

Background: Copper enters human cells through pores formed by trimeric hCTR1 transporters that require intramembrane methionines near the extracellular side.
Results: The copper transport rate is increased by mutations on the intracellular side of hCTR1.
Conclusion: hCTR1 elements on the intracellular side affect the copper transport rate and response to high copper.
Significance: The mutations provide unexpected insight into the hCTR1 transport mechanism.
Keywords: Copper, Membrane Proteins, Membrane Transport, Metals, Transport Metals
Abstract
Human copper transporter 1 (hCTR1) is a homotrimer of a 190-amino acid monomer having three transmembrane domains believed to form a pore for copper permeation through the plasma membrane. The hCTR1-mediated copper transport mechanism is not well understood, nor has any measurement been made of the rate at which copper ions are transported by hCTR1. In this study, we estimated the rate of copper transport by the hCTR1 trimer in cultured cells using 64Cu uptake assays and quantification of plasma membrane hCTR1. For endogenous hCTR1, we estimated a turnover number of about 10 ions/trimer/s. When overexpressed in HEK293 cells, a second transmembrane domain mutant of hCTR1 (H139R) had a 3-fold higher Km value and a 4-fold higher turnover number than WT. Truncations of the intracellular C-terminal tail and an AAA substitution of the putative metal-binding HCH C-terminal tripeptide (thought to be required for transport) also exhibited elevated transport rates and Km values when compared with WT hCTR1. Unlike WT hCTR1, H139R and the C-terminal mutants did not undergo regulatory endocytosis in elevated copper. hCTR1 mutants combining methionine substitutions that block transport (M150L,M154L) on the extracellular side of the pore and the high transport H139R or AAA intracellular side mutations exhibited the blocked transport of M150L,M154L, confirming that Cu+ first interacts with the methionines during permeation. Our results show that hCTR1 elements on the intracellular side of the hCTR1 pore, including the carboxyl tail, are not essential for permeation, but serve to regulate the rate of copper entry.
Introduction
Copper ions enter eukaryotic cells via high affinity membrane transporters consisting of homotrimeric Ctr1 proteins (1). Cells require copper ions as cofactors for enzymes that are involved in redox reactions, such as mitochondrial oxidative phosphorylation and dismutation of superoxides (2). In mammals, free copper ions are not abundant in the serum (3) and are not measureable inside cells (4). Copper ions are bound to and exchanged among proteins or small molecules in the serum, the cytoplasm, and intracellular compartments through which copper transits (5–7).
The human copper transporter 1 (hCTR1)3 gene was discovered by virtue of its ability to complement copper deficiency in yeast Ctr1Ctr3 double mutants, which lack high affinity copper uptake (8). hCTR1 also suppresses phenotypes of copper deficiency in Drosophila CTR mutants (9) and mediates copper transport when expressed in insect cells (10). Studies in cultured cells from mouse CTR1 knockouts indicate that CTR1 is the principal high affinity transporter of copper in mammals (11), although other pathways of copper entry clearly exist (12, 13). It is generally believed that hCTR1 transports Cu+ and not Cu2+ ions (14). For example, Ag+ inhibits copper transport by hCTR1, whereas divalent metals do not (12).
Structural studies have revealed that the three highly conserved membrane-spanning domains in CTR1 proteins form a channel or permeation path for copper ions through the plasma membrane. Low resolution cryoelectron microscopy studies of the 190-amino acid hCTR1 transporter show that the transporter is a homotrimer, in which the N terminus is extracellular, the transmembrane domains form the pore, and the short C-terminal tail is cytoplasmic (15, 16). Previous biochemical studies suggested this topology (17–19), and multiple studies suggest that the second transmembrane domain lines the channel or pore (10, 20, 21).
Two methods have been used for structure/function studies of hCTR1 transport: evaluating yeast Ctr1 mutants by their ability to assemble in the plasma membrane and/or to grow in low copper (20, 21) and radioactive copper uptake in cells expressing hCTR1 mutants. Using 64Cu uptake as a measure of hCTR1 function, mutational studies have identified amino acid residues important for transport. Methionines in the N terminus (Met-43, Met-45) and second transmembrane domain (Met-150, Met-154) on the extracellular side are required for efficient copper uptake by hCTR1 (10, 22, 23). Other residues important for transport were identified in our previous study of 64Cu uptake in insect cells expressing hCTR1 mutants. In cells expressing H139R, both Km and Vmax for copper transport were substantially increased (10). Because His-139 is located in the second transmembrane domain near the cytoplasmic side, it has been proposed that the introduction of a positive charge in the copper permeation pathway affected the conformation of the transporter, facilitating faster transit of Cu+ ions through the pore (10, 24). De Feo et al. (16) proposed a model wherein Met-150 and Met-154 (near the extracellular aspect of hCTR1) provide an initial binding site in the pore (possibly serving as a selectivity filter), and C-terminal HCH motifs receive the metal ion at the cytoplasmic face and transfer it to copper chaperones or small molecules. A recent all-atom model of hCTR1 (25) also emphasizes the roles of the methionine residues in coordinating the permeating copper ions.
These studies suggest that the influx of copper ions across the plasma membrane via mammalian CTR1 transporters could take place by: 1) transfer of ligand-bound copper ions to the extracellular portion of the CTR1 homotrimer; 2) binding to Met-150 and Met-154 residues in the pore near the extracellular side followed by transit of copper ions through a pore composed of CTR1 transmembrane domains, to a second binding site, such as the C-terminal HCH motif; and 3) ligand exchange of copper at the cytoplasmic face of hCTR1 to molecules such as intracellular copper chaperones or glutathione (14, 16, 24). Numerous other studies have contributed to this model of CTR1 transport (10, 15–17, 20, 22). An alternative hCTR1-mediated copper uptake mechanism has been proposed that involves endocytosis of copper bound to hCTR1 followed by intracellular release of the metal and subsequent degradation of hCTR1 (26, 27) To clearly distinguish between these or other possible transport mechanisms, more experimental evidence is needed.
In this study, we estimated, for the first time, that the turnover number (maximal rate of copper transported per hCTR1 trimer) for hCTR1 in human cells is about 10 per second. We also showed that mutations on the intracellular side of the transporter substantially increase the rate of transport. We showed that the H139R mutation in the second transmembrane segment and mutations in the intracellular C-terminal tail result in a higher rate of copper transport and higher Km values for copper. Experiments with double mutants confirm that the intracellular side mutants act at a step after copper interacts with the methionines on the extracellular side of the pore. The intracellular side mutants, unlike the WT (wild type) protein, are not endocytosed in the presence of elevated extracellular copper. Our results suggest that rather than being a necessary part of the permeation pathway, the C-terminal tail serves a regulatory function. The tail appears to act to limit transport through the pore by regulating the rate of exit of copper ions at the intracellular side. In addition, the C-terminal tail is important in the regulatory endocytosis of hCTR1 in high external copper.
EXPERIMENTAL PROCEDURES
hCTR1 WT and Mutant Expression Constructs
A WT hCTR1 cDNA clone was obtained from Dr. J. Gitschier, University of California, San Francisco (UCSF) (GenBankTM accession number U83460, HUGO Gene Nomenclature Committee (HGNC)). As described previously (28), an N-terminal FLAG tagged hCTR1 clone was constructed using this cDNA. The H139R and C-terminal mutants were derived from the FLAG-tagged hCTR1 clone pEM94 (Ref. 29, available on request). H139R was made as described previously (29) with a site-directed mutagenesis kit (Stratagene, Carlsbad CA). H139R primers are: forward, 5′-CTGCAAACAGTGCTGCGCATCATCCAGGTGGTC, and reverse: 5′-GACCACCTGGATGATGCGCAGCACTGTTTGCAG. hCTR1 C-terminal truncation mutants (numbered by the last retained amino acid) and a mutant in which the terminal 3 amino acids HCH were replaced by alanines (AAA)4 were amplified with PCR primers containing compatible restriction sites for insertion into the pcDNATM5/FRT/TO vector. Forward primer (pEM94) was: 5′-CTCTAGAACTAGTGGATCCACCATGGACTACAA, and reverse primers were: tr179, 5′-GCTCTCGAGTTGTCACTTCTTCCAGCTGAAGAGGAAGTATCCTGT; tr183, 5′-GCTCTCGAGTTGTCACACTACCACTGCCTTCTTCCAGCTGAAGAGGA; and HCH-AAA, 5′-GCTCTCGAGTTGTCATGCAGCTGCCTCTGTGATATCCACTACCACTGCCTTC. Fragments containing the mutant hCTR1 coding sequences were cloned into the pcDNATM5/FRT/TO cloning vector (Life Technologies). Oligonucleotides used to make the M150L,M154L double mutant were: forward primer: 5′-AGCTACTTCCTCCTGCTCATCTTCCTGACCTACAACGGGT, and reverse primer: 5′-ACCCGTTGTAGGTCAGGAAGATGAGCAGGAGGAAGTAGCT. Combination mutants (e.g. M150L,M154L; HCH-AAA) were made in sequential steps using the same methods used for each mutant alone.
Cell Culture
HEK293 Flp-InTM T-RExTM cells (Life Technologies) were cultured in Dulbecco's minimal essential medium (DMEM) (Life Technologies), 25 mm Hepes buffer, and tetracycline (tet)-free 10% fetal bovine serum (FBS, Atlanta Biologicals, Atlanta, GA) and maintained with selective antibiotics as described below. Caco-2 cells (ATCC) were cultured in Eagle's minimum essential medium with Earle's balanced salt solution and 2 mm l-glutamine (ATCC) and supplemented with 20% FBS. All cells were grown at 37 °C in 5% CO2. HEK293 cells were subcultured every 3–5 days. Caco-2 cells were grown on Transwell culture plates as described previously (30).
Cell lines containing tetracycline-regulated FLAG-tagged WT and mutant hCTR1 genes were created in HEK293 Flp-InTM T-RExTM cells using Invitrogen Flp-InTM T-RExTM vectors. The cells were transfected with hCTR1 constructs using Lipofectamine 2000 (Life Technologies). Transfected cells were selected in 12 μg/ml blasticidin S (RPI Corp., Mount Prospect, IL) and 400 μg/ml hygromycin (Life Technologies). Resistant colonies were pooled and tested for tetracycline-regulated expression.
64Cu Uptake Assays
64Cu uptake assays in HEK293 cells were performed in 12-well tissue culture plates. hCTR1 was overexpressed by adding medium containing 1 μg/ml tetracycline 48 h prior to the assay. One day prior to the assays, 12-well tissue culture plates were seeded with 1 × 106 cells/well in DMEM-10%. The following day, the cells were washed once with DMEM-10% (without antibiotics) and then incubated in 1 ml of DMEM-10% containing 2.0–20 μm added CuCl2 and trace amounts of 64Cu (2–4 × 106 cpm/well) for 5 min at room temperature or 30 min at 37 °C (64Cu uptakes from 5-min incubations were subtracted from 30-min incubations to correct for nonspecific binding). In experiments measuring the kinetics of copper uptake, no correction was made for initial nonspecific binding. Copper uptake was stopped by removing 64Cu and adding ice-cold buffer (28), after which the cells were washed twice with the cold buffer and then dissolved in 0.4% Triton X-100, 10 mm DTT, 2 mm EDTA, and 20 mm Tris, pH 8.0, and incubated for >10 min at 37 °C. Lysed cells were collected, and half the volume was counted in a Beckman LS 6500 scintillation counter. The amount of 64Cu taken up by cells in each experiment was converted from cpm values to pm copper (see Ref. 10). A portion of the lysed cells was used for protein determination. The copper uptake was calculated as pm copper/min/well or as pm copper/mg of cellular protein/min. Each condition was performed in 3–8 replicate wells and reported as the mean uptake ± S.D. Where 64Cu uptake was expressed as atoms of copper transported per hCTR1 trimer per minute, replicate wells not receiving 64Cu were mock-incubated, washed with ice-cold sample buffer, and biotinylated (see below) to obtain an estimate of the number of transporters per well.
64Cu uptake assays in Caco-2 cells were performed in 6-well Transwell plates. Caco-2 cells were plated on 24-mm polyester membrane Transwell inserts and cultured as described previously (30). Copper uptake measurements were performed at 2.5 μm CuCl2 on either apical or basolateral membrane of polarized Caco-2 cells as described previously (30) Duplicate Transwell culture plates were used for protein determination.
Cell Surface Biotinylation
Biotinylation of cell surface proteins was carried out using cell-impermeable, thiol-cleavable Sulfo-NHS-SS-biotin (Thermo-Fisher-Pierce). All of the biotinylation procedures were performed at 4 °C. Caco-2 cells were grown in Transwell culture plates until polarized, and either the apical or the basolateral membrane was biotinylated (30, 31).
PAGE and Western Blots
PAGE and subsequent Western blotting was performed as described previously (31). The following primary antibodies were used: rabbit anti-hCTR1 antibody against hCTR1 C-terminal tail (10), mouse anti-FLAG (GenScript, Piscataway, NJ), rabbit anti-FLAG (Sigma-Aldrich), mouse anti-Na,K-ATPase β1 (Affinity Bioreagents, Golden, CO), mouse anti-β-catenin (BD Biosciences), and mouse anti-α1 subunit of Na,K-ATPase (Affinity Bioreagents). Secondary antibodies were donkey anti-rabbit HRP (GE Healthcare) or goat anti-mouse HRP (Thermo-Fisher-Pierce). Western blot signals were obtained using luminol-based reagents (Thermo-Fisher-Pierce) and collected with a Chemi-Doc XRS system (Bio-Rad Laboratories). Relative band intensity was determined using Quantity One® software (Bio-Rad).
Calibration of hCTR1 from Individual Wells
To quantify the number of hCTR1 transporters in HEK293 and Caco-2 cells, we used purified hCTR1 protein expressed in yeast as a calibration standard on Western blots (a generous gift of C. De Feo and V. Unger). Biotinylated HEK293 cells from individual wells from 12-well culture plates or biotinylated Caco-2 cells from a single Transwell were prepared as described above and analyzed by SDS-PAGE, after which the gels were transferred to membranes for Western blot analysis. The purified hCTR1 protein was prepared at 2, 1, and 0.5 ng/μl in dilution buffer (150 mm NaCl, 50 mm NaP, pH 7, 0.2% n-dodecyl-β-d-maltoside) to which 2× SB containing 50 mm DTT was added. Transferred membranes were removed from the gels and placed on a filter paper wetted with transfer buffer prior to the dotting of 2, 1, and 0.5 ng/μl of purified hCTR1 protein, 4–5 dots for each concentration, along the edge of the membrane. The membranes were then dried at room temperature for >2 h, after which Western blot analysis was performed with rabbit anti-hCTR1 C-terminal antibody as described (31).
The Western blot signals from hCTR1 bands were quantified using Quantity One® software (Bio-Rad Laboratories). The concentration of the unknown hCTR1 protein was determined from the signal intensity of the known hCTR1 standards. Direct dotting of the protein was superior to running the purified protein in lanes of the same gel due to the propensity of purified unglycosylated hCTR1 protein to form ladders of multimeric species,5 which were difficult to quantitate in the imaging system. We note that direct application of the purified hCTR1 protein standard to the membrane would avoid losses expected to occur during biotinylation, recovery, and transfer of recovered hCTR1 from cells. This disparity would result in an underestimate of hCTR1 from the biotinylated cells. Thus, the calculated values of hCTR1 trimers/cell are considered a lower limit.
Normalizing Expression Levels of hCTR1-expressing Lines
For experiments involving 64Cu uptake assays in which several different lines expressing FLAG-tagged hCTR1 WT and mutants were compared, hCTR1 expression levels were normalized as described previously (28). Western blots from total membranes from each line were probed with FLAG or Na,K-ATPase antibodies and quantified as described (28), and copper uptake levels were adjusted for expression when expressed as the percentage of control uptake. Most lines had expression levels within 10% of WT control.
Cell Assay for Viability and Proliferation
Viability of cells was assayed using PrestoBlueTM (Life Technologies), a nontoxic fluorescence-based indicator that is reduced by metabolic activity of viable, growing cells. The reagent was added directly to cells grown in 24-well plates as directed by the manufacturer, the cells were incubated for 3–4 h, and supernatant fluorescence was subsequently quantified using a FLUOstar Omega plate reader (BMG Labtech, Cary, NJ).
Confocal Imaging
Cells were grown on glass coverslips and treated in 12-well culture plates prior to fixing and staining with antibodies, exactly as described previously (29). Images of HEK293 cells were collected using a Zeiss LSM 700 confocal microscopy system equipped with a 100× objective, controlled with ZenTM software. Instrument settings were not changed when switching between samples.
Chemicals and Protease Inhibitors
Standard reagent chemicals were from Fisher or Sigma-Aldrich. Protease inhibitor mixture tablets were from Roche Applied Science (Mannheim, Germany). 64Cu was produced at Washington University, St. Louis, MO.
Data Analysis
Data from 64Cu uptake experiments were analyzed using Prism 5 (GraphPad Software, La Jolla, CA). The Michaelis-Menten equation was used to determine Km and Vmax values for copper transport by WT and mutant hCTR1 transporters: pmol of copper transported = Vmax × [copper]/(Km + [copper]).
RESULTS
Copper Transport in tet-regulated Cells Expressing hCTR1
To determine the rate of copper transport by endogenous and overexpressed hCTR1 in tet-regulated HEK293 Flp-InTM cells, we used a copper uptake assay described previously (28). Cells were incubated in standard DMEM with 10% FBS and further supplemented with 2 μm CuCl2 containing trace amounts of 64Cu. In this medium, under these conditions, 70–85% copper uptake occurs via hCTR1 (11, 12, 30). We performed the assay in 12-well culture plates having confluent monolayers of cells, using 3–8 replicate wells for each condition. Replicate 12-well plates were prepared during each 64Cu uptake assay to count the number of cells per well and to determine the number of hCTR1 copper transporters per well in each experiment.
We measured the kinetics of copper uptake in HEK293 Flp-InTM cells with or without tet-induced overexpression of hCTR1 in multiple cell lines. As shown in Fig. 1A, copper uptake was linear over the first 30 min. These cell lines had undergone varying numbers of passages after creation or thawing from frozen stocks. The different slopes of each uptake assay reflected the level of overexpression of hCTR1. Fig. 1B shows the relative level of hCTR1 protein in purified plasma membranes from these lines. Long term passage of the same Flp-InTM cell line results in diminished expression from the tet-regulated site, seen as a reduction in slope of HEK293 line 1, comparing medium versus high passage (Fig. 1A). The rate of copper uptake in various cell lines correlated with the abundance of hCTR1 protein expressed in each line. In the absence of tet induction, uptake by endogenous hCTR1 in HEK293 cells is lower but measurable (Fig. 1A).
FIGURE 1.

Copper uptake and expression levels in HEK293 cells. A, 64Cu-uptake kinetics for cells expressing FLAG-tagged (WT) hCTR1 under tetracycline regulation. Line 2 was a newly created line assayed after four passages. Line 1 was a line that was assayed after about 15 (MP, medium passage) or 30 (HP, high passage) passages. B, plasma membranes (10 μg/lane) from tet-induced cell lines shown in A, detected with antibody raised against the hCTR1 C terminus (“C-term ab” (17)). Ovals at the left show monomeric and dimer hCTR1 proteins. C, rates of copper uptake in HEK293 cells with or without tet induction. Data are average of four experiments ± S.D. D, Western blot showing surface biotinylated proteins (detected with C-term ab from replicate wells used in uptake experiments to quantitate hCTR1 trimers/cell). Recovered proteins from 8 wells were loaded in the left lane (− tet) or from half of a single well in the right lane (+ tet). In addition to monomeric and dimeric hCTR1 in the − tet lane, nonspecific reactivity from other proteins (*) due to the large amount (>50 μg of protein) loaded was detected. At right is shown 1 or 2 ng of purified hCTR1 protein dotted on the Western blot membrane. hCTR1 Std, hCTR1 standard.
Rate of Copper Uptake by hCTR1 Transporters
To determine the rate of copper transport per hCTR1 trimer, a direct measurement of the amount of hCTR1 in cells whose uptake of copper was also measured was required. We measured the amount of uptake (as pm/min/well) by HEK293 Flp-InTM cells in 12-well culture plates. Cells were incubated with 64Cu for 5 or 30 min. Copper uptake after 5 min was subtracted from 30-min uptakes to control for nonspecific binding. tet markedly increased copper uptake, which varied between 0.9 and 3.0 pm/min/well (Fig. 1C). The level of endogenous hCTR1 is low in HEK293 cells, resulting in copper uptake averaging 0.2 pm/min/well (Fig. 1C). To determine the number of hCTR1 transporters per cell, we used purified recombinant hCTR1 protein expressed in yeast as a calibration standard on Western blots. To recover and quantify hCTR1 transporters, we used surface biotinylation of cells in replicate wells from 64Cu uptake assays. We previously showed that biotinylation under these conditions results in >95% recovery of hCTR1 in lysates of biotinylated cells (28).
Cells from replicate wells used in the 64Cu uptake assays were biotinylated as described under “Experimental Procedures,” and the recovered proteins were analyzed by Western blot. Biotinylated samples from HEK293 Flp-InTM cells containing endogenous or overexpressed hCTR1 are shown in Fig. 1D. We were unable to quantify endogenous (− tet) hCTR1 from HEK293 cells collected from individual wells as we did with overexpressed hCTR1, but we were able to visualize and quantitate endogenous hCTR1 by pooling multiple wells. As seen in Fig. 1D, pooling multiple wells resulted in heavy protein loading in a single lane, as well as recognition of non-hCTR1 proteins (*) not observed when biotinylated proteins from a single well (or less) were run in a single lane.
The hCTR1 signals from monomeric and dimeric hCTR1 polypeptides were quantified in replicate wells from 64Cu uptake experiments by calibration of the luminol-based Western blot signals with signals from known quantities of the purified hCTR1 protein from yeast Fig. 1D (inset). We estimated that HEK293 Flp-InTM cells not induced with tetracycline contain 5.4 × 108 hCTR1 transporters/well, corresponding to about 250 transporters/cell (Table 1). After induction with tetracycline, the cells in a low passage line (Fig. 1B) contained an average of 1.4 × 1010 transporters/well, or about 6700 transporters/cell (Table 1). Multiple independent experiments were performed with various HEK293 tet-inducible cell lines. In each case, hCTR1 was quantified in replicate wells and was subsequently used for only the 64Cu uptake experiment done in the same replicate set of wells.
TABLE 1.
Comparison of copper turnover rate (s−1) for hCTR1 transporters in Caco-2 and HEK293 cells over-expressing FLAG tagged hCTR1
Copper uptake experiments were performed at 2.5 μm copper and 2 μm copper in Caco-2 and HEK/hCTR1-FLAG cells, respectively. The determination of number of hCTR1 trimers from biotinylated protein was calculated from purified recombinant hCTR1 protein (see “Experimental Procedures”). Exp, experiment.
| Cells | hCTR1 trimers/wella | Trimers/cellb | pmol of copper/min/well | Copper ions/hCTR1/sc | Turnover rate of copper ions/sd |
|---|---|---|---|---|---|
| HEK293 − tet | |||||
| Exp 1 | 6.5 × 108 | 287 | 0.16 | 2.5 | 10.9 |
| Exp 2 | 4.6 × 108 | 218 | 0.22 | 4.7 | 19.3 |
| Exp 3 | 6.3 × 108 | 266 | 0.19 | 2.8 | 12.3 |
| Average | 5.8 (±0.9) × 108 | 253 (±36) | 0.19 (±0.03) | 3.5 (±0.38) | 14.2 (±3.7) |
| HEK293 + tet | |||||
| Exp 1 | 1.1 × 1010 | 5850 | 1.9 | 1.3 | 5.7 |
| Exp 2 | 1.6 × 1010 | 8000 | 2.3 | 1.4 | 6.0 |
| Exp 3 | 1.5 × 1010 | 6250 | 1.8 | 1.8 | 7.9 |
| Exp 4e | 6.5 × 109 | 3622 | 0.68 | 1.1 | 4.9 |
| Exp 5e | 4.9 × 109 | 2843 | 0.5 | 1.2 | 5.3 |
| Average | 1.4 (±0.2) × 1010 | 6700 (±117) | 2.1 (±0.2) | 1.4 (±0.24) | 6.0 (±1.0) |
| Caco-2 | |||||
| Exp 1 | 2.2 × 109 | 654 | 0.64 | 2.9 | 10.2 |
| Exp 2 | 2.3 × 109 | 740 | 0.98 | 4.6 | 16.4 |
| Exp 3 | 2.7 × 109 | 618 | 1.26 | 4.5 | 16.8 |
| Average | 2.4 (±0.2) × 109 | 671 (±63) | 1.20 (±0.19) | 4.0 (±0.8) | 14.5 (±4.15) |
a Calculated from Western blots using purified mass standard as described under “Experimental Procedures.” 1 ng of purified hCTR1 = 2.74 × 1010 hCTR1 monomers (9.12 × 109 trimers).
b Determined by counting cells in replicate wells (or Transwell culture plates for Caco-2 cells) used in 64Cu uptake experiments and dividing total trimers/well by average cells/well. For HEK293, the average cells/well was 2.1 × 106, and for Caco-2, the average cells/Transwell was 3.6 × 106.
c Calculated by converting pmol to ions, dividing copper ions/min/well by average trimers/well, and converting to seconds.
d Calculated using a Km of 4.4 μm (Fig. 3), and Vmax = 2 x velocity at Km. Copper uptakes were done in 2 μm (HEK293) or 2.5 μm (Caco-2), assuming a linear rate of uptake at values below Km.
e Experiments 4 and 5 were done with a high passage line (>20 passages), like that shown in Fig. 1A. tet-induced expression declines with high passage. The bolded values were not included in the averages below.
Based on the 64Cu uptake values and our estimates of the number of trimeric hCTR1 transporters per cell, the rate of copper transport per (overexpressed) hCTR1 trimer was 1.4 copper ions/second with 2 μm copper in DMEM. In HEK293 Flp-InTM cells not induced with tetracycline, the rate of copper transport per (endogenous) hCTR1 trimer was 3.5 copper ions/second. From these values, the turnover number (transport rate at Vmax) for endogenous hCTR1 was about 14 copper ions/s/trimer, and 6 copper ions/s/trimer for overexpressed hCTR1.
Rate of Copper Uptake and Copy Number of hCTR1 in Caco-2 Cells
We also estimated the rate of copper uptake per hCTR1 transporter on the basolateral side of polarized Caco-2 cells. We have previously shown that Caco-2 cells exhibit a relatively high level of hCTR1 expression on the basolateral surface when grown in Transwell culture plates. For this reason, we used Caco-2 basolateral hCTR1 as a second source of endogenous hCTR1 whose transport properties we previously characterized (30). Caco-2 cells were grown in Transwell culture plates until they formed tight junctions as determined by transepithelial resistance measurements (30). We measured 64Cu uptake in apical and basolateral compartments in separate Transwell culture plates. Basolateral copper uptake for Caco-2 cells was on average 1.2 pm/mg of protein/min (Fig. 2A). We determined the number of basolateral hCTR1 transporters by biotinylating cells from replicate Transwell culture plates used for 64Cu uptake assays. Polarization of Caco-2 cells was confirmed by the basolateral localization of the β1-subunit of Na,K-ATPase (Fig. 2B). The number of endogenous Caco-2 hCTR1 transporters was quantified by calibrating Western blot signal of Caco-2 hCTR1 (Fig. 2B) with purified yeast hCTR1 as described for HEK293 cells (30). We estimated that there were 2.3 × 109 hCTR1 trimers per well corresponding to an average of about 670 hCTR1 trimers per Caco-2 cell, which had a transport rate of about 4 Cu+ ions/s, at 2 μm copper or a turnover number of 14.5 Cu+ ions/s/trimer (Table 1). This rate agrees well with the value we estimated for endogenous hCTR1 in HEK293 cells.
FIGURE 2.

Cell surface biotinylation and copper uptake measurements in intestinal epithelial cells, Caco-2. Caco-2 cells were grown on Transwell culture plates until they had formed tight junctions as described under “Experimental Procedures.” A, 64Cu uptake assays performed on either the apical or the basolateral compartment of Caco-2 cells. Shown is the average of three experiments ± S.D. B, Western blot of biotinylated apical or basolateral (Baso.) membranes of Caco-2 cells, each from a single Transwell. Recovered protein was split between two lanes. Top: hCTR1 was detected using the hCTR1 C-term ab. Bottom: the same blot was probed with Na,K-ATPase β1 ab (Na,K-ATPase also localizes to the basolateral membrane).
H139R Mutant and Copper Uptake
Although previous studies of hCTR1 in mammalian cells have examined the role of residues in the N terminus and on the extracellular side of the pore (22, 23, 28, 29), little has been reported on the role of residues on the intracellular side of the pore. We next characterized an hCTR1 mutant in HEK293 cells having a histidine to arginine substitution at position 139 (H139R) in the second transmembrane segment, which we previously showed to have an elevated rate of copper transport when expressed in insect cells (10). We expressed H139R in HEK293 Flp-InTM cells and measured the rate of copper uptake using 64Cu (Fig. 3). As shown in Fig. 3, the H139R mutant expressed in HEK293 cells had an elevated Vmax and Km when compared with WT transporter. Using biotinylation of cells expressing the H139R mutant calibrated with purified yeast protein, we determined copy number and copper transported in several experiments, resulting in a turnover number (using a Km of 12.9 μm) of 18.4 ± 5.9 copper ions/s/trimer, versus 4.4 ± 1.8 for WT protein in the same experiment (Fig. 3).
FIGURE 3.

Michaelis-Menten plots of the concentration dependence of copper uptake in HEK293 cells expressing WT hCTR1 or H139R mutant. Shown are the results from one of three experiments. Below are shown the values of the Km and turnover numbers for WT or H139R. Error bars indicate ± S.D.
C-terminal Mutants
We also characterized mutants with substitutions or truncations of the hCTR1 (cytoplasmic) C-terminal tail that had copper transport phenotypes similar to that of H139R. We analyzed a mutant in which the last three C-terminal amino acids (HCH, a putative metal-binding motif (16)) were changed to alanines, hereafter referred to as AAA. We also analyzed two truncations, lacking 7 or 11 C-terminal residues, hereafter referred to as tr183 and tr179, respectively (Fig. 4A). The three mutants all exhibit elevated copper uptake rates when compared with WT protein, particularly at higher copper concentrations (Fig. 4B). The truncation mutants had high 64Cu uptake despite being relatively poorly expressed (from the same expression site in the HEK293 cells used for WT or AAA hCTR1 proteins.). As with the H139R mutant, Km and turnover for the AAA and tr179 mutants were elevated (Fig. 5). The estimated turnover number for the AAA mutant was 17.4 copper ions/s/trimer. In the case of tr179, we could not directly quantitate the levels of biotinylated mutant protein with the anti-C-terminal antibody used with other hCTR1 proteins (because it lacks the amino acids in the immunizing peptide), but based on relative expression levels (Fig. 4), we estimate 30–40 copper ions/transporter/s.
FIGURE 4.

hCTR1 C-terminal mutants and copper uptake. A, amino acid sequence of the hCTR1 C terminus in WT and three mutants. B, 64Cu uptake in the mutants shown in A at 5 and 20 μm added copper. Error bars indicate ± S.D. C, Western blot showing the relative expression level of wild type and the mutants. 10 μg of plasma membrane protein was run in each lane. Anti-FLAG antibody shows the ∼35-kDa monomer and ∼70-kDa dimer forms of overexpressed hCTR1 (FLAG-tagged) proteins. Na,K-ATPase α-subunit was used as a loading control.
FIGURE 5.

Michaelis-Menten plots of the concentration dependence of copper uptake in HEK293 cells expressing hCTR1 AAA (A) or tr179 (B) mutants. Km and turnover numbers are shown below. Error bars in panels A and B indicate ± S.D.
Metal Ion Selectivity of C-terminal Mutants
The C-terminal mutants exhibit increases in the Km for copper transport, suggesting a reduced affinity for the metal. This could conceivably also result in a change in the metal ion selectivity of hCTR1. The selectivity has previously been investigated by adding excess competitor metals in 64Cu transport assays. WT hCTR1 is significantly inhibited by Ag+, but not by divalent metals (12). We tested cells expressing WT, AAA, and tr179 hCTR1 proteins for inhibition of 64Cu transport by three metals (Fig. 6). We found that Zn2+ and Cd2+ had no significant effect on 64Cu transport, whereas Ag+ did inhibit transport. Thus, the C-terminal mutants do not appear to have a relaxed selectivity for metals.
FIGURE 6.
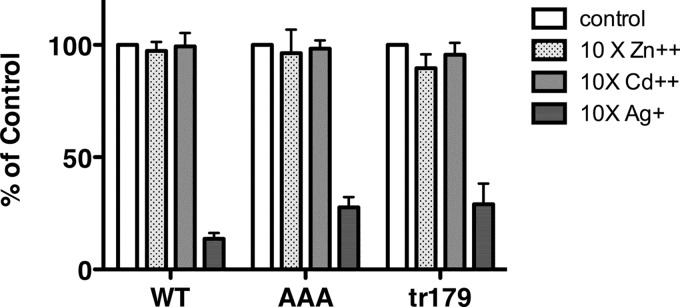
Inhibition of hCTR1 64Cu uptake by competition with other metals. HEK293 cells overexpressing WT, AAA, or tr179 hCTR1 proteins were incubated in media containing 2 μm (WT) or 10 μm (AAA and tr179) copper, trace amounts of 64Cu, and a 10-fold excess of Zn2+, Cd2+, or Ag+. 64Cu uptake is expressed as the percentage of uptake in cells without a competitor metal. Values are the mean of three separate experiments ± S.D.
hCTR1 Mutants Define Separate Steps in the Copper Permeation Mechanism
It was shown previously that two highly conserved methionines in the second transmembrane domain near the extracellular side of hCTR1 (Met-150 and Met-154) are required for copper uptake (22). In contrast to these extracellular side methionine mutations, the mutants we describe on the intracellular side of hCTR1 have higher rates of copper transport and a higher Km than WT protein. We used the elevated transport rate phenotype of the intracellular side mutants to test the hypothesis that the Met-150 and Met-154 mutants are essential for the first step in Cu+ permeation through the pore. If so, mutations on the cytoplasmic side that elevate copper transport should have no effect when combined with the M150L,M154L substitutions. We made mutant combinations that include M150L,M154L (MM) and either H139R or AAA and compared copper uptake between cells expressing these mutants and the “single mutants” (MM or AAA are considered as single mutants). As previously observed (22), cells expressing MM had greatly reduced 64Cu uptake in comparison with WT (Fig. 7A). The rate of copper uptake in cells expressing the double mutants (MM + H139R or AAA) was the same as cells expressing M150L,M154L alone (Fig. 7A.). We also measured copper uptake in cells expressing hCTR1 combination mutants having both H139R and the AAA mutations. The double mutant uptake was similar to either single mutant alone (Fig. 7B).
FIGURE 7.

Copper uptake of hCTR1 mutants. A, hCTR1 WT, MM, H139R, and AAA and combination mutants described under “Results” were expressed in HEK293 Flp-InTM T-RExTM cells and induced with tetracycline. 64Cu uptake assays were done in 10 μm total copper. Uptake values were normalized to cells overexpressing WT, which was set to 100%. Normalized values (“Experimental Procedures”) from three independent experiments are shown ± S.D. B, hCTR1 WT, H139R, H139R AAA, and AAA mutants were assayed for 64Cu uptake exactly as in panel A.
Mutants Affecting the Cytoplasmic Side of hCTR1 Lack Copper-dependent Endocytosis
We used the elevated rate of copper uptake in cells expressing H139R to test a model for hCTR1-mediated copper transport in which it was proposed that hCTR1 contains extracellular binding sites for copper, which serve as a vehicle for copper transport after endocytosis of the hCTR1-copper complex (26, 27), analogous to iron uptake by the transferrin receptor (32). The model is based on the observation that preincubation of cells in high copper conditions stimulates endocytosis of hCTR1 (26, 27, 31). If endocytosis is required for copper transport, then the process should occur at an elevated rate in cells expressing H139R when compared with cells expressing WT, reflecting the relative rates of copper uptake. We preincubated cells expressing the H139R mutant and WT hCTR1 in media containing 0, 20, or 100 μm copper for 30 min. The cells were then biotinylated, and cell surface hCTR1 was detected by Western blot analysis using hCTR1 C-terminal antibody (Fig. 8A) and quantified as described under “Experimental Procedures.” As a loading control, hCTR1 was normalized to the β1-subunit of Na,K-ATPase. There was between 35 and 60% decrease in WT cell surface hCTR1 after exposure to 20–100 μm copper (Fig. 8B), in agreement with our previous study (31). However, there was no change in cell surface level of hCTR1 in H139R mutant cells after the addition of extracellular copper (Fig. 8B). Although the H139R mutant has an elevated rate of copper transport, it does not undergo detectable endocytosis. This shows that copper transport does not occur via the endocytosis of the hCTR1-copper complex.
FIGURE 8.

Effect of extracellular copper on cell surface hCTR1 in WT and HEK-H139R expressing cells. A, Western blot of surface-biotinylated proteins was detected using hCTR1 C-term ab or Na,K-ATPase β1 ab. B, quantification of Western blots like that shown in A. Relative amount hCTR1 in cells treated in high copper (gray and black bars) is given as the percentage of the level in cells with no added copper in the media. Values are the mean of three separate experiments ± S.D.
Because the C-terminal mutants and H139R resulted in a similar phenotype (increased rate and higher Km for copper uptake, Figs. 3 and 5), we were curious about whether the AAA and tr179 mutants also did not respond to elevated copper with regulatory endocytosis. Even 100 μm copper pretreatment did not result in endocytosis of the AAA and tr179 mutants. In these same experiments, biotinylated plasma membrane WT hCTR1 was reduced by 45–50% (Fig. 9).
FIGURE 9.
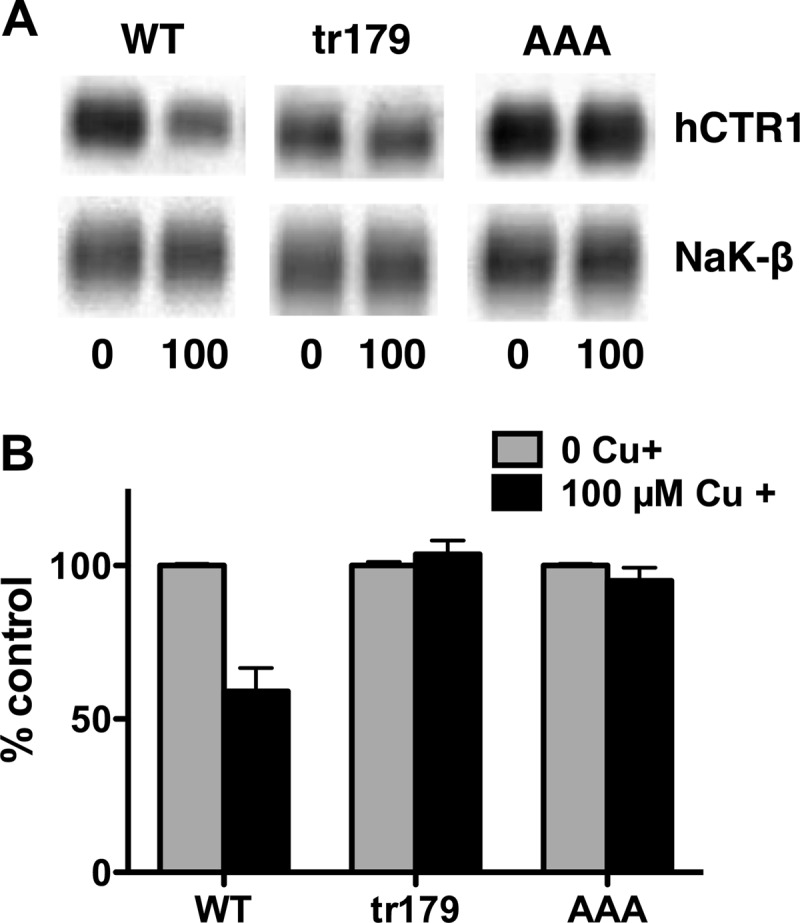
Effect of extracellular copper on cell surface hCTR1 in WT and C-terminal mutant expressing cells. Cells were treated with 100 μm copper for 1 h prior to being biotinylated. A, Western blots of biotinylated proteins were probed with anti-FLAG (overexpressed hCTR1 proteins) and anti-Na,K-ATPase β1-subunit antibodies. B, quantification of Western blots. Relative amount hCTR1 in cells treated in high copper (black bars) is given as the percentage of the level in cells with no added copper in the media. Values are the mean of three separate experiments ± S.D.
We also performed confocal imaging on cells overexpressing WT or AAA hCTR1 (FLAG-tagged) proteins treated with or without copper (Fig. 10). As seen when comparing Fig. 10, A–C (no added copper) with Fig. 10, D–F (incubation for 60 min with 25 μm added copper), a significant portion of the signal from the anti-FLAG antibody detecting WT hCTR1 underwent a clear redistribution to relatively large punctate structures inside the cells after copper treatment (Fig. 10D, arrows), no longer co-localized with the β-catenin membrane marker staining. In contrast, there was no similar change in the distribution of the hCTR1 signal in cells overexpressing the AAA mutant (Fig. 10, G–I versus J–L). Smaller, less intense puncta (seen in some cells overexpressing hCTR1) appeared more abundant in some AAA mutant calls treated with copper (Fig. 10J, arrow), but there was no redistribution to the large vesicles or significant loss of signal from the membrane, as seen in the WT cells treated with copper. Most of the hCTR1 signal in AAA mutant cells treated with copper overlapped β-catenin staining.
FIGURE 10.

Confocal imaging of cells treated with copper. Cells were treated with normal media or media containing 25 μm copper for 60 min prior to fixation and staining with mouse anti-FLAG (green, detects hCTR1) and rabbit anti-β-catenin (red, β-catenin is primarily found in membranes). In each panel, cells were overexpressing the following: A–C, WT hCTR1, no added copper in the media; D–F, WT hCTR1, 25 μm added copper in media; G–I, hCTR1 AAA mutant, no added copper in the media; J–L, hCTR1 AAA mutant, 25 μm added copper in media.
Elevated Copper Toxicity in Cells Expressing AAA Mutant
Because the C-terminal mutants exhibited an increase in copper uptake rates and failed to undergo endocytosis in response to high copper, we considered the possibility that cells expressing these mutants might be more sensitive to the toxic effects of copper. We tested the sensitivity of cells expressing the WT hCTR1or the AAA mutant for sensitivity to copper toxicity. As shown in Fig. 11A, cells overexpressing WT hCTR1 (or endogenous hCTR1 in the case of cells not induced by tetracycline) exhibited a slight but consistent decrease in viability with increasing copper in the medium. In contrast, overexpression of the AAA mutant resulted in a consistently greater decrease in viability when compared with WT or uninduced cells. Both cell lines with or without tet induction had equivalent sensitivity to cadmium, a toxic divalent metal (Fig. 11B) not transported by hCTR1.
FIGURE 11.

Metal toxicity in cells expressing WT hCTR1or AAA mutant hCTR1 proteins. A, PrestoBlueTM fluorometric viability/proliferation assay in HEK293 cells with or without tetracycline induction of hCTR1 WT (WT) or C-terminal AAA mutant (AAA), with indicated concentrations of CuCl2 in the media for 12 h prior to assay. 100% fluorescence is the value in arbitrary units of cells not treated with excess copper. B, as in A, with indicated concentrations of CdCl2 in the media for 12 h prior to assay. Values are the mean of three separate experiments ± S.D.
DISCUSSION
The experiments we report here provide new insights into hCTR1 copper transport and its regulation. We estimated the turnover number of hCTR1 in cultured cells (number of copper ions transported per unit of time per trimer at saturating concentrations) by overexpressed and endogenous hCTR1 transporters. We found that hCTR1 transports about 5–15 copper atoms per second in saturating conditions. To our knowledge, these are the first such estimates of the rate of copper transport mediated by hCTR1. We further estimated that the number of endogenous hCTR1 transporters per cell is in the hundreds.
In addition, we characterized hCTR1 mutations affecting structural elements on the intracellular side of the trimer that increase both Km and the turnover number of copper transport. These mutants include H139R, located near the intracellular side of hCTR1 in the pore-forming second transmembrane segment (10), truncations of the short cytoplasmic C-terminal tail, and AAA substitution of the putative copper-binding motif HCH at the C terminus of the hCTR1 polypeptide. Unlike methionine mutants (M150L,M154L) on the extracellular side of hCTR1 pore that abolish copper transport (22), each of the mutants we describe elevates the rate of transport, suggesting that elements on the intracellular side of the transporter function to reduce the rate of transport. Finally, we showed that cells expressing the mutants on the cytoplasmic side of hCTR1 do not respond to elevated copper in the medium by endocytic down-regulation of hCTR1, as do cells expressing WT hCTR1.
Turnover Numbers for Copper Transport
To date, there have been no estimates of the rate of copper transport by hCTR1, which has an unusual homotrimeric structure that includes a pore formed by membrane-spanning segments from each monomer (15, 16). Under the conditions of our assay, overexpressed hCTR1 trimers in tet-regulated HEK293 transported 1–2 copper ions per second in standard DMEM with 10% FBS containing 2–2.5 μm added copper, corresponding to a turnover rate of 4–5 copper atoms per second per hCTR1 trimer (assuming a Km of 4–5 μm, Table 1). Endogenous hCTR1 in uninduced HEK293 or CaCo-2 cells had turnover numbers of about 14 copper atoms per second, respectively, in similar media (Table 1). It seems likely that this difference reflects uncertainties in the quantitation of low abundance endogenous transporters. Loss of hCTR1 during biotinylation, purification, and gel transfer could reduce the fractional recovery of endogenous hCTR1 to a greater extent than the more abundant overexpressed protein. This would result in a lower relative estimate of copy number, and subsequently, a higher rate per transporter as we observed for endogenous hCTR1 transporters. In any case, the Km measured for both overexpressed and endogenous hCTR1 has been consistent in several different studies in mammalian cells, between 3 and 5 μm (Refs. 12, 31, and 23 and this work).
The estimated turnover number for endogenous or overexpressed hCTR1 is therefore between about 5 and 15 ions per second (Table 1), a rate within the range of transporters in mammalian cells (33–37). For example, the mouse GABA transporter GAT4 has a turnover rate of about 1.5/s in frog oocytes at 20 °C, −50 mV, with an estimated physiological turnover rate of 15–20/s, similar to the turnover we estimate for hCTR1 (36).
The Roles of Intracellular hCTR1 Residues in Copper Permeation
Previous lines of evidence suggest that the pore formed by hCTR1 trimers allows the permeation of Cu+ ions by a series of ligand-exchange reactions as the metal ion passes through rings of stabilizing methionine residues, Met-150 and Met-154, on the extracellular side of hCTR1 (16, 38). Entry is presumably followed by membrane permeation via coordinating steps with intra-pore residues and subsequent delivery to an intracellular acceptor (14, 38). We characterized several mutants on the intracellular side of hCTR1 that elevate both the rate and the Km value of copper transport. We previously identified a transmembrane mutation (H139R) near the intracellular side of the hCTR1 that elevated both Km and Vmax (10) when this mutant was expressed in insect cells (27 °C). In this work, we found that the mutant also has an elevation of Km and turnover number when compared with WT when expressed in HEK293 cells at 37 °C (Fig. 4). To date, this is the only mutation in the pore that elevates copper transport and affects the Km of transport. We and others (10, 24) have speculated that the introduction of positively charged arginine residues near the intracellular exit site in the pore might cause distortion of the pore and facilitate exit of Cu+ ions from the transporter.
We also investigated the role of the C-terminal tail in hCTR1 copper permeation. Our experiments suggest that the tail functions in a manner that is distinctly different from that previously supposed. Replacement of the C-terminal three amino acids (HCH to AAA) or removal of the terminal 7 or 11 amino acids that comprise the hCTR1 C terminus resulted in a significant elevation of turnover numbers and of Km (Figs. 4 and 5). The elevated rate of copper transport in the AAA mutant as well as in truncation mutants of the hCTR1 C-terminal tail was unexpected, given previous speculation that the HCH motif is essential in permeation (16, 24, 38). Our results are incompatible with an essential role of the terminal three amino acids HCH in copper permeation, and instead imply that the highly conserved C-terminal tail and HCH putative metal-binding motif play a different role, perhaps as a plug or a valve at the exit site, transiently halting or slowing the progress of permeation out of the pore. This idea is supported by the observation that truncation of the tail had caused an even greater elevation of copper transport than the AAA mutant. Surprisingly, the C-terminal tail mutations affected hCTR1 copper uptake in the same manner as the H139R mutation, increasing the rate of uptake and raising the Km (Figs. 3 and 5). Furthermore, copper uptake in cells expressing the double mutant (H139R and AAA) was similar to either single mutant (Fig. 7B). The lack of additivity of the double mutant implied that both mutations affected the same function, permeation through the latter parts of the pore.
The elevated copper uptake rates of the H139R and C-terminal intracellular side mutants allowed us to test the hypothesis that Met-150 and Met-154 are essential at an early step in transport, a model supported by the mutant phenotype (22) structural/modeling data (15, 16, 21, 24, 25), and extended x-ray absorption fine structure measurements of Cu+-bound hCTR1 (16). We constructed hCTR1 multiple mutants having MM and either H139R or the AAA and measured copper uptake in cells expressing WT and the various mutants and mutant combinations. As seen previously, copper uptake in cells expressing the MM mutant was severely reduced in comparison with cell expressing WT protein (Fig. 7A). Cells expressing the multiple mutants (MM; H139R or MM; AAA) had the same reduced copper uptake as the M150L,M154L-expressing cells (Fig. 7A). These data indicate separate and sequential roles of Met-150 and Met-154 and H139R/C-terminal mutants in copper transport. These data also show that distortions of or changes in the intracellular aspect of hCTR1 do not affect the role of Met-150 and Met-154. Further experiments using the intracellular side mutants (and other related mutations) will be useful for future detailed studies on the mechanism of permeation.
hCTR1 Responsiveness to Extracellular Copper and Regulatory Endocytosis
Our work also demonstrated that the intracellular side mutants that exhibit higher transport rates also fail to respond to high copper in the medium via regulatory endocytosis. This is inconsistent with a previously proposed mechanism in which copper transport occurs by extracellular copper binding to hCTR1, internalization of hCTR1, and internal release of copper before recycling of the transporter to the surface (31) or release following transporter degradation (26, 27). Our demonstration that multiple mutants having markedly elevated rates of copper transport do not internalize at all in response to elevated external copper rules out any endocytosis/degradation mechanism for hCTR1 copper transport in mammalian cells. Recent work on yeast Ctr1 also failed to provide evidence in support of the endocytosis and degradation pathway in homeostatic copper acquisition (39).
The failure of the C-terminal and H139R mutants to respond to high external copper by endocytosis adds to previous experimental data about structures/residues required for the process. Previous work showed that the N terminus does not seem to be essential for endocytosis because mutation of histidine and methionine clusters thought to bind copper does not inhibit copper-stimulated endocytosis (40). In addition, loss of the first 30 amino acids of the hCTR1 N terminus also did not inhibit endocytic down-regulation (28). On the other hand, Met-150 and Met-154, which line the extracellular opening of the pore and are required for permeation, were shown to be required for endocytic down-regulation of hCTR1 (40). This suggests that either copper permeation or occupancy of the Met-150 and Met-154 copper-binding site(s), or both, is necessary for copper-stimulated endocytosis.
The loss of copper-stimulated endocytosis in the H139R and C-tail mutants we describe here demonstrates that both the cytoplasmic tail portion of hCTR1 and His-139 are also essential for endocytic regulation. The mutations affect different structural elements of hCTR1, but share an elevated rate of copper transport and a failure to exhibit copper-stimulated endocytosis. To explain these observations, we consider alternate explanations. There could be a kinetic effect, where the more rapidly transporting mutants traverse too rapidly through a critical intermediate to allow interaction with the endocytic machinery that normally occurs with WT hCTR1. There is evidence that copper binding to hCTR1 changes its conformation (17), as well as a modeling study predicting conformational changes during transport that include a role for His-139 (24). Alternatively, structural features that are important for interaction with the endocytic machinery are distorted or absent in both types (pore and C-terminal tail, respectively) of intracellular side mutants. Further investigation will be necessary to distinguish these and other possibilities. Our newly identified mutants that are defective in the endocytic process will be useful in such studies.
Concluding Comments
Our results advance the understanding of the mechanism of hCTR1 transport and provide new information about endocytic down-regulation of hCTR1 in high copper conditions. It is still largely unknown how copper transits the hCTR1 pore, but our experiments with the C-terminal tail mutants show that exit from the hCTR1 transporter on the intracellular side does not require participation of the putative HCH metal-binding motif. The increase in transport rate in the tail mutants suggests that the tail and HCH motif might instead act as a barrier, regulating or limiting the rate of copper exit from the pore.
There are striking parallels between our observations and related studies on the yeast Ctr1 from Saccharomyces cerevisiae (39). The intracellular C terminus of yeast Ctr1 has about 125 amino acids, including 5 cysteine residues. This compares with hCTR1 having a nonhomologous C terminus of only 15 amino acids and a single cysteine. Truncation of the C-terminal tail or substitution of the C-terminal Cys residues in yeast Ctr1 results in cells that are more susceptible to copper toxicity as well as having elevated copper transport rates and elevated Km of transport when compared with WT cells. Thus, the function of the C-terminal tails in human and yeast CTR1 copper transporters appears very similar based on the effects of analogous mutations in the two proteins. It seems likely that general aspects of the mechanism and regulation of CTR1 copper transport have been evolutionarily conserved utilizing different protein sequences/structures, an idea that is supported by the rescue of yeast Ctr1/Ctr3 mutants by hCTR1 (8). Our work, together with studies on yeast Ctr1 (39), suggests that the CTR1 family of copper transporters comprises permeation pores formed from homotrimers and clusters of C termini on the cytoplasmic side of the pore that regulate the rate of copper exit from the transporter.
Acknowledgments
We thank Drs. C. De Feo and V. Unger for purified hCTR1 protein.
This work was supported, in whole or in part, by National Institutes of Health Grant P01 GM067166 (to J. H. K.).
The following multiple mutant designations are used throughout: AAA, H188A,C189A,H190A; MM, M150L,M154L.
E. Maryon and J. Kaplan, unpublished results.
- hCTR1
- human Ctr1 transporter
- tet
- tetracycline
- DMEM
- Dulbecco's minimal essential medium
- ab
- antibody.
REFERENCES
- 1. Gupta A., Lutsenko S. (2009) Human copper transporters: mechanism, role in human diseases, and therapeutic potential. Future Med. Chem. 1, 1125–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. MacPherson I. S., Murphy M. E. (2007) Type-2 copper-containing enzymes. Cell. Mol. Life Sci. 64, 2887–2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McMillin G. A., Travis J. J., Hunt J. W. (2009) Direct measurement of free copper in serum or plasma ultrafiltrate. Am. J. Clin. Pathol. 131, 160–165 [DOI] [PubMed] [Google Scholar]
- 4. Rae T. D., Schmidt P. J., Pufahl R. A., Culotta V. C., O'Halloran T. V. (1999) Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science 284, 805–808 [DOI] [PubMed] [Google Scholar]
- 5. Wirth P. L., Linder M. C. (1985) Distribution of copper among components of human serum. J. Natl. Cancer Inst. 75, 277–284 [PubMed] [Google Scholar]
- 6. Huffman D. L., O'Halloran T. V. (2001) Function, structure, and mechanism of intracellular copper trafficking proteins. Annu. Rev. Biochem. 70, 677–701 [DOI] [PubMed] [Google Scholar]
- 7. Cabrera A., Alonzo E., Sauble E., Chu Y. L., Nguyen D., Linder M. C., Sato D. S., Mason A. Z. (2008) Copper binding components of blood plasma and organs, and their responses to influx of large doses of 65Cu, in the mouse. Biometals 21, 525–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou B., Gitschier J. (1997) hCTR1: a human gene for copper uptake identified by complementation in yeast. Proc. Natl. Acad. Sci. U.S.A. 94, 7481–7486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hua H., Georgiev O., Schaffner W., Steiger D. (2010) Human copper transporter Ctr1 is functional in Drosophila, revealing a high degree of conservation between mammals and insects. J. Biol. Inorg. Chem. 15, 107–113 [DOI] [PubMed] [Google Scholar]
- 10. Eisses J. F., Kaplan J. H. (2005) The mechanism of copper uptake mediated by human CTR1: a mutational analysis. J. Biol. Chem. 280, 37159–37168 [DOI] [PubMed] [Google Scholar]
- 11. Lee J., Petris M. J., Thiele D. J. (2002) Characterization of mouse embryonic cells deficient in the Ctr1 high affinity copper transporter: Identification of a Ctr1-independent copper transport system. J. Biol. Chem. 277, 40253–40259 [DOI] [PubMed] [Google Scholar]
- 12. Lee J., Peña M. M., Nose Y., Thiele D. J. (2002) Biochemical characterization of the human copper transporter Ctr1. J. Biol. Chem. 277, 4380–4387 [DOI] [PubMed] [Google Scholar]
- 13. Zimnicka A. M., Ivy K., Kaplan J. H. (2011) Acquisition of dietary copper: a role for anion transporters in intestinal apical copper uptake. Am. J. Physiol. Cell Physiol. 300, C588–C599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maryon E. B., Molloy S. A., Zimnicka A. M., Kaplan J. H. (2007) Copper entry into human cells: progress and unanswered questions. Biometals 20, 355–364 [DOI] [PubMed] [Google Scholar]
- 15. Aller S. G., Unger V. M. (2006) Projection structure of the human copper transporter CTR1 at 6-Ä resolution reveals a compact trimer with a novel channel-like architecture. Proc. Natl. Acad. Sci. U.S.A. 103, 3627–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. De Feo C. J., Aller S. G., Siluvai G. S., Blackburn N. J., Unger V. M. (2009) Three-dimensional structure of the human copper transporter hCTR1. Proc. Natl. Acad. Sci. U.S.A. 106, 4237–4242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eisses J. F., Kaplan J. H. (2002) Molecular characterization of hCTR1, the human copper uptake protein. J. Biol. Chem. 277, 29162–29171 [DOI] [PubMed] [Google Scholar]
- 18. Klomp A. E., Juijn J. A., van der Gun L. T., van den Berg I. E., Berger R., Klomp L. W. (2003) The N-terminus of the human copper transporter 1 (hCTR1) is localized extracellularly, and interacts with itself. Biochem. J. 370, 881–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Klomp A. E., Tops B. B., Van Denberg I. E., Berger R., Klomp L. W. (2002) Biochemical characterization and subcellular localization of human copper transporter 1 (hCTR1). Biochem. J. 364, 497–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aller S. G., Eng E. T., De Feo C. J., Unger V. M. (2004) Eukaryotic CTR copper uptake transporters require two faces of the third transmembrane domain for helix packing, oligomerization, and function. J. Biol. Chem. 279, 53435–53441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. De Feo C. J., Mootien S., Unger V. M. (2010) Tryptophan scanning analysis of the membrane domain of CTR-copper transporters. J. Membr. Biol. 234, 113–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Puig S., Lee J., Lau M., Thiele D. J. (2002) Biochemical and genetic analyses of yeast and human high affinity copper transporters suggest a conserved mechanism for copper uptake. J. Biol. Chem. 277, 26021–26030 [DOI] [PubMed] [Google Scholar]
- 23. Liang Z. D., Stockton D., Savaraj N., Tien Kuo M. (2009) Mechanistic comparison of human high-affinity copper transporter 1-mediated transport between copper ion and cisplatin. Mol. Pharmacol. 76, 843–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schushan M., Barkan Y., Haliloglu T., Ben-Tal N. (2010) Cα-trace model of the transmembrane domain of human copper transporter 1, motion and functional implications. Proc. Natl. Acad. Sci. U.S.A. 107, 10908–10913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tsigelny I. F., Sharikov Y., Greenberg J. P., Miller M. A., Kouznetsova V. L., Larson C. A., Howell S. B. (2012) An all-atom model of the structure of human copper transporter 1. Cell Biochem. Biophys. 63, 223–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Petris M. J., Smith K., Lee J., Thiele D. J. (2003) Copper-stimulated endocytosis and degradation of the human copper transporter, hCtr1. J. Biol. Chem. 278, 9639–9646 [DOI] [PubMed] [Google Scholar]
- 27. Holzer A. K., Katano K., Klomp L. W., Howell S. B. (2004) Cisplatin rapidly down-regulates its own influx transporter hCTR1 in cultured human ovarian carcinoma cells. Clin. Cancer Res. 10, 6744–6749 [DOI] [PubMed] [Google Scholar]
- 28. Maryon E. B., Molloy S. A., Kaplan J. H. (2007) O-linked glycosylation at threonine 27 protects the copper transporter hCTR1 from proteolytic cleavage in mammalian cells. J. Biol. Chem. 282, 20376–20387 [DOI] [PubMed] [Google Scholar]
- 29. Maryon E. B., Zhang J., Jellison J. W., Kaplan J. H. (2009) Human copper transporter 1 lacking O-linked glycosylation is proteolytically cleaved in a Rab9-positive endosomal compartment. J. Biol. Chem. 284, 28104–28114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zimnicka A. M., Maryon E. B., Kaplan J. H. (2007) Human copper transporter hCTR1 mediates basolateral uptake of copper into enterocytes: implications for copper homeostasis. J. Biol. Chem. 282, 26471–26480 [DOI] [PubMed] [Google Scholar]
- 31. Molloy S. A., Kaplan J. H. (2009) Copper-dependent recycling of hCTR1, the human high affinity copper transporter. J. Biol. Chem. 284, 29704–29713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mayle K. M., Le A. M., Kamei D. T. (2012) The intracellular trafficking pathway of transferrin. Biochim. Biophys. Acta 1820, 264–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brahm J. (1977) Temperature-dependent changes of chloride transport kinetics in human red cells. J. Gen. Physiol. 70, 283–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brahm J. (1983) Kinetics of glucose transport in human erythrocytes. J. Physiol. 339, 339–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jørgensen P. L. (1986) Structure, function and regulation of Na,K-ATPase in the kidney. Kidney Int. 29, 10–20 [DOI] [PubMed] [Google Scholar]
- 36. Karakossian M. H., Spencer S. R., Gomez A. Q., Padilla O. R., Sacher A., Loo D. D., Nelson N., Eskandari S. (2005) Novel properties of a mouse γ-aminobutyric acid transporter (GAT4). J. Membr. Biol. 203, 65–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fang Y., Kolmakova-Partensky L., Miller C. (2007) A bacterial arginine-agmatine exchange transporter involved in extreme acid resistance. J. Biol. Chem. 282, 176–182 [DOI] [PubMed] [Google Scholar]
- 38. Pope C. R., Flores A. G., Kaplan J. H., Unger V. M. (2012) Structure and function of copper uptake transporters. Curr. Top. Membr. 69, 97–112 [DOI] [PubMed] [Google Scholar]
- 39. Wu X., Sinani D., Kim H., Lee J. (2009) Copper transport activity of yeast Ctr1 is down-regulated via its C terminus in response to excess copper. J. Biol. Chem. 284, 4112–4122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guo Y., Smith K., Lee J., Thiele D. J., Petris M. J. (2004) Identification of methionine-rich clusters that regulate copper-stimulated endocytosis of the human Ctr1 copper transporter. J. Biol. Chem. 279, 17428–17433 [DOI] [PubMed] [Google Scholar]