

Abstract
Objective
Autoantibodies against melanoma differentiation-associated protein 5 (MDA5) have been described in several Asian dermatomyositis (DM) cohorts, often associated with amyopathic DM and rapidly progressive interstitial lung disease (ILD). A recent study of a DM cohort seen at a US dermatology clinic reports that MDA5 autoantibodies are associated with a unique cutaneous phenotype. Given the widening spectrum of clinical findings, we evaluated the clinical features of anti-MDA5-positive patients seen at a US myositis referral center.
Methods
160 DM patients were screened for MDA5 autoantibodies by immunoprecipitation and antibody titers were analyzed in longitudinal serum samples. Anti-MDA5 positive patients were evaluated for the presence of additional myositis autoantibodies. Patient clinical characteristics were compared by retrospective chart review.
Results
MDA5 was targeted in 11/160 (6.9%) patients with DM. Of these, nine presented with a symmetric polyarthropathy, six demonstrated overt clinical myopathy and eight had ILD. Eight anti-MDA5-positive patients exhibited the clinical attributes of the antisynthetase syndrome in the absence of Jo-1 or other anti-synthetase autoantibodies. MDA5 autoantibody titers did not correlate with clinical course.
Conclusions
MDA5 autoantibodies are found in DM patients presenting with a symmetric polyarthritis, clinically similar to rheumatoid arthritis. These patients often have features of the antisynthetase syndrome, but in the absence of antisynthetase autoantibodies. Most anti-MDA5 positive patients had overt clinical myopathy and ILD. The latter, while occasionally severe, typically resolved with immunosuppressive therapy. In this cohort, the MDA5 phenotype is frequently a clinical mimic of the antisynthetase syndrome and is not associated with rapidly progressive ILD.
DM is a systemic autoimmune disease that affects muscle, lungs and skin to varying extents in different patients. Like many other systemic autoimmune diseases, DM patients frequently have specific autoantibodies which are strongly associated with distinct clinical phenotypes, making autoantibodies useful for disease diagnosis and prognosis (1). For example, autoantibodies which recognize Mi-2 are associated with a more severe cutaneous form of DM which responds favorably to therapy (2–4), while antibodies against the aminoacyl tRNA synthetases are associated with a clinical phenotype termed the “antisynthetase syndrome”, consisting of myopathy, fever, ILD, Raynaud’s phenomenon, non-erosive arthritis and mechanics hands (4–6).
Autoantibodies against the interferon (IFN)-inducible antigen MDA5, have recently been described in 10–20% of Japanese DM patients. Anti-MDA5 antibody-positive patients had predominantly amyopathic DM and a high risk for ILD, including rapidly progressive ILD which was frequently fatal (7–11) (reviewed in (12)). To date, only one US cohort of DM patients has been systematically evaluated with regard to prevalence of MDA5 autoantibodies and the associated clinical attributes (13). These patients were drawn from an academic dermatology practice, and similar to the Japanese experience, 13% of DM patients had MDA5 autoantibodies. Consistent with previous reports, these patients were more likely to be amyopathic and have ILD. In addition, they demonstrated a characteristic cutaneous phenotype consisting of skin ulceration, tender palmar papules, or both.
We sought to determine the prevalence of MDA5 autoantibodies in a cohort of 160 DM patients evaluated at a tertiary referral US myositis specialty center, and to define the clinical features of these patients. In this cohort, anti-MDA5 antibody positive patients often demonstrated hallmark features of the antisynthetase syndrome in the absence of tRNA synthetase autoantibodies. ILD was less severe than previously reported (7–11), and was absent completely in some patients over years of follow-up. Thus, anti-MDA5-associated myositis should be strongly considered when a patient with features of the antisynthetase syndrome is negative for anti-synthetase antibodies.
MATERIALS AND METHODS
Patients and sera
160 consecutive patients with a Bohan and Peter diagnosis of definite or probable DM (14, 15) or a diagnosis of amyopathic or hypomyopathic DM by Sontheimer’s criteria (16) were evaluated. Patients underwent routine clinical care at the Johns Hopkins Myositis Center between 2006 and 2012, and provided serum samples for research, which were stored at −80°C. Normal sera from 32 donors were also used for this study. Informed consent was obtained from all subjects, and all samples were obtained under the auspices of Johns Hopkins Medicine Institutional Review Board-approved protocols. Clinical information was retrospectively retrieved from all patients by medical record review.
Assessment of muscle disease
Muscle disease was evaluated clinically by strength assessment using the Medical Research Council 5-point scale in addition to electrophysiologic testing, radiographic assessment by muscle MRI, and laboratory testing for serum muscle enzymes, and when deemed clinically appropriate, by muscle biopsy. All patients with longitudinal follow-up were subsequently re-assessed by the same physician.
Assessment of interstitial lung disease
Patients with MDA5 autoantibodies were evaluated for the presence of radiographic findings consistent with ILD including radiologist documented ground glass opacities, reticulation or honeycombing (17). Pulmonary function testing (PFT), when available, was reviewed for the presence of restriction (FVC<80% predicted in the absence of obstruction) and decreased DLCO (<80% predicted). Patients were considered to have ILD if defining features on CT were present.
Immunoprecipitation using 35S-methionine labeled proteins generated by in vitro transcription and translation (IVTT)
cDNAs encoding full length human MDA5, Mi2, NXP2 and Ro60 were used to generate 35S-methionine-labeled proteins by IVTT, per the manufacturer’s protocol (Promega). Immunoprecipitations using these products were performed as described (13) and the immunoprecipitates were electrophoresed on 10% SDS-polyacrylamide gels and visualized by fluorography. For densitometry, X-ray films were scanned using an AGFA Arucs II scanner and densities were quantified using BioRad Quantity One software.
Immunoprecipitation from radiolabeled HeLa cell extracts
HeLa cells were cultured using standard protocols. In some cases, cells were treated with purified leukocyte IFNα(Sigma) [1000U/ml] for 22 hours before beginning the radiolabeling protocol. HeLa cells were radiolabeled for 2 hr with 35S-methionine/cysteine; where appropriate, IFNα was also added to the radiolabeling incubations (bringing the total treatment time with IFNα to 24 hours). Cells were lysed in RIPA buffer (50 mM Tris pH7.4, 150 mM NaCl, 5 mM EDTA, 0.5% Nonidet P40, 0.5% sodium deoxycholate, 0.1% SDS), and precleared with immobilized Protein A agarose (Thermo Scientific). Immunoprecipitations were performed by adding 1 μl of patient serum to the lysate (1 hr at 4°C), followed by Protein A agarose (20 mins, 4°C), electrophoresis on 10% SDS-polyacrylamide gels and visualization of radiolabeled immunoprecipitates by fluorography. An important difference between immunoprecipitations performed using IVTT proteins and radiolabeled cell lysates is that the latter detects all antibody specificities present in the patient serum (e.g. anti-synthetases, MDA5, Ro52 etc), whereas the former reads out antibodies only against the single input protein.
ELISA
Ro52 and Jo-1 antibodies were assessed by ELISA, using commercially available kits (INOVA Diagnostics), according to the manufacturer’s protocol.
Statistical analysis
Χ2 (and, where appropriate, Fisher’s exact test) and independent 2-sample t-tests were used for comparisons of frequencies. p-values of ≤ 0.05 were considered statistically significant. Each clinical feature was tested separately with the statistical methods mentioned such that the correction for multiple comparisons was not performed. Comparison of MDA5-positive cohorts was performed using a one-sample test of proportion.
RESULTS
Determination of MDA5 autoantibody prevalence and association with known myositis autoantigens
In this cohort, patients were referred with a confirmed or suspected diagnosis of myositis and presented with muscle weakness, rash, and/or ILD. The demographics of the 160 DM patients, at the time of presentation, are shown in Table 1. To determine MDA5 autoantibody prevalence we performed immunoprecipitation (IP) of IVTT-generated proteins using patient sera. IP of IVTT radiolabeled proteins is a sensitive method to determine the presence of defined autoantibodies (13, 18–20) since autoantibody detection by immunoblotting typically underestimates actual antibody frequency (21). MDA5 antibodies were detected in 11/160 (6.9%) patients with DM and 0/32 control sera (Fig. 1A; all anti-MDA5 antibody positive sera are shown (DM 1–11), and 2 representative anti-MDA5 antibody negative sera (DM 12 & 13) and 2 controls). Of note, these antibodies were high titer in 9/11 sera, whereas the levels in the remaining two (DM4 and DM8) were low. Longer autoradiographic exposures (not shown) demonstrated clearly that the levels of IVTT 35S-MDA5 immunoprecipitated by these two sera were above the background obtained using control sera or anti-MDA5 antibody negative sera.
Table 1.
Demographic characteristics of DM patients
| N (%) | |
|---|---|
| Gender | |
| Male | 42 (26.3) |
| Female | 118 (73.8) |
| Race | |
| Caucasian | 132 (82.5) |
| Native American | 1 (0.6) |
| Hispanic or Latino | 3 (1.9) |
| Pacific Islander | 0 (0) |
| Asian | 5 (3.1) |
| African American | 19 (11.9) |
| Mean (S.D.) age at diagnosis, y | 44.7 (15.9) |
| Median disease duration, m | 8 (range: 1 m – 28.8 y) |
y = years; m = months.
Figure 1. Detection of MDA5 antibodies using two different immunoprecipitation (IP) methods.

(A) IP using 35S-methionine labeled MDA5 generated by in vitro transcription and translation (IVTT). Sera from 160 DM patients and 32 controls were assayed by IVTT IP. All 11 anti-MDA5 antibody-positive sera (DM 1–11) are shown here, as well as 2 representative anti-MDA5 antibody negative DM sera (DM 12 & 13), and 2 normal controls (C1, C2). On longer autoradiographic exposures, the signals detected by DM 4 and 8 were robust and well above negative controls (not shown). (B) Untreated (lanes denoted “−”) and IFNα-treated (lanes denoted “+”) HeLa cells were radiolabeled and cell lysates were immunoprecipitated with 4 different anti-MDA5 antibody-positive sera. Representative results from DM 8, 9, 10 and 11 are shown. Open triangles denote MDA5 and filled triangles denote Ro52.
To define whether MDA5 autoantibodies occur in association with other prominent myositis autoantibodies, we screened anti-MDA5 antibody positive patient sera for antibodies against Jo-1 and Ro52 by ELISA, and Mi-2, NXP2 and Ro60 by IP of appropriate IVTT products. Jo-1 antibodies, which are strongly associated with the anti-synthetase syndrome, were not detected in any of the MDA5 autoantibody-positive patients, whereas Ro52 antibodies were detected in 3 of 11 (27.3%) patients. Antibodies against Mi-2, which is found in 10–20% of DM patients (4, 6), were absent in all anti-MDA5 antibody positive patients, as were Ro60 antibodies. Low titers of anti-NXP2 antibodies (22) were detected in one patient.
As an alternate method to assess whether other antibody specificities are detected in the sera of anti-MDA5 antibody positive patients, we immunoprecipitated lysates of radiolabeled HeLa cells (Fig 1B, lanes denoted “−”). This assay is a useful for identifying antibody specificities (23) (e.g. anti-tRNA synthetases) present in patient sera. Interestingly, the majority of MDA5 antibody positive sera were monospecific, with only one patient (DM 10) demonstrating another antibody specificity (~82 kDa) that was detected in untreated HeLa lysates. Given the association of anti-synthetase antibodies with ILD (4–6), we tested whether this antigen was an aminoacyl tRNA synthetase by immunoprecipitating lysates of radiolabeled HeLa cells with reference sera from patients with confirmed tRNA synthetase autoantibodies and comparing the migration patterns of DM10 and these standards. The unidentified 82 kDa antigen did not co-migrate with the aminoacyl tRNA synthetases (not shown).
Since MDA5 is an IFN-inducible protein which senses dsRNA in the cytoplasm of cells (24, 25), we investigated whether antibodies against other IFN-inducible proteins were found in the anti-MDA5 antibody positive sera. Sera from anti-MDA5 antibody-positive patients were screened by IP of lysates from IFNα-treated, radiolabeled HeLa cells and the profiles were compared to those obtained in the absence of IFNα treatment. MDA5 autoantibodies were not detected in the absence of IFNα treatment (Fig. 1B, lanes denoted “−”) but were readily detected in lysates of IFNα treated cells (Fig. 1B, lanes denoted “+”). Similar to MDA5, Ro52 antibodies were undetectable in the absence of IFNα treatment, but were detected when patient sera were screened against lysates of IFNα-treated cells (Fig. 1B). No additional IFN-induced specificities were seen in the MDA5 antibody-positive patients. Immunoblots performed on unlabeled lysates incubated in the absence or presence of IFNα using a rabbit polyclonal anti-MDA5 antibody confirmed robust biochemical levels of MDA5 only in the latter lysates (not shown). This likely explains our inability to readily detect these antibodies using untreated radiolabeled HeLa lysates as source material for the immunoprecipitations. Thus, MDA5 autoantibodies are found in the absence of antibodies against aminoacyl tRNA synthetases, but can occur in association with Ro52 autoantibodies.
Clinical characteristics of MDA5 autoantibody-positive patients
The first symptom in the majority of anti-MDA5 antibody positive patients was the appearance of the characteristic DM rash (14, 15). Palmar papules and/or a livedoid appearance were noted in 2 of the anti-MDA5 antibody positive patients (this data was unavailable in 1 patient), and ulcerations (mouth, nails, fingers, knuckles, thigh and/or buttocks) were present in 9 (Table 2). Interestingly, 8 anti-MDA5 antibody positive patients had a phenotype similar to that of the antisynthetase syndrome with three or more clinical features of this syndrome (Table 2). 3 patients had all 6 clinical features of this syndrome. Calcinosis was present in 3 patients, with one having calcinosis universalis in the absence of other features of myositis with the exception of having a heliotrope rash (Table 2). Two patients presented with elevated liver function tests out of proportion to muscle enzyme elevation in the absence of use of hepatotoxic therapy; however, liver biopsy, when performed, revealed no specific abnormalities, and frequently LFT elevations normalized over time.
Table 2.
Clinical features of anti-MDA5 antibody positive DM patients
| Patient ID | ILD | Weakness | High CK | High Aldo | Arthritis | Fever | Raynauds | Calcinosis | Ulcerations | Mouth pain | Mechanic’s Hands | Gottron’s | Heliotrope | Palmar erythema | Medications* |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DM 1 | Y | Y | Y | U | Y | Y | Y | N | Y | Y | Y | Y | Y | N | MTX, Pred, MMF |
| DM 2 | Y | Y | Y | Y | Y | N | N | N | Y | N/A | N | Y | Y | U | MTX, Pred, HCQ |
| DM 3 | Y | N | N | N | Y | Y | N | N | N | N | Y | Y | N | N | MTX, Pred, AZA |
| DM 4 | N | Y | N | Y | Y | Y | N | N | Y | N | N | Y | Y | N | Pred, HCQ |
| DM 5 | Y | N | Y | Y | N | N | N | N | N | Y | Y | Y | Y | N | Pred, MMF, HCQ |
| DM 6 | Y | Y | N | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | MTX, MMF, Pred, Imuran |
| DM 7 | N | N | N | N | Y | N | Y | N | Y | N | Y | Y | Y | N | MTX, IVIG, HCQ |
| DM 8 | N | N | N | N | N | N | N | Y | Y | N | Y | Y | Y | N | MMF, Pred, IVIG, MTX |
| DM 9 | Y | Y | N | N | Y | N | Y | N | Y | Y | Y | Y | Y | Y | Pred, MMF, RTX, Imuran, IVIG |
| DM 10 | Y | N | N | Y | Y | N | N | Y | Y | N | Y | Y | Y | N | Pred, MMF, IVIG, MTX |
| DM 11 | Y | Y | Y | Y | Y | Y | Y | N | Y | Y | Y | Y | N | N | MMF, Pred, MTX, AZA, Tacro |
|
| |||||||||||||||
| Total %positive | 72.7 | 54.5 | 36.4 | 60 | 81.8 | 45.5 | 45.5 | 27.3 | 81.8 | 50 | 81.8 | 100 | 81.8 | 18.2 | |
Aldo = aldolase; AZA = azathioprine; HCQ = hydroxychloroquine; IVIG = intravenous immunoglobulin; MMF = mycophenylate mofetil; MTX = methotrexate; Pred = prednisone; RTX = rituximab;Tacro = tacrolimus
Medication use is indicated for the entire course of follow-up
Y = yes; N = no; U = unknown
We compared the demographics and clinical features of the eleven anti-MDA5 antibody positive patients and 149 anti-MDA5 antibody-negative patients (Table 3). There were no statistically significant differences in gender, race, age at diagnosis, or disease duration. While some clinical features (e.g. Gottron’s sign, Raynaud’s phenomenon) did not differ, there were several significant clinical differences between these groups. Anti-MDA5 antibody positive patients exhibited an increased prevalence of mechanics hands (81.8% vs. 19.0%, p<0.001), inflammatory arthritis (81.8% vs. 26.7%, p<0.001), and fevers (45.5% vs. 16.4%, p=0.030). The arthritis observed in the anti-MDA5 positive patients was frequently symmetric, present in the small joints of the hands and associated with morning stiffness, clinically similar to rheumatoid arthritis. These patients had undergone laboratory testing for rheumatoid factor and anti-CCP antibodies due to suspicion of rheumatoid arthritis. Of these, one patient had an elevated rheumatoid factor and one had anti-CCP antibodies. Only one of these patients fulfilled the 2010 ACR diagnostic criteria for RA. A small erosion on the radial site of the 2nd metacarpal head was seen in one patient (DM10) but only by hand MRI. Conventional X-rays did not demonstrate erosive disease in any of the patients with arthritis.
Table 3.
Demographics and Clinical Characteristics of MDA5 Positive and Negative DM Patients
| MDA5-positive (N = 11) | MDA5-negative (N = 149) | p-value | |
|---|---|---|---|
| N (%) | N (%) | ||
| Demographics | |||
| Gender | 0.94 | ||
| Male | 3 (27.3) | 39 (26.2) | |
| Female | 8 (72.7) | 110 (73.8) | |
| Race | 0.24 | ||
| Caucasian | 8 (72.7) | 124 (83.2) | |
| African American | 2 (18.2) | 17 (11.4) | |
| Asian | 0 (0) | 6 (4.0) | |
| Other | 1 (9.1) | 2 (1.3) | |
| Mean age at diagnosis, y | 41.4 | 44.9 | 0.48 |
| Median disease duration, m | 24.5 | 26.3 | 0.9 |
| Clinical Features | |||
| Gottron’s Papules/Sign | 11(100) | 111 (75)† | 0.055 |
| Heliotrope rash | 9 (81.8) | 71 (48.0)† | 0.03 |
| Weakness | 6 (54.5) | 138 (93.2)† | <0.001 |
| Fever | 5 (45.5) | 24 (16.4)¥ | 0.017 |
| Inflammatory Arthropathy | 9 (81.8) | 39 (26.7)¥ | <0.001 |
| Raynaud’s Phenomenon | 5 (45.5) | 44 (30.3)§ | 0.3 |
| Mechanic’s Hands | 9 (81.8) | 28 (19.0)‡ | <0.001 |
| Interstitial Lung Disease | 8 (72.7) | 17 (11.4) | <0.001 |
| Calcinosis | 3 (27.3) | 18 (12.1) | 0.15 |
Clinical data was not available for every MDA5-negative patient. Where indicated, n is less than 149.
n = 148;
n= 147;
n = 146;
n = 145
ILD was enriched in anti-MDA5 antibody positive patients (72.7% vs. 11.4%, p<0.001), with 8 of 11 patients having evidence of ILD by radiographic criteria (Tables 2 & 3). Of the 8 patients with radiographic evidence of lung involvement on chest CT scan, 6 had reticular changes, 1 had ground glass opacities only and 1 had atelectasis. The initial pulmonary function testing (PFT) of the cohort of anti-MDA5 patients is shown in Figure 2A. The median value for FVC was 89.7% predicted; TLC was 87.7% predicted and DLCo was 70.7% predicted. Serial PFTs were available on 6 patients. Of those with ILD, the majority stabilized or improved with regard to their lung disease once treated with immunosuppressive medications (Figure 2B–E). Only two patients to date have demonstrated progressive ILD on immunosuppressive therapy. Both of these patients were found to have Ro52 autoantibodies. Figure 2F shows a patient representative of this declining course. One additional anti-MDA5 antibody positive patient had Ro52 autoantibodies, but did not have evidence of ILD.
Figure 2. Pulmonary function testing in the anti-MDA5 positive cohort.
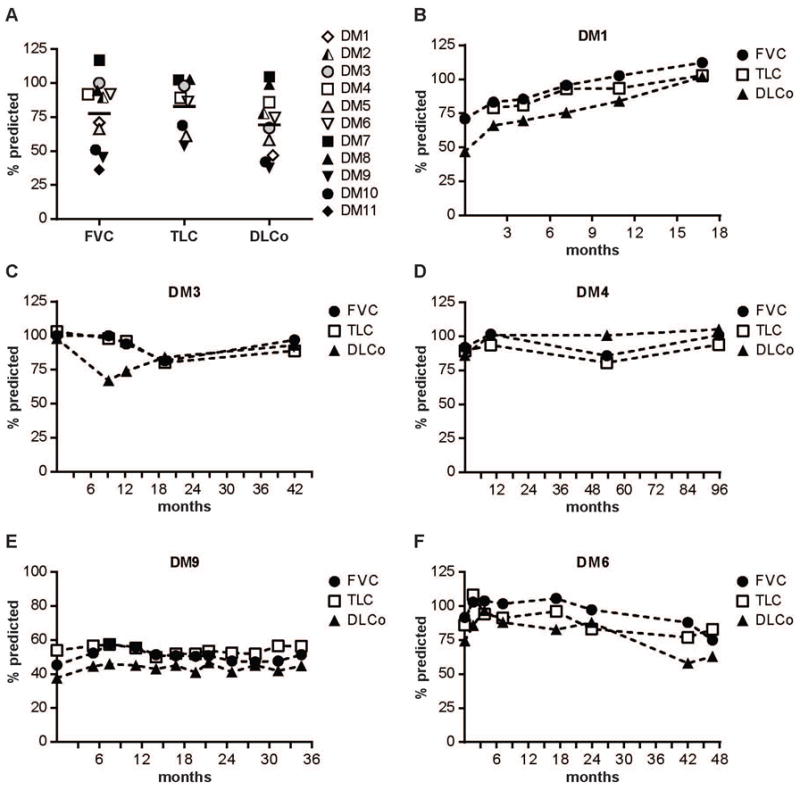
(A) Results of initial PFTs (where available) performed at or prior to the first clinic visit for all 11 anti-MDA5-positive patients. FVC, TLC and DLCo for each individual are shown as % predicted. The median value for each test is shown by the thick black bar. (B–F) Longitudinal PFTs for 5 anti-MDA5-positive patients demonstrating (B) lung function improvement; (C, D) a stable pattern within the normal range; (E) a stable pattern in the moderate to severely reduced range; and (F) declining lung function. The X-axis represents months of follow-up.
Anti-MDA5 antibody positive patients demonstrated less weakness (54.5% vs. 93.2%, p<0.001) than the rest of the cohort, however these patients demonstrated overt clinical myopathy. Muscle enzymes were elevated in 7 of 11 patients, with 3 of these patients exhibiting an isolated elevated aldolase with a normal CK (Table 2). Of those with elevated muscle enzymes, 6 were clinically weak. Five patients with elevated muscle enzymes had a STIR sequence bilateral thigh muscle MRI performed, and one had a deltoid MRI. Four MRIs demonstrated muscle edema while two were normal but they were performed over 2 years after the initial disease presentation. EMG studies were performed on 9 of 11 patients; 4 patients had an irritable myopathy, 3 had a non-irritable myopathy, and 2, who were characterized as amyopathic, showed no evidence of myopathy by EMG testing.
Correlation between MDA5 autoantibody titers and disease activity
Longitudinal serum samples were available from 6 of the 11 anti-MDA5 antibody positive patients. These ranged from 2 to 5 samples, over a follow-up period of 1 to 5 years. To determine whether MDA5 autoantibody levels fluctuated and if they correlated with disease activity, the samples were assayed by IP of 35S-methionine-labeled MDA5 generated by IVTT. All IPs using sera from the same patient were electrophoresed on one gel for the purposes of comparison. The images were scanned and density values were calculated for each time point and plotted on an arbitrary scale of anti-MDA5 units (Figure 3). In general, patients with low titer MDA5 autoantibodies had less severe disease. For each patient tested, MDA5 autoantibody titers did not vary much over the entire course of follow-up. In contrast, disease course, as determined by a composite score of rash, muscle weakness, ILD, number of immunosuppressive medications, and prednisone dose, generally improved over time. While all patients received immunosuppressive therapy, the treatments varied, with azathioprine and mycophenolate being most common (Table 2). Nine of the 11 patients had clear clinical resolution of their symptoms (rash, myositis, and/or lung disease) enabling a decrease in dose or cessation of prednisone, although length of therapy was variable. All eleven patients were able to reduce their steroid dose by 50% or greater by the time of their last visit with 4 patients taking no prednisone at all. Three of those four were also taking no other immunosuppressive medications at the time of their last visit.
Figure 3. Longitudinal analysis of MDA5 autoantibody titers.
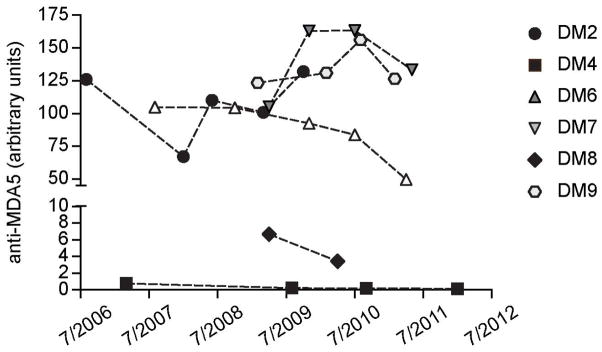
Anti-MDA5 antibody-positive patients with multiple banked serum samples were screened for MDA5 autoantibodies by IVTT IP. Data were scanned and density values are presented as arbitrary units. Antibody titers at each visit are shown. The X-axis represents the date of sampling.
DISCUSSION
Phenotype is closely linked with autoantibody status among patients with the idiopathic inflammatory myopathies, including DM (1). It is therefore important that as new DM-specific antibodies are described, the associated phenotype(s) need to be carefully defined. Reports of MDA5 autoantibodies, found in 10–20% of Japanese DM patients, have defined an association with clinically amyopathic disease and a high incidence of ILD; the latter was often rapidly progressive and fatal (7–11). Additional reports have confirmed the association with rapidly progressive ILD, but have shown that these antibodies occur in patients with myopathic disease (26). More recently, MDA5 autoantibodies were identified in 13% of patients in a DM cohort seen at the Stanford University Dermatology clinic (13). The association with an increased prevalence of ILD and amyopathic disease was confirmed in this US cohort. Prominent skin manifestations, including ulceration and tender palmar papules, were also noted in these patients (11, 13).
Our study of predominantly Caucasian (8/11, 72.7%) anti-MDA5 antibody positive DM patients from a US Myositis tertiary referral Center contrasts in some regards with previously reported anti-MDA5 phenotypes ascertained in Asian cohorts, but is similar in many aspects to the report from a dermatology cohort in the United States (7, 10, 13). The frequency of MDA5 autoantibodies in our cohort was lower (6.9%, compared to 13% (13), p = 0.212); this difference is noteworthy because it exists even when identical methodologies were used for assaying anti-MDA5 antibodies. Compared to the Japanese cohorts our patients more often had clinical myopathy (6/11, 54.5%) which required a potent combination immunosuppressive treatment regimen (Table 2). Additionally, ILD was generally mild in our cohort of anti-MDA5 antibody positive patients, and even those with a relatively fulminant clinical course of ILD were often able to attain sustained clinical remission.. Various factors may contribute to these discrepancies, including ethnic differences and subspecialty referral bias. It is conceivable, for example, that amyopathic DM patients are more likely to be evaluated by a dermatologist, given the prominent cutaneous features of the disease, and not referred on for further evaluation at a rheumatology specialty center. Conversely, DM patients presenting with prominent arthritis will likely be evaluated by a rheumatologist rather than a dermatologist, accounting for the potential differences in the clinical spectrum. Additionally, a treatment bias may exist, owing to earlier immunomodulatory therapies (e.g. methotrexate, mycophenolate, or rituximab) in patients with symptomatic polyarthritis and/or myositis, leading to a better prognosis of concomitant ILD.
In our experience, there is a broad spectrum of clinical disease associated with MDA5 antibodies. Many patients were initially labeled as a rheumatologic “overlap” phenotype owing to their impressive synovitis in combination with Raynaud’s phenomenon and myositis. Several were incorrectly diagnosed with rheumatoid arthritis upon presentation of their disease when arthritis was a prominent component. Our patients expressed many of the defining features of the antisynthetase syndrome including arthritis, fevers, Raynaud’s phenomenon, mechanics hands, and ILD in the absence of antibodies against amino-acyl tRNA synthetases. These patients may be clinically indistinguishable from patients with the antisynthetase syndrome. There is even heterogeneity of clinical features and outcomes among antisynthetase patients with anti-Jo-1 patients reported to have more severe myositis, joint impairment and increased risk of cancer, while anti-PL7/PL12 antibody positive patients had earlier and more severe ILD, and gastrointestinal complications (27). It is unclear why autoantibodies directed against distinct cellular proteins (e.g. tRNA synthetases and MDA5) can manifest in similar phenotypes. Perhaps the antibodies readout pathways which drive pathogenesis (for example, MDA5 and Ro52 are both IFN-induced proteins). Defining the expression of tRNA synthetases and MDA5 in lung tissue from patients with ILD may provide important clues into the relationship between these seemingly unrelated pathways, and are a priority for future studies.
Interestingly, 27% of the anti-MDA5 antibody positive patients in our cohort also had Ro52 autoantibodies; a similar percentage was reported in the Fiorentino study (of note, Jo-1 antibodies were not found in any of the anti-MDA5 antibody positive sera in the Fiorentino study, consistent with our current findings). Both Ro52 and MDA5 are cytoplasmic proteins which are highly induced by IFN. MDA5 is a member of the RIG-I-like receptor (RLR) family of proteins which sense viral dsRNA structures in the cytoplasm of cells and induce the production of type I IFNs through activation of IRF3 (24). Recent studies have shown that RLR family members RIG-I and MDA5 can also be activated by ubiquitin-induced oligomerization to produce type I IFN (28). For RIG-I, this process is mediated via the ubiquitin E3 ligase TRIM25 (29). Ro52 (TRIM21) is a member of the TRIM family of proteins, and has documented ubiquitin E3 ligase activity (30). Ro52 has been shown to regulate IFN-signaling through interactions with IRF3 (31). Perhaps interactions between Ro52 and MDA5 induce the formation of novel complexes which are particularly immunogenic (32), possibly explaining the co-existence of these antibodies in a subset of DM patients.
The prognosis and associated clinical features for patients with anti-MDA5 antibodies has been variable in the medical literature – ranging from those developing rapidly progressive ILD associated with a high mortality to those with more cutaneous features (ulceration, etc.) – both with attenuated muscle disease. In our cohort, anti-MDA5 antibody positive patients often presented with an inflammatory arthritis that was clinically similar in pattern to rheumatoid arthritis. MDA5 autoantibodies should be added to the differential diagnosis when the antisynthetase hallmark features are present in addition to the classic DM rash – especially when antisynthetase autoantibodies are not detected. We have found that those patients with a relatively fulminant clinical course with regard to ILD, myositis and cutaneous arthritis were often able to obtain sustained clinical remission, in some cases even after discontinuation of immunosuppression. In our experience, while ILD is an associated feature, patients may be able to have complete resolution of their pulmonary disease, suggesting that in at least some anti-MDA5 antibody positive patients, the prognosis may be more favorable than both the antisynthetase syndrome and the previous reports of anti-MDA5 antibody positive patients.
SIGNIFICANCE AND INNOVATIONS.
Individuals with MDA5 autoantibodies typically present with a symmetric inflammatory polyarthropathy, which was frequently clinically indistinguishable from rheumatoid arthritis. A majority of MDA5 antibody-positive patients had a clinical myopathy, and ILD, when present, typically resolved with treatment.
A majority of MDA5 antibody-positive patients exhibited three or more clinical features of the antisynthetase syndrome, with many demonstrating all six clinical features (fever, non-erosive arthritis, myopathy, Raynaud’s phenomenon, mechanics hands and ILD).
Jo-1 antibodies, which are strongly associated with the antisynthetase syndrome, were not detected in any MDA5 autoantibody-positive patients. However, Ro52 antibodies, which are frequently found in anti-Jo-1 positive patients, were detected in 3/11 (27%) of anti-MDA5 autoantibody-positive patients.
MDA5 antibody titers did not vary significantly over time, nor did they track with clinical course.
Acknowledgments
Funding: JCH is funded by NIH R37 DE12354-12S1. LCR is funded by NIH RO1 AR-44684. SD is supported by ACR WOR Grant. The Johns Hopkins Rheumatic Diseases Research Core Center, where the assays were performed, is supported by the NIH grant P30-AR-053503. This study was also supported by the Huayi and Siuling Zhang Discovery Fund.
We thank Po-Han Chen, ScM, from the Johns Hopkins Biostatistics, Epidemiology and Data Management (BEAD) Core, Johns Hopkins Bayview Medical Center, for assistance with the statistical analyses.
Footnotes
The authors declare they have received no benefits from commercial sources for the work reported in this manuscript, nor do they have any other financial interests which could create a potential conflict of interest or the appearance thereof.
References
- 1.Gunawardena H, Betteridge ZE, McHugh NJ. Myositis-specific autoantibodies: Their clinical and pathogenic significance in disease expression. Rheumatology (Oxford) 2009;48:607–12. doi: 10.1093/rheumatology/kep078. [DOI] [PubMed] [Google Scholar]
- 2.Targoff IN, Reichlin M. The association between mi-2 antibodies and dermatomyositis. Arthritis Rheum. 1985;28:796–803. doi: 10.1002/art.1780280711. [DOI] [PubMed] [Google Scholar]
- 3.Ghirardello A, Zampieri S, Iaccarino L, Tarricone E, Bendo R, Gambari PF, et al. Anti-mi-2 antibodies. Autoimmunity. 2005;38:79–83. doi: 10.1080/08916930400022681. [DOI] [PubMed] [Google Scholar]
- 4.Mammen AL. Autoimmune myopathies: Autoantibodies, phenotypes and pathogenesis. Nat Rev Neurol. 2011;7:343–54. doi: 10.1038/nrneurol.2011.63. [DOI] [PubMed] [Google Scholar]
- 5.Nishikai M, Reichlin M. Heterogeneity of precipitating antibodies in polymyositis and dermatomyositis. characterization of the jo-1 antibody system. Arthritis Rheum. 1980;23:881–8. doi: 10.1002/art.1780230802. [DOI] [PubMed] [Google Scholar]
- 6.Targoff IN. Laboratory testing in the diagnosis and management of idiopathic inflammatory myopathies. Rheum Dis Clin North Am. 2002;28:859, 90, viii. doi: 10.1016/s0889-857x(02)00032-7. [DOI] [PubMed] [Google Scholar]
- 7.Sato S, Hirakata M, Kuwana M, Suwa A, Inada S, Mimori T, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum. 2005;52:1571–6. doi: 10.1002/art.21023. [DOI] [PubMed] [Google Scholar]
- 8.Sato S, Hoshino K, Satoh T, Fujita T, Kawakami Y, Fujita T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: Association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009;60:2193–200. doi: 10.1002/art.24621. [DOI] [PubMed] [Google Scholar]
- 9.Nakashima R, Imura Y, Kobayashi S, Yukawa N, Yoshifuji H, Nojima T, et al. The RIG-I-like receptor IFIH1/MDA5 is a dermatomyositis-specific autoantigen identified by the anti-CADM-140 antibody. Rheumatology (Oxford) 2010;49:433–40. doi: 10.1093/rheumatology/kep375. [DOI] [PubMed] [Google Scholar]
- 10.Hamaguchi Y, Kuwana M, Hoshino K, Hasegawa M, Kaji K, Matsushita T, et al. Clinical correlations with dermatomyositis-specific autoantibodies in adult japanese patients with dermatomyositis: A multicenter cross-sectional study. Arch Dermatol. 2011;147:391–8. doi: 10.1001/archdermatol.2011.52. [DOI] [PubMed] [Google Scholar]
- 11.Koga T, Fujikawa K, Horai Y, Okada A, Kawashiri SY, Iwamoto N, et al. The diagnostic utility of anti-melanoma differentiation-associated gene 5 antibody testing for predicting the prognosis of japanese patients with DM. Rheumatology (Oxford) 2012;51:1278–84. doi: 10.1093/rheumatology/ker518. [DOI] [PubMed] [Google Scholar]
- 12.Chaisson NF, Paik J, Orbai AM, Casciola-Rosen L, Fiorentino D, Danoff S, et al. A novel dermato-pulmonary syndrome associated with MDA-5 antibodies: Report of 2 cases and review of the literature. Medicine (Baltimore) 2012;91:220–8. doi: 10.1097/MD.0b013e3182606f0b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fiorentino D, Chung L, Zwerner J, Rosen A, Casciola-Rosen L. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): A retrospective study. J Am Acad Dermatol. 2011;65:25–34. doi: 10.1016/j.jaad.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts) N Engl J Med. 1975;292:344–7. doi: 10.1056/NEJM197502132920706. [DOI] [PubMed] [Google Scholar]
- 15.Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts) N Engl J Med. 1975;292:403–7. doi: 10.1056/NEJM197502202920807. [DOI] [PubMed] [Google Scholar]
- 16.Sontheimer RD. Dermatomyositis: An overview of recent progress with emphasis on dermatologic aspects. Dermatol Clin. 2002;20:387–408. doi: 10.1016/s0733-8635(02)00021-9. [DOI] [PubMed] [Google Scholar]
- 17.American Thoracic Society, European Respiratory Society. American thoracic Society/European respiratory society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. this joint statement of the american thoracic society (ATS), and the european respiratory society (ERS) was adopted by the ATS board of directors, june 2001 and by the ERS executive committee, june 2001. Am J Respir Crit Care Med. 2002;165:277–304. doi: 10.1164/ajrccm.165.2.ats01. [DOI] [PubMed] [Google Scholar]
- 18.Ulanet DB, Wigley FM, Gelber AC, Rosen A. Autoantibodies against B23, a nucleolar phosphoprotein, occur in scleroderma and are associated with pulmonary hypertension. Arthritis Rheum. 2003;49:85–92. doi: 10.1002/art.10914. [DOI] [PubMed] [Google Scholar]
- 19.Pillemer SR, Casciola-Rosen L, Baum BJ, Rosen A, Gelber AC. Centromere protein C is a target of autoantibodies in sjogren’s syndrome and is uniformly associated with antibodies to ro and la. J Rheumatol. 2004;31:1121–5. [PubMed] [Google Scholar]
- 20.Mammen AL, Chung T, Christopher-Stine L, Rosen P, Rosen A, Doering KR, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum. 2011;63:713–21. doi: 10.1002/art.30156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Casciola-Rosen LA, Pluta AF, Plotz PH, Cox AE, Morris S, Wigley FM, et al. The DNA mismatch repair enzyme PMS1 is a myositis-specific autoantigen. Arthritis Rheum. 2001;44:389–96. doi: 10.1002/1529-0131(200102)44:2<389::AID-ANR58>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 22.Ichimura Y, Matsushita T, Hamaguchi Y, Kaji K, Hasegawa M, Tanino Y, et al. Anti-NXP2 autoantibodies in adult patients with idiopathic inflammatory myopathies: Possible association with malignancy. Ann Rheum Dis. 2012;71:710–3. doi: 10.1136/annrheumdis-2011-200697. [DOI] [PubMed] [Google Scholar]
- 23.Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum. 2010;62:2757–66. doi: 10.1002/art.27572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175:2851–8. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- 25.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–5. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 26.Kang EH, Nakashima R, Mimori T, Kim J, Lee YJ, Lee EB, et al. Myositis autoantibodies in korean patients with inflammatory myositis: Anti-140-kDa polypeptide antibody is primarily associated with rapidly progressive interstitial lung disease independent of clinically amyopathic dermatomyositis. BMC Musculoskelet Disord. 2010;11:223. doi: 10.1186/1471-2474-11-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marie I, Josse S, Decaux O, Dominique S, Diot E, Landron C, et al. Comparison of long-term outcome between anti-Jo1- and anti-PL7/PL12 positive patients with antisynthetase syndrome. Autoimmun Rev. 2012;11:739–45. doi: 10.1016/j.autrev.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 28.Jiang X, Kinch LN, Brautigam CA, Chen X, Du F, Grishin NV, et al. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity. 2012;36:959–73. doi: 10.1016/j.immuni.2012.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–20. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 30.Wada K, Kamitani T. Autoantigen Ro52 is an E3 ubiquitin ligase. Biochem Biophys Res Commun. 2006;339:415–21. doi: 10.1016/j.bbrc.2005.11.029. [DOI] [PubMed] [Google Scholar]
- 31.Higgs R, Ni Gabhann J, Ben Larbi N, Breen EP, Fitzgerald KA, Jefferies CA. The E3 ubiquitin ligase Ro52 negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. J Immunol. 2008;181:1780–6. doi: 10.4049/jimmunol.181.3.1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosen A, Casciola-Rosen L, Ahearn J. Novel packages of viral and self-antigens are generated during apoptosis. J Exp Med. 1995;181:1557–61. doi: 10.1084/jem.181.4.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]