

Background: The role of HDL in sepsis remains to be clarified.
Results: ApoA-I-KO mice are susceptible but apoA-I-tg mice are resistant to CLP-induced sepsis. Lack of HDL leads to less LPS neutralization, less LPS clearance, impaired leukocyte recruitment, and reduced corticosterone generation in sepsis.
Conclusion: HDL exerts multiple protective effects in polymicrobe-induced sepsis.
Significance: Our study supports efforts to raise HDL levels as a therapeutic approach for sepsis.
Keywords: Dyslipidemia, High Density Lipoprotein (HDL), Immunology, Lipopolysaccharide (LPS), Sepsis, Apolipoprotein A-I, SCARB1, Scavenger Receptor BI, Cecal Ligation and Puncture
Abstract
HDL has been considered to be a protective factor in sepsis; however, most contributing studies were conducted using the endotoxic animal model, and evidence from clinically relevant septic animal models remains limited and controversial. Furthermore, little is known about the roles of HDL in sepsis other than LPS neutralization. In this study, we employed cecal ligation and puncture (CLP), a clinically relevant septic animal model, and utilized apoA-I knock-out (KO) and transgenic mice to elucidate the roles of HDL in sepsis. ApoA-I-KO mice were more susceptible to CLP-induced septic death as shown by the 47.1% survival of apoA-I-KO mice versus the 76.7% survival of C57BL/6J (B6) mice (p = 0.038). ApoA-I-KO mice had exacerbated inflammatory cytokine production during sepsis compared with B6 mice. Further study indicated that serum from apoA-I-KO mice displayed less capacity for LPS neutralization compared with serum from B6 mice. In addition, apoA-I-KO mice had less LPS clearance, reduced corticosterone generation, and impaired leukocyte recruitment in sepsis. In contrast to apoA-I-KO mice, apoA-I transgenic mice were moderately resistant to CLP-induced septic death compared with B6 mice. In conclusion, our findings reveal multiple protective roles of HDL in CLP-induced sepsis. In addition to its well established role in neutralization of LPS, HDL exerts its protection against sepsis through promoting LPS clearance and modulating corticosterone production and leukocyte recruitment. Our study supports efforts to raise HDL levels as a therapeutic approach for sepsis.
Introduction
Sepsis is a major health issue that claims over 215,000 lives and costs $16.7 billion per year in the United States (1–4). The prognosis for sepsis remains grim, with a mortality rate exceeding 30%, due to the poor understanding of the disease (5). Identifying molecules involved in sepsis, especially endogenous protective modulators, may provide new insights for efficient therapies.
Disorders of lipid metabolism are critical issues in septic patients. Plasma lipoproteins, especially HDL, are markedly reduced during sepsis, and a number of clinical studies have pointed out that low plasma HDL cholesterol is a poor prognostic factor for severe sepsis (6–8). It has been speculated that HDL plays a protective role in sepsis and that raising circulating HDL levels may be an important therapeutic approach for the treatment of sepsis, given the roles of HDL in binding and neutralizing LPS (9–16) and in the anti-inflammatory response (17).
As a major protein component of HDL, apoA-I is a key determinant for HDL formation (18). ApoA-I levels are highly correlated with HDL cholesterol levels, and a lack of apoA-I results in a gross deficiency of HDL (19, 20). Clinical studies indicated that decreased apoA-I levels correlate with sepsis and adverse outcomes among intensive care unit patients (21). Thus, numerous efforts have been made to raise circulating HDL levels by using reconstituted HDL made from apoA-I or by using apoA-I mimetic peptide and to test whether an increase in circulating HDL levels provides protection against sepsis. Tests in the endotoxic animal model have provided encouraging results. Administration of reconstituted HDL or apoA-I mimetic peptide to LPS-challenged animals alleviates LPS-induced inflammatory cytokine production, improves cardiac function, and reduces endotoxic animal death (22–25). Administration of reconstituted HDL to humans challenged with low dose LPS is also followed by a reduction in inflammatory cytokine production and a decrease in membrane CD14 expression on monocytes (26, 27). In addition to the reconstituted HDL and apoA-I mimetic peptide approaches, mice overexpressing apoA-I show a 2-fold increase in circulating HDL levels, and this is associated with protection against the LPS-induced inflammatory response and endotoxic animal death (28), again pointing to a potentially beneficial role of HDL in endotoxemia.
However, when looking at more clinically relevant models of sepsis, the data on the protective role of HDL are less clear. It is important to recognize that although LPS-induced endotoxemia is widely used for the study of sepsis, the endotoxic animal model does not fully mimic the changes observed in sepsis (29). For example, Tlr4 mutant mice are completely resistant to LPS-induced endotoxic death but are still highly susceptible to bacteria-induced septic death (30–32). To determine the role of HDL in a septic animal model, Zhang et al. (33) tested the effect of apoA-I mimetic peptide in rats using cecal ligation and puncture (CLP)2 and showed that apoA-I mimetic peptide treatment suppresses inflammatory responses and improves survival by 28% in CLP-treated rats. Unfortunately, survival was monitored for only 2 days in that study. As there are profound septic animal deaths between 3 and 7 days post-CLP (34), it is unclear whether the treatment still provides efficient protection against sepsis beyond 2 days. Furthermore, a Gram-negative bacteria-induced septic animal model showed that reconstituted HDL suppresses inflammatory cytokine production in canines but paradoxically causes more animal deaths (35).
In light of the distinct differences between endotoxemia and sepsis, the limited studies that aimed to clarify the significance of HDL in sepsis, and the poor understanding of the roles of HDL in sepsis beyond LPS neutralization, we employed CLP, a clinically relevant sepsis animal model (34), to elucidate the role of HDL in sepsis. As mentioned above, apoA-I knock-out (KO) and transgenic (tg) mice are excellent models to study the effects of deficiency and abundance of HDL, respectively. In this study, we employed these unique animals to assess the roles of HDL in sepsis. We found that apoA-I-KO mice were more susceptible to CLP-induced death and had exacerbated inflammatory cytokine production during sepsis. We also found that serum from apoA-I-KO mice displayed less LPS neutralization compared with C57BL/6J (B6) control mice. ApoA-I-KO mice also displayed less LPS clearance, reduced corticosterone generation, and impaired recruitment of neutrophils/monocytes to the peritoneal cavity in sepsis. In contrast, apoA-I-tg mice were moderately resistant to CLP-induced septic death compared with B6 mice. Our findings demonstrate multiple protective roles of HDL in sepsis and suggest that raising HDL levels may provide a therapeutic approach for sepsis.
EXPERIMENTAL PROCEDURES
Materials
LPS (Escherichia coli serotype K-12) was from InvivoGen. Low endotoxin FBS was from HyClone. ELISA kits for quantifying TNF-α and IL-6 were from eBioscience. The LPS kit was from Charles River. The competitive ELISA kit for quantifying mouse corticosterone was from Cayman.
Animals
ApoA-I-KO and apoA-I-tg mice on a B6 background and B6 mice were obtained from The Jackson Laboratory. The animals were bred at the animal facility of the University of Kentucky. The animals were fed a standard laboratory diet. Animal care and experiments were approved by the Institutional Animal Care and Use Committee of the University of Kentucky.
CLP Septic Animal Model
CLP was performed on 10–12-week-old mice as described previously (36). Mice on a B6 background are very susceptible to sepsis. Thus, a relatively mild CLP (21-gauge needle, half-ligation) was employed in this study. Survival was monitored for a 7-day period.
Gram-positive Bacterial Infection
ApoA-I-KO and B6 mice (8–10 weeks old) were intraperitoneally injected with 4.8 × 108 cfu of mouse Staphylococcus aureus (ATCC 25923), and survival was monitored for 7 days.
Lipoprotein Profiling by FPLC
Serum (50 μl) was resolved by gel filtration chromatography using an FPLC system equipped with a Superose 6 column (GE Healthcare). The column was eluted at a flow rate of 0.5 ml/min in buffer containing 150 mm NaCl, 10 mm Tris-HCl (pH 7.4), and 0.01% sodium azide, and 0.5 ml/fraction was collected. 100 μl of sample was mixed with an equal volume of 2× assay reagent (Wako Chemicals) to determine the cholesterol content of fractions.
Neutralization of LPS-induced Inflammatory Response by Serum
We used the HEK-BlueTM cell system (InvivoGen) to analyze neutralization of the LPS-induced inflammatory response by serum. HEK-Blue cells stably express TLR4, CD14, MD2, and a NF-κB reporter. The HEK-Blue cell system is very sensitive; as low as subnanogram amounts of LPS can be detected (37). Instead of using isolated HDL, we utilized serum in this assay because 1) the mouse is a HDL animal with >90% of its lipoproteins as HDL and 2) the serum contains cofactors of LPS, such as LPS-binding protein, which are required for LPS-induced inflammatory signaling. Briefly, HEK-Blue cells were cultured in a 96-well plate in complete DMEM containing 10% low endotoxin FBS to 90% confluency. The cells were treated with 0.5 ng/ml LPS for 16 h in the presence and absence of 5% mouse serum isolated from apoA-I-KO or B6 mice. 100 μl of the culture supernatant was mixed with 100 μl of HEK-Blue detection medium and incubated at 37 °C for 1 h. LPS binding to TLR4 results in induction of NF-κB reporter expression, which catalyzes the HEK-Blue detection medium to turn blue. The blue color was quantified by measuring absorption at 650 nm.
Analysis of Endotoxicity of Serum from CLP-challenged Mice
HEK-Blue cells were cultured in a 96-well plate in complete DMEM containing 10% low endotoxin FBS to 90% confluency. The cells were treated for 16 h with 5% mouse serum isolated from apoA-I-KO or B6 mice that were challenged with CLP for 8 h. The activation of TLR4/NF-κB was quantified by measuring absorption at 650 nm as described above.
Analysis of Serum LPS
The serum LPS levels in CLP-challenged mice were quantified with the LPS ELISA kit following the manufacturer's instructions.
Analysis of Leukocyte Recruitment to Peritoneum
Leukocyte recruitment to the peritoneal cavity was analyzed by flow cytometry as described (38, 39). Briefly, 8–10-week-old apoA-I-KO and B6 mice were CLP- or sham-treated for 6 h, and the peritoneal fluids were collected. Peritoneal neutrophils (polymorphonuclear leukocytes (PMNs); CD11bhi Ly6Chi) and inflammatory monocytes (IMs; CD11bint Ly6Chi) were gated by CD11b and Ly6C expression on CD45+ cells. Ly6G expression was confirmed to be high in gated PMNs and low in gated IMs.
Biochemical Assays
Mice (10–12 weeks old) were killed by CO2 inhalation 2, 4, and 8 h following CLP. Blood was obtained by cardiac puncture. Serum TNF-α, IL-6, or corticosterone was quantified with a corresponding kit.
Statistical Analysis
p values for survival curves were determined from the Kaplan-Meier survival curves with the log-rank test using SAS software. Significance in experiments comparing two groups was determined by two-tailed Student's t test. Means were considered different at p < 0.05.
RESULTS
Increased Susceptibility to CLP-induced Septic Death in ApoA-I-KO Mice
ApoA-I is a major protein component in HDL. ApoA-I-KO mice had an 80% decrease in plasma cholesterol levels and were grossly deficient in HDL as shown by lipoprotein profile assay (Fig. 1, A and B). Thus, apoA-I-KO mice have been widely employed as a HDL-deficient model to determine the role of HDL in vivo (40–42). We determined the role of HDL in sepsis using a clinically relevant septic animal model: CLP. Considering the B6 background of apoA-I-KO mice, we utilized a relatively mild CLP (21-gauge needle, half-ligation). As shown in Fig. 1C, apoA-I-KO mice were more susceptible to CLP-induced septic death as shown by the 47.1% survival of apoA-I-KO mice versus the 76.7% survival of B6 control mice (p = 0.038).
FIGURE 1.
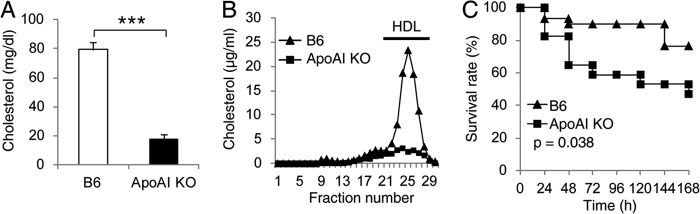
ApoA-I-KO mice are more susceptible to CLP-induced septic death. A, plasma cholesterol concentration. Data are means ± S.E. (n = 4 per group with duplicate measurements) ***, p < 0.001 versus B6. B, lipoprotein profile analyzed by FPLC. Representative data are shown. C, survival analysis. ApoA-I-KO (n = 17) and B6 (n = 30) mice (10–12 weeks old) were subjected to CLP (21-gauge needle, half-ligation), and survival was monitored for 7 days. Data are expressed as the percentage of mice surviving at the indicated times and analyzed by the log-rank x2 test.
Exacerbated Inflammatory Cytokine Generation in ApoA-I-KO Mice during Sepsis
To understand why apoA-I-KO mice are susceptible to septic death, we subjected apoA-I-KO and B6 mice to CLP and determined the serum cytokine level. As shown in Fig. 2, apoA-I-KO mice generated more TNF-α and IL-6 compared with B6 mice as shown by a 3.6-fold increase in the IL-6 level 4 h post-CLP and a 1.7-fold increase in the TNF-α level 8 h post-CLP.
FIGURE 2.
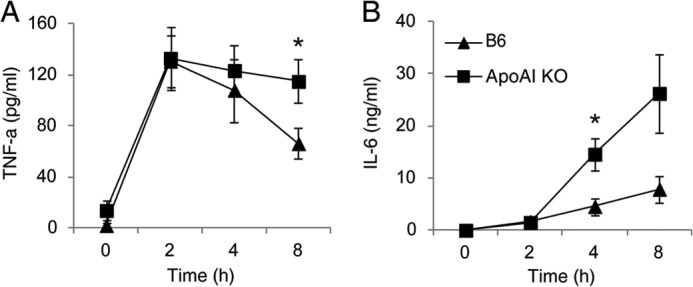
Exacerbated inflammatory cytokine generation in apoA-I-KO mice during sepsis. ApoA-I-KO and B6 mice were subjected to CLP for 2, 4, and 8 h, and the serum concentrations of TNF-α (A) and IL-6 (B) were quantified. Data are means ± S.E. (n = 7–11 per group with duplicate measurements). *, p < 0.05 versus B6.
Impaired LPS Neutralization and Clearance in ApoA-I-KO Mice
Previous reports have shown that HDL is capable of neutralizing LPS (15, 16). ApoA-I-KO mice were grossly deficient in HDL (Fig. 1A). Thus, we speculated that the HDL deficiency in apoA-I-KO mice leads to a decreased capability to neutralize LPS. To test this, we took two approaches. First, we assessed LPS neutralization by serum isolated from apoA-I-KO and B6 mice using the HEK-Blue cell system. In the absence of B6 mouse serum, we observed a profound activation of TLR4/NF-κB by LPS (Fig. 3A). In the presence of B6 mouse serum, the activation of TLR4/NF-κB by LPS was effectively suppressed. As expected, serum from apoA-I-KO mice was significantly less capable of neutralizing LPS compared with serum from B6 mice as shown by a 3-fold increase at A650 nm (Fig. 3B). Second, we isolated serum from CLP-challenged apoA-I-KO and B6 mice and tested TLR4/NF-κB activation using the HEK-Blue cell system. As shown in Fig. 3C, serum from CLP-challenged apoA-I-KO mice induced 3-fold more TLR4/NF-κB activation than serum from CLP-challenged B6 mice.
FIGURE 3.
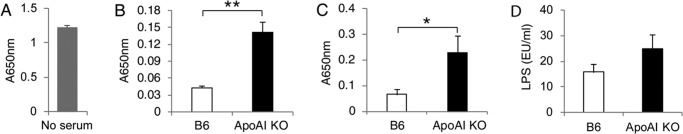
Impaired LPS neutralization and clearance in apoA-I-KO mice. A, activation of TLR4/NF-κB by LPS in the absence of mouse serum. HEK-Blue cells were treated with LPS at 0.5 ng/ml for 16 h, and the activation of TLR4/NF-κB was analyzed. B, less LPS neutralization by serum from apoA-I-KO mice. HEK-Blue cells were treated with LPS at 0.5 ng/ml for 16 h in the presence of 5% serum from apoA-I-KO and B6 mice. Data are means ± S.E. (n = 4 per group with duplicate measurements). **, p < 0.01. C, serum from CLP-treated apoA-I-KO mice induced a greater inflammatory response in HEK-Blue cells than that from wild-type mice. Data are means ± S.E. (n = 7–11 per group with duplicate measurements). *, p < 0.05 versus B6. D, serum LPS concentration 18 h post-CLP. Data are means ± S.E. (n = 9–11 per group with duplicate measurements; p = 0.18 versus B6).
A body of evidence indicates that LPS gets cleared via scavenger receptor class B, type I (SR-BI), as SR-BI binds LPS and mediates the uptake of LPS in vitro (43), and SR-BI null mice display impaired LPS clearance in circulation in LPS-induced endotoxemia or CLP-induced sepsis (37, 44). Interestingly, the previous study also showed that HDL promotes SR-BI-mediated LPS uptake in vitro (43). Considering that LPS associates with HDL to form HDL-LPS complexes and that almost all LPSs exist as HDL-LPS complexes in circulating blood (15, 16), we speculated that HDL functions as a LPS transporter that facilitates LPS clearance. If so, HDL deficiency in apoA-I mice may impair LPS clearance. To test this possibility, we quantified circulating LPS levels 18 h following CLP. ApoA-I-KO mice had a moderate increase in LPS concentration in serum compared with B6 control mice (Fig. 3D). Taken together, the decreased capacity of LPS neutralization and impaired LPS clearance in apoA-I-KO mice likely contribute to the increase in fatality and greater inflammatory cytokine production in sepsis.
To determine whether HDL provides protection other than LPS neutralization and clearance, we infected apoA-I-KO and B6 mice with the Gram-positive bacterium S. aureus and monitored survival for 7 days. ApoA-I-KO mice exhibited a moderately decreased survival upon S. aureus infection as shown by the 20% fatality of apoA-I-KO mice compared with the 6.7% fatality of B6 mice (p = 0.296) (supplemental Fig. I). These data suggest that HDL exerts its protection in addition to LPS neutralization and clearance.
Reduced Corticosterone Generation in ApoA-I-KO Mice during Sepsis
An important function of HDL is to provide cholesterol for glucocorticoid synthesis in adrenal glands (37, 44, 45). HDL deficiency in apoA-I-KO mice likely impairs glucocorticoid synthesis in response to septic stress. To test this, we measured serum corticosterone, the major type of glucocorticoid in circulation. As shown in Fig. 4, there was no significant difference in serum corticosterone concentration in sham-treated animals. However, 4 h following CLP, apoA-I-KO mice produced significantly less corticosterone compared with B6 control mice.
FIGURE 4.
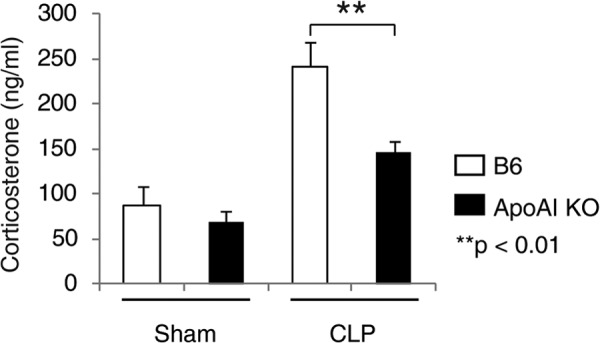
Impaired corticosterone generation in apoA-I-KO mice during sepsis. ApoA-I-KO and B6 mice were subjected to CLP or sham-treated for 4 h, and the serum corticosterone concentration was quantified with an ELISA kit. Data are means ± S.E. (n = 5–11 per group with duplicate measurements).
Moderately Impaired Recruitment of Leukocytes into Peritoneum
HDL has been shown to regulate the expression of adhesion molecules (46), which may affect the mobilization of leukocytes to the peritoneal cavity following CLP. We quantified PMNs and IMs in CLP/sham-treated apoA-I-KO and B6 mice by flow cytometry. Almost no PMNs and IMs were observed in the sham-treated animals (Fig. 5A). 6 h post-CLP, a significant number of PMNs and IMs were recruited to the peritoneum (Fig. 5B), and apoA-I-KO mice displayed a 2-fold decrease in the number of PMNs and IMs (Fig. 5C). The difference was not statistically significant due to the relatively low number of mice used.
FIGURE 5.

Impaired leukocyte recruitment into peritoneum in apoA-I-KO mice during sepsis. ApoA-I-KO and B6 mice were subjected to CLP or sham-treated for 6 h, and the peritoneal fluids were collected. PMNs (CD11bhi Ly6Chi) and IMs (CD11bint Ly6Chi) were gated by CD11b and Ly6C expression on CD45+ cells. Ly6G expression was confirmed to be high in gated PMNs and low in gated IMs. A, non-CLP-treated apoA-I-KO and B6 mice. Almost no PMNs and IMs were observed in the peritoneum of non-CLP-treated mice. B and C, CLP-treated apoA-I-KO and B6 mice. A similar percentage of PMNs and IMs in CD45+ cells was detected in CLP-treated apoA-I-KO and B6 mice (B), and a 2-fold increase in peritoneal PMN and IM numbers was detected in CLP-treated apoA-I mice compared with CLP-treated B6 mice (C). Data are means ± S.E. (n = 6 per group).
Overexpression of ApoA-I Provides Moderate Protection against CLP-induced Septic Death
Finally, we asked whether overexpression of apoA-I in vivo provides protection against CLP-induced septic death. To test this, we utilized apoA-I-tg mice. ApoA-I-tg mice had a 2-fold increase in plasma cholesterol levels (Fig. 6A), which is consistent with a previous study showing that apoA-I-tg mice have 2-fold increases in apoA-I and HDL cholesterol levels in circulation (47). As shown in Fig. 6B, CLP induced 23.3% fatality in B6 mice, but only 10% fatality in apoA-I-tg mice. Although the difference in survival was not statistically significant, this finding suggests that an elevation in the HDL level provides moderate protection against septic death.
FIGURE 6.
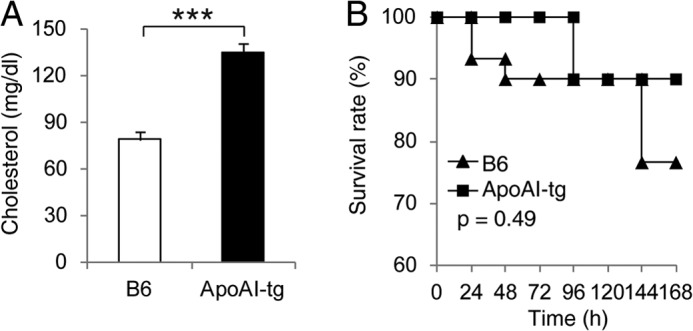
Moderately improved CLP survival in mice overexpressing apoA-I. A, plasma cholesterol concentration. Data are means ± S.E. (n = 4 per group with duplicate measurements). ***, p < 0.001 versus B6. B, survival analysis. ApoA-I-tg (n = 20) and B6 (n = 30) mice (10–12 weeks old) were subjected to CLP (21-gauge needle, half-ligation), and survival was monitored for 7 days. The data are expressed as the percentage of mice surviving at the indicated times and analyzed by the log-rank x2 test.
DISCUSSION
ApoA-I is a major protein component of HDL and a key determinant of HDL formation. ApoA-I-KO mice are grossly deficient in HDL, and apoA-I-tg mice have a 2-fold increase in HDL cholesterol levels. Thus, apoA-I-KO and apoA-I-tg mice present unique animal models to assess HDL function in vivo (40, 48). In this study, we used CLP (a clinically relevant septic animal model) to investigate a role of HDL in sepsis. We found that apoA-I-KO mice are more susceptible to CLP-induced death and that apoA-I-tg mice have moderate protection against septic death.
HDL is well known for its role in binding and neutralizing LPS (15, 16). As expected, we found that serum from apoA-I-KO mice exhibits decreased LPS neutralization compared with serum from B6 control mice. In addition to LPS neutralization, emerging evidence suggests that HDL may play a role in LPS clearance. 1) Almost all LPSs exist as HDL-LPS complexes in circulation (15, 16). 2) SR-BI, a HDL receptor, binds and mediates the uptake of LPS, and HDL promotes SR-BI-mediated LPS uptake in vitro (43, 49). 3) SR-BI null mice display impaired LPS clearance in circulation in LPS-induced endotoxemia and CLP-induced sepsis (37, 44). We speculated that HDL functions as a LPS transporter that promotes LPS clearance via SR-BI. Indeed, we found that CLP-treated apoA-I-KO mice have impaired LPS clearance, which provides support to this speculation.
Using a Gram-positive bacterial infection animal model, we found that apoA-I-KO mice are moderately more susceptible to S. aureus infection, which suggests that HDL exerts its protection in addition to LPS neutralization and clearance. HDLs are multifunctional particles. HDL provides cholesterol for glucocorticoid synthesis via SR-BI-mediated intracellular cholesterol uptake (37, 44, 45). A previous report showed a moderate decrease in corticosterone generation in ACTH-treated apoA-I-KO mice compared with wild-type mice (50). Thus, HDL deficiency in apoA-I-KO mice may impair glucocorticoid production in CLP-induced sepsis. As expected, we observed a significant decrease in inducible corticosterone generation in CLP-challenged apoA-I-KO mice. The impaired glucocorticoid production during sepsis may contribute to an exacerbated inflammatory response in CLP-challenged apoA-I-KO mice; HDL has been shown to regulate the expression of adhesion molecules (46). Indeed, we found that apoA-I-KO mice have a 2-fold decrease in the number of PMNs and IMs. This altered leukocyte recruitment may impair innate immunity, which renders apoA-I-KO mice more susceptible to sepsis.
In summary, our findings reveal multiple protective roles of HDL in CLP-induced sepsis. In addition to the well established role of HDL in neutralization of LPS, HDL exerts its protection against sepsis through promoting LPS clearance, modulating leukocyte recruitment, and regulating corticosterone production. Our study supports efforts to raise HDL levels as a therapeutic approach for sepsis.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants R01 GM085231, R01 GM085231-2S1, and R01 GM085231-5S1 from NIGMS (to X.-A. L.). This work was also supported by a grant from the Children's Miracle Network.

This article contains supplemental Fig. I.
- CLP
- cecal ligation and puncture
- KO
- knock-out
- tg
- transgenic
- B6
- C57BL/6J
- PMN
- polymorphonuclear leukocyte
- IM
- inflammatory monocyte
- SR-BI
- scavenger receptor class B, type I.
REFERENCES
- 1. Bone R. C. (1991) The pathogenesis of sepsis. Ann. Intern. Med. 115, 457–469 [DOI] [PubMed] [Google Scholar]
- 2. Angus D. C., Linde-Zwirble W. T., Lidicker J., Clermont G., Carcillo J., Pinsky M. R. (2001) Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit. Care Med. 29, 1303–1310 [DOI] [PubMed] [Google Scholar]
- 3. Martin G. S., Mannino D. M., Eaton S., Moss M. (2003) The epidemiology of sepsis in the United States from 1979 through 2000. N. Engl. J. Med. 348, 1546–1554 [DOI] [PubMed] [Google Scholar]
- 4. Riedemann N. C., Guo R. F., Ward P. A. (2003) The enigma of sepsis. J. Clin. Invest. 112, 460–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sessler C. N., Perry J. C., Varney K. L. (2004) Management of severe sepsis and septic shock. Curr. Opin. Crit. Care 10, 354–363 [DOI] [PubMed] [Google Scholar]
- 6. Chien J. Y., Jerng J. S., Yu C. J., Yang P. C. (2005) Low serum level of high-density lipoprotein cholesterol is a poor prognostic factor for severe sepsis. Crit. Care Med. 33, 1688–1693 [DOI] [PubMed] [Google Scholar]
- 7. Tsai M. H., Peng Y. S., Chen Y. C., Lien J. M., Tian Y. C., Fang J. T., Weng H. H., Chen P. C., Yang C. W., Wu C. S. (2009) Low serum concentration of apolipoprotein A-I is an indicator of poor prognosis in cirrhotic patients with severe sepsis. J. Hepatol. 50, 906–915 [DOI] [PubMed] [Google Scholar]
- 8. van Leeuwen H. J., Heezius E. C., Dallinga G. M., van Strijp J. A., Verhoef J., van Kessel K. P. (2003) Lipoprotein metabolism in patients with severe sepsis. Crit. Care Med. 31, 1359–1366 [DOI] [PubMed] [Google Scholar]
- 9. Murch O., Collin M., Hinds C. J., Thiemermann C. (2007) Lipoproteins in inflammation and sepsis. I. Basic science. Intensive Care Med. 33, 13–24 [DOI] [PubMed] [Google Scholar]
- 10. Munford R. S. (2005) Detoxifying endotoxin: time, place and person. J. Endotoxin Res. 11, 69–84 [DOI] [PubMed] [Google Scholar]
- 11. Read T. E., Harris H. W., Grunfeld C., Feingold K. R., Calhoun M. C., Kane J. P., Rapp J. H. (1993) Chylomicrons enhance endotoxin excretion in bile. Infect. Immun. 61, 3496–3502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harris H. W., Grunfeld C., Feingold K. R., Rapp J. H. (1990) Human very low density lipoproteins and chylomicrons can protect against endotoxin-induced death in mice. J. Clin. Invest. 86, 696–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harris H. W., Grunfeld C., Feingold K. R., Read T. E., Kane J. P., Jones A. L., Eichbaum E. B., Bland G. F., Rapp J. H. (1993) Chylomicrons alter the fate of endotoxin, decreasing tumor necrosis factor release and preventing death. J. Clin. Invest. 91, 1028–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee R. P., Lin N. T., Chao Y. F., Lin C. C., Harn H. J., Chen H. I. (2007) High-density lipoprotein prevents organ damage in endotoxemia. Res. Nurs. Health 30, 250–260 [DOI] [PubMed] [Google Scholar]
- 15. Ulevitch R. J., Johnston A. R., Weinstein D. B. (1979) New function for high density lipoproteins. Their participation in intravascular reactions of bacterial lipopolysaccharides. J. Clin. Invest. 64, 1516–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ulevitch R. J., Johnston A. R., Weinstein D. B. (1981) New function for high density lipoproteins. Isolation and characterization of a bacterial lipopolysaccharide-high density lipoprotein complex formed in rabbit plasma. J. Clin. Invest. 67, 827–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mineo C., Shaul P. W. (2012) Novel biological functions of high-density lipoprotein cholesterol. Circ. Res. 111, 1079–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ji A., Wroblewski J. M., Cai L., de Beer M. C., Webb N. R., van der Westhuyzen D. R. (2012) Nascent HDL formation in hepatocytes and role of ABCA1, ABCG1, and SR-BI. J. Lipid Res. 53, 446–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eisenberg S. (1984) High density lipoprotein metabolism. J. Lipid Res. 25, 1017–1058 [PubMed] [Google Scholar]
- 20. Williamson R., Lee D., Hagaman J., Maeda N. (1992) Marked reduction of high density lipoprotein cholesterol in mice genetically modified to lack apolipoprotein A-I. Proc. Natl. Acad. Sci. U.S.A. 89, 7134–7138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pavlou E., Makris K., Palaiologou A., Kaldis B., Vrioni G., Economou E., Eforakopoulou M., Zerva L., Drakopoulos I., Ioannidou E. (2008) Decreased apolipoprotein A1 levels correlate with sepsis and adverse outcome among ICU patients. Crit. Care 12, P201 [Google Scholar]
- 22. Dai L., Datta G., Zhang Z., Gupta H., Patel R., Honavar J., Modi S., Wyss J. M., Palgunachari M., Anantharamaiah G. M., White C. R. (2010) The apolipoprotein A-I mimetic peptide 4F prevents defects in vascular function in endotoxemic rats. J. Lipid Res. 51, 2695–2705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hubsch A. P., Powell F. S., Lerch P. G., Doran J. E. (1993) A reconstituted, apolipoprotein A-I containing lipoprotein reduces tumor necrosis factor release and attenuates shock in endotoxemic rabbits. Circ. Shock 40, 14–23 [PubMed] [Google Scholar]
- 24. Cué J. I., DiPiro J. T., Brunner L. J., Doran J. E., Blankenship M. E., Mansberger A. R., Hawkins M. L. (1994) Reconstituted high density lipoprotein inhibits physiologic and tumor necrosis factor α responses to lipopolysaccharide in rabbits. Arch. Surg. 129, 193–197 [DOI] [PubMed] [Google Scholar]
- 25. Wang L., Chen W.-Z., Wu M.-P. (2010) Apolipoprotein A-I inhibits chemotaxis, adhesion, activation of THP-1 cells and improves the plasma HDL inflammatory index. Cytokine 49, 194–200 [DOI] [PubMed] [Google Scholar]
- 26. Pajkrt D., Doran J. E., Koster F., Lerch P. G., Arnet B., van der Poll T., ten Cate J. W., van Deventer S. J. (1996) Antiinflammatory effects of reconstituted high-density lipoprotein during human endotoxemia. J. Exp. Med. 184, 1601–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Parker T. S., Levine D. M., Chang J. C., Laxer J., Coffin C. C., Rubin A. L. (1995) Reconstituted high-density lipoprotein neutralizes Gram-negative bacterial lipopolysaccharides in human whole blood. Infect. Immun. 63, 253–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Levine D. M., Parker T. S., Donnelly T. M., Walsh A., Rubin A. L. (1993) In vivo protection against endotoxin by plasma high density lipoprotein. Proc. Natl. Acad. Sci. U.S.A. 90, 12040–12044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eskandari M. K., Bolgos G., Miller C., Nguyen D. T., DeForge L. E., Remick D. G. (1992) Anti-tumor necrosis factor antibody therapy fails to prevent lethality after cecal ligation and puncture or endotoxemia. J. Immunol. 148, 2724–2730 [PubMed] [Google Scholar]
- 30. Poltorak A., He X., Smirnova I., Liu M. Y., Van Huffel C., Du X., Birdwell D., Alejos E., Silva M., Galanos C., Freudenberg M., Ricciardi-Castagnoli P., Layton B., Beutler B. (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088 [DOI] [PubMed] [Google Scholar]
- 31. Echtenacher B., Freudenberg M. A., Jack R. S., Männel D. N. (2001) Differences in innate defense mechanisms in endotoxemia and polymicrobial septic peritonitis. Infect. Immun. 69, 7271–7276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vazquez-Torres A., Vallance B. A., Bergman M. A., Finlay B. B., Cookson B. T., Jones-Carson J., Fang F. C. (2004) Toll-like receptor 4 dependence of innate and adaptive immunity to Salmonella: importance of the Kupffer cell network. J. Immunol. 172, 6202–6208 [DOI] [PubMed] [Google Scholar]
- 33. Zhang Z., Datta G., Zhang Y., Miller A. P., Mochon P., Chen Y. F., Chatham J., Anantharamaiah G. M., White C. R. (2009) Apolipoprotein A-I mimetic peptide treatment inhibits inflammatory responses and improves survival in septic rats. Am. J. Physiol. Heart Circ. Physiol. 297, H866–H873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rittirsch D., Huber-Lang M. S., Flierl M. A., Ward P. A. (2009) Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc. 4, 31–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Quezado Z. M., Natanson C., Banks S. M., Alling D. W., Koev C. A., Danner R. L., Elin R. J., Hosseini J. M., Parker T. S., Levine D. M. (1995) Therapeutic trial of reconstituted human high-density lipoprotein in a canine model of Gram-negative septic shock. J. Pharmacol. Exp. Ther. 272, 604–611 [PubMed] [Google Scholar]
- 36. Feng H., Guo L., Song Z., Gao H., Wang D., Fu W., Han J., Li Z., Huang B., Li X. A. (2010) Caveolin-1 protects against sepsis by modulating inflammatory response, alleviating bacterial burden, and suppressing thymocyte apoptosis. J. Biol. Chem. 285, 25154–25160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Guo L., Song Z., Li M., Wu Q., Wang D., Feng H., Bernard P., Daugherty A., Huang B., Li X. A. (2009) Scavenger receptor BI protects against septic death through its role in modulating inflammatory response. J. Biol. Chem. 284, 19826–19834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ocuin L. M., Bamboat Z. M., Balachandran V. P., Cavnar M. J., Obaid H., Plitas G., DeMatteo R. P. (2011) Neutrophil IL-10 suppresses peritoneal inflammatory monocytes during polymicrobial sepsis. J. Leukoc. Biol. 89, 423–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yurdakul P., Dalton J., Beattie L., Brown N., Erguven S., Maroof A., Kaye P. M. (2011) Compartment-specific remodeling of splenic micro-architecture during experimental visceral leishmaniasis. Am. J. Pathol. 179, 23–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Suzuki M., Pritchard D. K., Becker L., Hoofnagle A. N., Tanimura N., Bammler T. K., Beyer R. P., Bumgarner R., Vaisar T., de Beer M. C., de Beer F. C., Miyake K., Oram J. F., Heinecke J. W. (2010) High-density lipoprotein suppresses the type I interferon response, a family of potent antiviral immunoregulators, in macrophages challenged with lipopolysaccharide. Circulation 122, 1919–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sorci-Thomas M. G., Zabalawi M., Bharadwaj M. S., Wilhelm A. J., Owen J. S., Asztalos B. F., Bhat S., Thomas M. J. (2012) Dysfunctional HDL containing L159R apoA-I leads to exacerbation of atherosclerosis in hyperlipidemic mice. Biochim. Biophys. Acta 1821, 502–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wilhelm A. J., Zabalawi M., Grayson J. M., Weant A. E., Major A. S., Owen J., Bharadwaj M., Walzem R., Chan L., Oka K., Thomas M. J., Sorci-Thomas M. G. (2009) Apolipoprotein A-I and its role in lymphocyte cholesterol homeostasis and autoimmunity. Arterioscler. Thromb. Vasc. Biol. 29, 843–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vishnyakova T. G., Bocharov A. V., Baranova I. N., Chen Z., Remaley A. T., Csako G., Eggerman T. L., Patterson A. P. (2003) Binding and internalization of lipopolysaccharide by Cla-1, a human orthologue of rodent scavenger receptor B1. J. Biol. Chem. 278, 22771–22780 [DOI] [PubMed] [Google Scholar]
- 44. Cai L., Ji A., de Beer F. C., Tannock L. R., van der Westhuyzen D. R. (2008) SR-BI protects against endotoxemia in mice through its roles in glucocorticoid production and hepatic clearance. J. Clin. Invest. 118, 364–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hoekstra M., Meurs I., Koenders M., Out R., Hildebrand R. B., Kruijt J. K., Van Eck M., Van Berkel T. J. (2008) Absence of HDL cholesteryl ester uptake in mice via SR-BI impairs an adequate adrenal glucocorticoid-mediated stress response to fasting. J. Lipid Res. 49, 738–745 [DOI] [PubMed] [Google Scholar]
- 46. Barter P. J., Nicholls S., Rye K.-A., Anantharamaiah G. M., Navab M., Fogelman A. M. (2004) Antiinflammatory properties of HDL. Circ. Res. 95, 764–772 [DOI] [PubMed] [Google Scholar]
- 47. Rubin E. M., Ishida B. Y., Clift S. M., Krauss R. M. (1991) Expression of human apolipoprotein A-I in transgenic mice results in reduced plasma levels of murine apolipoprotein A-I and the appearance of two new high density lipoprotein size subclasses. Proc. Natl. Acad. Sci. U.S.A. 88, 434–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yvan-Charvet L., Pagler T., Gautier E. L., Avagyan S., Siry R. L., Han S., Welch C. L., Wang N., Randolph G. J., Snoeck H. W., Tall A. R. (2010) ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 328, 1689–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Acton S., Rigotti A., Landschulz K. T., Xu S., Hobbs H. H., Krieger M. (1996) Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 271, 518–520 [DOI] [PubMed] [Google Scholar]
- 50. Plump A. S., Erickson S. K., Weng W., Partin J. S., Breslow J. L., Williams D. L. (1996) Apolipoprotein A-I is required for cholesteryl ester accumulation in steroidogenic cells and for normal adrenal steroid production. J. Clin. Invest. 97, 2660–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.