp815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R, Ishioka C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci U S A. 2003;100:8424–8429. doi: 10.1073/pnas.1431692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Marcel V, Hainaut P. p53 isoforms - a conspiracy to kidnap p53 tumor suppressor activity? Cell Mol. Life Sci. 2009;66:391–406. doi: 10.1007/s00018-008-8336-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wang Y, Arlt VM, Roufosse CA, McKim KL, Myers MB, Phillips DH, Parsons BL. ACB-PCR measurement of H-ras codon 61 CAA-->CTA mutation provides an early indication of aristolochic acid I carcinogenic effect in tumor target tissues. Environ. Mol. Mutagen. 2012;53:495–504. doi: 10.1002/em.21710. [DOI] [PubMed] [Google Scholar]
- [45].McDaniel LP, Elander ER, Guo X, Chen T, Arlt VM, Mei N. Mutagenicity and DNA adduct formation by aristolochic acid in the spleen of Big Blue(R) rats. Environ. Mol. Mutagen. 2012;53:358–368. doi: 10.1002/em.21696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Xing G, Qi X, Chen M, Wu Y, Yao J, Gong L, Nohmi T, Luan Y, Ren J. Comparison of the mutagenicity of aristolochic acid I and aristolochic acid II in the gpt delta transgenic mouse kidney. Mutat. Res. 2012;743:52–58. doi: 10.1016/j.mrgentox.2011.12.021. [DOI] [PubMed] [Google Scholar]
- [47].Hergenhahn M, Luo JL, Hollstein M. p53 designer genes for the modern mouse. Cell Cycle. 2004;3:738–741. [PubMed] [Google Scholar]
- [48].Liu Z, Hergenhahn M, Schmeiser HH, Wogan GN, Hong A, Hollstein M. Human tumor p53 mutations are selected for in mouse embryonic fibroblasts harboring a humanized p53 gene. Proc. Natl. Acad. Sci. U. S. A. 2004;101:2963–2968. doi: 10.1073/pnas.0308607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].vom Brocke J, Schmeiser HH, Reinbold M, Hollstein M. MEF immortalization to investigate the ins and outs of mutagenesis. Carcinogenesis. 2006;27:2141–2147. doi: 10.1093/carcin/bgl101. [DOI] [PubMed] [Google Scholar]
- [50].Nedelko T, Arlt VM, Phillips DH, Hollstein M. TP53 mutation signature supports involvement of aristolochic acid in the aetiology of endemic nephropathy-associated tumours. Int. J. Cancer. 2009;124:987–990. doi: 10.1002/ijc.24006. [DOI] [PubMed] [Google Scholar]
- [51].Reinbold M, Luo JL, Nedelko T, Jerchow B, Murphy ME, Whibley C, Wei Q, Hollstein M. Common tumour p53 mutations in immortalized cells from Hupki mice heterozygous at codon 72. Oncogene. 2008;27:2788–2794. doi: 10.1038/sj.onc.1210932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Whibley C, Odell AF, Nedelko T, Balaburski G, Murphy M, Liu Z, Stevens L, Walker JH, Routledge M, Hollstein M. Wild-type and Hupki (human p53 knock-in) murine embryonic fibroblasts: p53/ARF pathway disruption in spontaneous escape from senescence. J Biol. Chem. 2010;285:11326–11335. doi: 10.1074/jbc.M109.064444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hollstein M, Marion MJ, Lehman T, Welsh J, Harris CC, Martel-Planche G, Kusters I, Montesano R. p53 mutations at A:T base pairs in angiosarcomas of vinyl chloride- exposed factory workers. Carcinogenesis. 1994;15:1–3. doi: 10.1093/carcin/15.1.1. [DOI] [PubMed] [Google Scholar]
- [54].Weihrauch M, Lehnert G, Kockerling F, Wittekind C, Tannapfel A. p53 mutation pattern in hepatocellular carcinoma in workers exposed to vinyl chloride. Cancer. 2000;88:1030–1036. [PubMed] [Google Scholar]
- [55].Rothberg JM, Hinz W, Rearick TM, Schultz J, Mileski W, Davey M, Leamon JH, Johnson K, Milgrew MJ, Edwards M, Hoon J, Simons JF, Marran D, Myers JW, Davidson JF, Branting A, Nobile JR, Puc BP, Light D, Clark TA, Huber M, Branciforte JT, Stoner IB, Cawley SE, Lyons M, Fu Y, Homer N, Sedova M, Miao X, Reed B, Sabina J, Feierstein E, Schorn M, Alanjary M, Dimalanta E, Dressman D, Kasinskas R, Sokolsky T, Fidanza JA, Namsaraev E, McKernan KJ, Williams A, Roth GT, Bustillo J. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475:348–352. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- [56].Rosenquist TA. Genetic loci that affect aristolochic acid-induced nephrotoxicity in the mouse. Am. J Physiol Renal Physiol. 2011;300:F1360–F1367. doi: 10.1152/ajprenal.00716.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Stiborova M, Frei E, Arlt VM, Schmeiser HH. Metabolic activation of carcinogenic aristolochic acid, a risk factor for Balkan endemic nephropathy. Mutat. Res. 2008;658:55–67. doi: 10.1016/j.mrrev.2007.07.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.