

Background: Endogenous oncogenic Kras induces a highly penetrant acute T-cell lymphoblastic leukemia/lymphoma (T-ALL).
Results: Up-regulation of NOTCH1 signaling, through either overexpression of surface NOTCH1 or acquired gain-of-function mutations, is involved in both T-ALL initiation and progression.
Conclusion: Notch1 mutations contribute to leukemogenic transformation of normal T-cells.
Significance: Our data provide a rationale to target both NOTCH1 and RAS signaling for T-ALL treatment.
Keywords: Leukemia, Lymphoma, Notch, Oncogene, Wnt Pathway, KRAS, Leukemia-initiating Cells
Abstract
Acute T-cell lymphoblastic leukemia/lymphoma (T-ALL) is an aggressive hematopoietic malignancy affecting both children and adults. Previous studies of T-ALL mouse models induced by different genetic mutations have provided highly diverse results on the issues of T-cell leukemia/lymphoma-initiating cells (T-LICs) and potential mechanisms contributing to T-LIC transformation. Here, we show that oncogenic Kras (Kras G12D) expressed from its endogenous locus is a potent inducer of T-ALL even in a less sensitized BALB/c background. Notch1 mutations, including exon 34 mutations and recently characterized type 1 and 2 deletions, are detected in 100% of Kras G12D-induced T-ALL tumors. Although these mutations are not detected at the pre-leukemia stage, incremental up-regulation of NOTCH1 surface expression is observed at the pre-leukemia and leukemia stages. As secondary genetic hits in the Kras G12D model, Notch1 mutations target CD8+ T-cells but not hematopoietic stem cells to further promote T-ALL progression. Pre-leukemia T-cells without detectable Notch1 mutations do not induce T-ALL in secondary recipient mice compared with T-ALL tumor cells with Notch1 mutations. We found huge variations in T-LIC frequency and immunophenotypes of cells enriched for T-LICs. Unlike Pten deficiency-induced T-ALL, oncogenic Kras-initiated T-ALL is not associated with up-regulation of the Wnt/β-catenin pathway. Our results suggest that up-regulation of NOTCH1 signaling, through either overexpression of surface NOTCH1 or acquired gain-of-function mutations, is involved in both T-ALL initiation and progression. Notch1 mutations and Kras G12D contribute cooperatively to leukemogenic transformation of normal T-cells.
Introduction
Acute T-cell lymphoblastic leukemia/lymphoma (T-ALL)3 is an aggressive hematopoietic malignancy that accounts for 10–15% of pediatric and 25% of adult ALL cases (1). The tumor cells isolated from T-ALL patients are usually TdT+ (terminal deoxynucleotidyltransferase) with variable expression of CD1A, CD2, CD3, CD4, CD5, CD7, and CD8 (2). These expression patterns usually reflect transformation or a differentiation blockade at distinct stages of intrathymic T-cell development (3, 4).
Numerous genetic alterations have been identified in the pathogenesis of T-ALL (5). Gain-of-function NOTCH1 mutations are the most common ones, being present in 50–70% of T-ALL patients (6, 7). These mutations often occur within the heterodimerization domain and/or the proline-, glutamic acid-, serine-, and threonine-rich (PEST) domain and render increased levels of intracellular C-terminal NOTCH1 protein. In addition, elevation of RAS signaling, which plays an important role in T-cell development through mediating T-cell receptor (TCR) complex signaling and inducing cytokine gene production (8), is detected in ∼50% of T-ALL cases (9). Recently, activating mutations in two RAS isoforms, NRAS and KRAS, have been identified in patients with a subtype of T-ALL termed early T-cell precursor ALL (10).
It remains controversial whether T-ALL is maintained by a minor subpopulation of tumor cells called T-cell leukemia/lymphoma-initiating cells (T-LICs). The answer to this question has fundamental implications for cancer therapy. If leukemogenic cells reside in small minority populations of T-LICs, improved anticancer therapies should be identified based on the ability to kill T-LICs rather than the bulk non-tumorigenic cancer cells. Alternatively, if cells with leukemogenic potential are common, it would be impossible to eradicate cancer by focusing on small minority subpopulations. Rare T-LICs enriched in CD3+ cKitmid cells have been identified to maintain Pten deletion-initiated T-ALL (11), whereas in T-ALL initiated by overexpression of oncogenic NRAS, a majority of tumor cells have tumorigenic capability (12). Collectively, these results suggest that the presence of T-LICs might be genetic alteration-dependent.
Consistent with results from human studies, we and others have found that oncogenic Kras (Kras G12D) expressed from its endogenous locus promotes T-ALL development with a high penetrance in bone marrow-transplanted C57BL/6 recipient mice, which are known to be highly sensitized to T-ALL development. In this model, hematopoietic stem cells (HSCs) expressing KRAS G12D are required to initiate T-ALL (13). Our previous results suggest that secondary genetic hits might target downstream T-cells and transform them to maintain T-ALL phenotypes (14). It remains unknown, however, whether tumorigenic activity is present only in a small population of T-LICs or in bulk T-ALL cells.
In a search for genes or pathways that are involved in tumor cell transformation, we and others have reported that CD44 is overexpressed in 100% of Kras G12D-induced T-ALL tumors (14, 15). Although loss of CD44 significantly prolongs the survival of T-ALL mice, it does not prevent T-ALL development or its transplantability to subsequent recipient mice, suggesting that CD44 plays a minor role in tumor cell transformation (13). In addition, ∼50% of Kras G12D-induced T-ALL tumors carry Notch1 mutations in the PEST domain of exon 34 (14–17). Although Notch1 mutations are weak tumor initiators, they accelerate Kras G12D-initiated T-ALL (18). Given its incomplete penetrance in the Kras G12D model, it is unclear whether up-regulation of NOTCH1 signaling represents a common mechanism contributing to leukemia cell transformation.
Here, we show that Kras G12D is a potent inducer of T-ALL not only in the C57BL/6 (B6) background but also in the BALB/c background, which is less sensitized for T-ALL. All Kras G12D-induced T-ALL tumors contain various Notch1 mutations, including exon 34 mutations and the recently characterized type 1 and 2 deletions (19). Although these mutations are not detected at the pre-leukemia stage, incremental up-regulation of NOTCH1 surface expression is observed at the pre-leukemia and leukemia stages. Consistent with our previous hypothesis, Notch1 mutations target T-lineage-committed precursor cells instead of HSCs. Huge variations are observed in T-LIC frequency and immunophenotypes of cells enriched for T-LICs. Unlike Pten deficiency-induced T-ALL, oncogenic Kras-initiated T-ALL is not associated with up-regulation of the Wnt/β-catenin pathway. Our results suggest that up-regulation of NOTCH1 signaling, through either overexpression of surface NOTCH1 or acquired gain-of-function mutations, is involved in both T-ALL initiation and progression. Notch1 mutations contribute to transformation of CD8+ T-cells to leukemia cells.
MATERIALS AND METHODS
Mice
All mouse lines (LSL Kras G12D/+, Mx1-Cre, and LSL Kras G12D/+;Mx1-Cre/+) were maintained in a pure B6 or BALB/c genetic background (>N10). All data were obtained from mice in the B6 background unless specified otherwise. Genotyping of Kras G12D and Mx1-Cre was done as described previously (14). CD45.1+ B6 recipient mice were purchased from NCI, whereas BALB/c recipient mice were obtained from The Jackson Laboratory.
To induce CRE expression, 5–7-week-old mice were injected intraperitoneally with 250 μg of poly(I-C) (Sigma-Aldrich) every other day for two doses. All experiments were performed 2 days after the second injection of poly(I-C). All experiments were conducted with the ethical approval of the International Association for Assessment and Accreditation of Laboratory Animal Care at the University of Wisconsin-Madison.
Bone Marrow Transplantation
Bone marrow transplantation experiments were performed as described previously (17) using 2.5 × 105 Kras G12D bone marrow cells along with the same number of congenic competitor/helper cells in individual lethally irradiated mice. In serial transplantation experiments, 1 × 106 T-ALL cells were transplanted into individual sublethally irradiated mice as described (14). Fractionated and/or diluted T-ALL cells were transplanted with (donor cell number = 104 and below) or without 2 × 105 congenic (CD45.1+) whole spleen carrier cells into individual sublethally irradiated mice. Recipient mice were monitored for 16–20 weeks for T-ALL development.
Flow Cytometric Analysis
Control thymocytes and T-ALL cells were analyzed using flow cytometry at 4-week intervals after bone marrow transplantation. Stained samples were analyzed on a FACSCalibur or LSR II flow cytometer (BD Biosciences). The data were analyzed using FlowJo software.
Intracellular staining of unphosphorylated β-catenin in thymocytes was carried out using monoclonal antibody 8E4 as described previously (11). Samples were analyzed on a FACSCalibur. The data were analyzed using CellQuest software.
Characterization of Notch1 Mutations
Genomic DNAs were isolated from thymus using the Puregene® genomic DNA purification kit (Qiagen). Total RNAs were extracted from thymus using the RNeasy mini kit (Qiagen). First-strand cDNAs were synthesized using the SuperScript first-strand synthesis system (Invitrogen). Detection of Notch1 mutations was performed essentially as described previously (13).
Analysis of Rag1/2 Expression
Genomic DNA and total RNA were extracted using the AllPrep DNA/RNA mini kit (Qiagen). Reverse transcription was performed using the SuperScript first-strand synthesis system according to the manufacturer's instructions. The PCR primers used were described previously (15). PCRs were performed under the following conditions: 94 °C for 30 s and 35 cycles at 50 °C for 30 s and 72 °C for 30 s.
RESULTS
Kras G12D Induces a Fully Penetrant T-ALL in the BALB/c Genetic Background
We transferred the LSL Kras G12D;Mx1-Cre model from the B6 background (14) to a pure BALB/c background to determine whether Kras G12D efficiently induces T-ALL even in a less sensitized genetic background. In primary non-transplanted Kras G12D mice, changing to a different genetic background did not significantly affect the survival of and HSC depletion and aberrant GM-CSF signaling in Kras G12D mice (supplemental Fig. S1) (13, 20). In recipient mice transplanted with Kras G12D bone marrow cells, although BALB/c mice survived significantly longer than B6 mice (Fig. 1A), the disease penetrance and phenotypes in the BALB/c background were very similar to those in the B6 background (Fig. 1, B–D), suggesting that Kras G12D is a potent inducer of T-ALL in both genetic backgrounds. Because of the ease in tracing donor-derived leukemias and more rapid disease development in the B6 genetic background, the data presented below were obtained from Kras G12D B6 mice.
FIGURE 1.

Kras G12D induces a highly penetrant T-ALL in the BALB/c genetic background. Lethally irradiated mice were transplanted with 2.5 × 105 total bone marrow cells from control or Kras G12D mice in a pure B6 or BALB/c background along with same number of competitor cells. A, Kaplan-Meier survival curves of reconstituted mice. Cumulative survival is plotted against days after transplantation. The p value was determined by the log-rank test. B, disease distribution patterns in recipient mice transplanted with Kras G12D cells. C, flow cytometric analysis of total thymocytes from representative mice with control cells and T-ALL mice with Kras G12D cells. D, flow cytometric analysis of peripheral blood from representative mice with control cells and myeloproliferative neoplasm (MPN) mice with Kras G12D cells using antibodies against Mac-1 (CD11b) and Gr-1 (Ly6C/Ly6G). The percentages of cells enriched for monocytes (upper left quadrant) and granulocytes (upper right quadrant) are indicated on the plots.
Notch1 Mutations Are Identified in 100% of Kras G12D-induced T-ALL Tumors
To better understand the mechanisms of Kras G12D-initiated T-ALL, we characterized recipient animals transplanted with Kras G12D bone marrow cells at 4 or 8 weeks or at a moribund stage (∼12 weeks) after transplantation (Fig. 2). Four weeks after transplantation, <50% of thymocytes were donor-derived in all of the recipient mice we examined, and their thymus size was comparable with that of controls (Fig. 2A). Five of eight recipient mice showed >99% donor-derived thymocytes 8 weeks after transplantation (pre-leukemia stage), and their thymus weight was moderately but significantly increased (Fig. 2, A and B). At this stage, thymi in mutant borrow recipients displayed variable expression patterns of CD4 and CD8 but unanimously up-regulated CD44 expression (Fig. 2C). At a moribund stage ∼12 weeks after transplantation, all recipient mice developed T-ALL with a markedly enlarged thymus filled with donor-derived cells (Fig. 2, A and B). These T-ALL cells were mostly CD2+ CD5+/− CD3− CD4+ CD8+ CD44+ TdT+, representative of T-ALL developed at a precursor stage (Fig. 2D) (14, 17). In the subsequent studies, our analysis was focused on the five recipient mice with >99% of thymocytes contributed from donor cells at the pre-leukemia stage and moribund mice with fully developed T-ALL.
FIGURE 2.

Evaluation of thymi from recipient mice transplanted with Kras G12D bone marrow cells at different stages. Bone marrow cells expressing endogenous KRAS G12D were transplanted together with the same number of congenic competitor cells into lethally irradiated mice. Recipient mice were killed at 4 weeks (1 month) and 8 weeks (2 month) or at a moribund stage (around 3 month). A, representative results of donor contribution from thymi of recipient mice at different stages. B, thymus weights in recipient mice at different stages. Results are presented as scatter plots of the thymus weight of individual animals with mean ± S.D. Student's t test was performed: *, p < 0.05. mon, months. C and D, flow cytometric analysis of total thymocytes from representative mice with control cells or Kras G12D cells at 8 weeks (C) or at a moribund stage (D).
We examined various types of Notch1 mutations at different stages of T-ALL. The heterozygous type 1 deletion was detectable in all of the moribund recipient mice (n = 13) but not in the mice at the pre-leukemia stage (n = 5) (Fig. 3A). Consistent with a previous report (19), the deletion occurred in the proximal promoter and exon 1 region of Notch1 and at the conserved sites recognized by RAG recombinases (Fig. 3B). Some repetitive sequences were inserted into the deleted regions, which led to different sizes of the PCR products (e.g. clones M-7 and M-8). Type 1 deletions were also detected in 100% of Nras G12D-induced T-ALL tumors (supplemental Fig. S2). Unlike type 1 deletions, type 2 deletions were found only in 2 of 12 mice at the moribund stage (Fig. 3C). In addition, mutations in the PEST domain of exon 34 were identified in 10 of 13 moribund mice (Fig. 3E) but not in the five mice at the pre-leukemia stage (Fig. 3D). Our data suggest that Notch1 mutations occur between the pre-leukemia and leukemia stages and might play an important role in T-ALL progression.
FIGURE 3.

Notch1 mutations are identified in 100% of moribund Kras G12D-induced T-ALL mice. Recipient mice transplanted with control or Kras G12D cells were killed at 8 weeks (2 month) or at a moribund stage (∼12 weeks). A, the type 1 deletion and its corresponding wild-type allele were examined in thymocytes of recipient mice 2 months after transplantation or at a moribund stage. B, schematic representation of type 1 deletions identified in different T-ALL tumors. Numbers indicate the nucleotide positions on mouse chromosome 2 according to the UCSC Genome Browser. C, the type 2 deletion was examined in thymocytes from recipient mice 2 months after transplantation or at a moribund stage. PC, positive control. D and E, the status of Notch1 mutations is summarized for recipient mice 2 months after transplantation (D) or for moribund recipient mice (E). N.A., not applicable.
Because the type 1 deletion at the Notch1 locus is mediated by RAG recombinases, we examined the expression levels of these enzymes at different stages of T-ALL development to determine whether elevated Rag1/2 levels contribute to the prevalent type 1 deletion (Fig. 4A). We found that the expression levels of Rag1/2 in Kras G12D thymocytes were comparable with those in controls at the pre-leukemia stage but were generally down-regulated in fully developed T-ALL tumors, consistent with partially blocked T-cell differentiation in these tumor cells (14). In contrast, surface expression of NOTCH1 was significantly elevated at both the pre-leukemia and moribund stages in an incremental manner (Fig. 4B), suggesting that up-regulation of NOTCH1 signaling by overexpression of surface NOTCH1 might be involved in both T-ALL initiation and progression.
FIGURE 4.
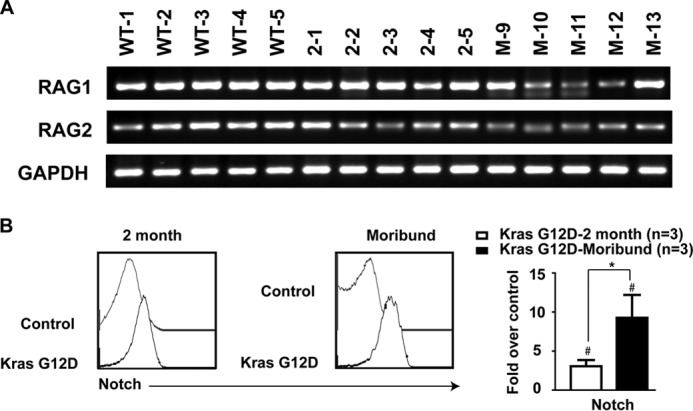
Kras G12D thymocytes overexpress surface NOTCH1 but not Rag1/2 at the pre-leukemia stage. Recipient mice transplanted with control or Kras G12D cells were killed at 8 weeks (2 month) or at a moribund stage (∼12 weeks). A, semiquantitative RT-PCR was performed to detect Rag1 and Rag2 levels in control and Kras G12D thymocytes. B, representative histograms of surface expression of NOTCH1 in control and Kras G12D CD4+ CD8+ thymocytes at different T-ALL stages. Quantified data are mean ± S.D. over control cells. #, p < 0.05 (compared with controls); *, p < 0.05 (compared between different stages of T-ALL).
Notch1 Mutations Target Kras G12D CD8+ Cells That Contain Leukemogenic Activity
The fully penetrant Notch1 type 1 deletion provides a great opportunity to test our previous hypothesis that, in the Kras G12D model, secondary genetic event(s) occur in lineage-restricted progenitor/precursor cells rather than in HSCs (14). We analyzed type 1 deletions in three recipient mice that simultaneously developed T-ALL and a myeloproliferative neoplasm (Fig. 5A). As we expected, type 1 deletions were detectable only in the thymic T-ALL cells but not in the bone marrow myeloid cells isolated from the same animals, suggesting that the Notch1 mutation occurs after Kras G12D HSCs differentiate into T-lineage cells. To further exclude the possibility that type 1 deletions pre-exist in Kras G12D bone marrow cells and are selected during leukemogenesis, we performed a high-sensitivity PCR and did not detect the type 1 deletion in these cells (supplemental Fig. S3). Furthermore, Kras G12D thymocytes isolated at the pre-leukemia stage (without detectable Notch1 mutations) did not induce T-ALL in secondary recipient mice, whereas T-ALL tumor cells (with Notch1 mutations) did (Fig. 5B). These data suggest that Notch1 mutations target T-ALL cells as secondary genetic hits and contribute to their malignant transformation.
FIGURE 5.

Notch1 mutations target CD8+ T-cells that contain leukemogenic activity. Recipient mice transplanted with Kras G12D cells were killed at a moribund stage. A, evaluation of the Notch1 type 1 deletion in thymocytes (Thy) and purified bone marrow myeloid cells (Mye) of moribund mice with both T-ALL and myeloproliferative neoplasm. B, recipient mice transplanted with Kras G12D cells were killed at the pre-leukemia stage (8 weeks after transplantation) or at a moribund stage. Thymocytes (1 × 105) were further transplanted into sublethally irradiated secondary recipient mice. Kaplan-Meier survival curves are plotted against days after transplantation. The p value was determined by the log-rank test. C, HSCs expressing KRAS G12D were sorted and transplanted into primary recipient mice (1st). Subsequently, 1 × 106 T-ALL cells were transplanted into sublethally irradiated recipient mice (2nd–5th). Black lines represent the average survival of T-ALL mice. D, limiting dilution analysis of the frequency of T-LICs. Sublethally irradiated CD45.1 recipient mice were transplanted with various numbers of T-ALL cells. The percentage of recipient mice free of T-ALL for up to 20 weeks post-transplant is plotted. The frequency of T-LICs was calculated using L-Calc software.
As the first step toward identifying leukemogenic cells in our model, we determined that the leukemogenic activity of T-ALL cells is sustained and self-renewable in vivo using a serial transplantation approach (Fig. 5C). Next, we used a limiting dilution approach to estimate the frequency of these leukemia-initiating cells, which varied from 1 of 7 to 1 of 16,000 (Fig. 5D), indicating that Kras G12D-induced T-ALL is maintained by either a small population of tumor cells called T-LICs or bulk T-ALL tumor cells. We further fractionated T-ALL cells into different populations based on their immunophenotypes and found that only CD8+ cells but not CD8− cells contain T-LIC activity (supplemental Fig. S4A). Consistent with our hypothesis that Notch1 mutations contribute to malignant transformation of T-cells, only CD8+ cells, including both CD8+ CD4+ and CD8+ CD4− cells, but not CD8− CD4− cells were positive for the Notch1 type 1 deletion (supplemental Fig. S4B).
Due to the heterogeneity of T-ALL tumors, we were unable to identify a single population of tumor cells that are enriched for T-LICs in all of the tumors (supplemental Fig. S5). In some T-ALL tumors (Group I), T-LICs were enriched only in the distinct CD8+ cKit+ cells, whereas in other tumors (Group II), T-LICs were detected primarily in CD8+ cKit− cells. The identities of these T-LICs could be transferred to the subsequent recipient mice. Additional fractionation based on expression of CD34 or Sca1 yielded similar, highly variable results. Our data indicate that the frequency and identity of leukemogenic cells are highly variable in Kras G12D-initiated T-ALL.
The Wnt/β-Catenin Pathway Is Down-regulated in Kras G12D-induced T-ALL Cells
Previous studies showed that β-catenin stabilization predisposes thymocytes to malignant transformation (21) and is one of the mechanisms involved in Pten deficiency-induced T-ALL (11). Therefore, we further examined whether the Wnt/β-catenin pathway is dysregulated in our T-ALL model (Fig. 6). Consistent with a previous report (11), ∼18% of control thymocytes expressed the unphosphorylated (activated) form of β-catenin. However, expression of unphosphorylated β-catenin was almost completely absent in Kras G12D-induced T-ALL tumors, indicating that up-regulation of the Wnt/β-catenin pathway is not associated with Kras G12D-induced T-ALL.
FIGURE 6.
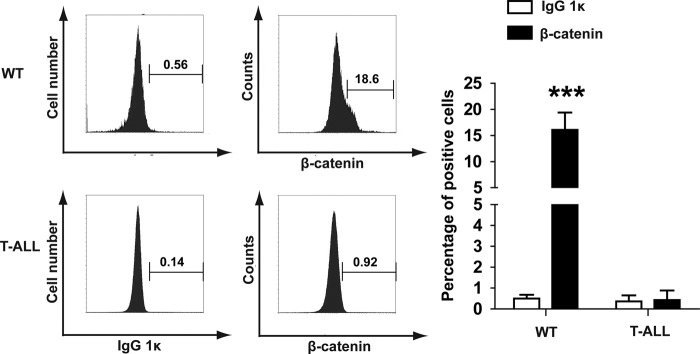
Kras G12D suppresses the Wnt/β-catenin pathway in T-ALL cells. WT thymocytes and bulk T-ALL cells were stained for the unphosphorylated (active) form of β-catenin. Purified mouse IgG1κ was used as an isotype control. Representative histograms from three independent experiments are shown. Positive cells are gated as illustrated. The percentages of positive cells are indicated on each plot. ***, p < 0.001.
DISCUSSION
In this work, we investigated whether Kras G12D-initiated T-ALL is maintained by rare T-LICs and whether up-regulation of NOTCH1 signaling represents a common mechanism contributing to malignant transformation of normal T-cells in this model. Our results show that Kras G12D-initiated T-ALL is maintained by either a small subset of or bulk CD8+ cells. As secondary genetic hits, Notch1 mutations are detected in 100% of T-ALL tumors but not at the pre-leukemia stage. In contrast, leukemogenesis is not associated with up-regulation of the Wnt/β-catenin pathway. Combined with human T-cell precursor ALL sequencing results, our data provide a rationale to target both NOTCH1 and RAS signaling for T-cell precursor ALL treatment.
Kras G12D Is a Potent Inducer of T-ALL in Both B6 and BALB/c Genetic Backgrounds
Tumorigenesis is greatly influenced by genetic backgrounds (22). It is well known that the B6 background is more sensitized for T-ALL, whereas the BALB/c background is more supportive for development of myeloid diseases. Overexpression of a number of oncogenes, including all the RAS isoforms and BCR-ABL, in these two genetic backgrounds yields very different disease spectrums, with T-cell malignancies dominant in the B6 background and highly pure myeloid malignancies dominant in the BALB/c background (23, 24). Although endogenous Kras G12D promotes a highly penetrant T-ALL in the B6 background (14, 15), we were uncertain whether this is greatly influenced by the specific genetic background or is due mainly to the Kras G12D function. Therefore, we transferred the Kras G12D model from B6 to BALB/c. To our surprise, Kras G12D also induced a highly penetrant T-ALL in the BALB/c background, and the phenotypes of both T-ALL and myeloproliferative neoplasm were indistinguishable between the two genetic backgrounds (Fig. 1). Our data indicate that endogenous Kras G12D is a functionally potent inducer of T-ALL. In support of our finding, activating mutations in KRAS have been identified in pediatric T-ALL patients (10, 25).
A Subset of CD8+ Cells or Bulk T-ALL Cells Contain Leukemogenic Activity
It remains controversial whether T-ALL is maintained by rare T-LICs or bulk tumor cells. Highly variable results have been reported not only in T-ALL models induced by different genetic mutations (11, 12) but also in T-ALL tumors containing a common initiating mutation (e.g. Kras G12D). One possibility is that the observation of T-LICs is associated with tumor development at an early/intermediate stage. Once tumorigenesis progresses to a more advanced stage, for example, by accumulation of more malignant genetic mutations, bulk tumor cells become tumorigenic.
In the Pten deficiency-induced T-ALL model (11), T-LICs appear to be enriched in CD3+ cKitmid cells in all tumors. However, in the Kras G12D model, T-LICs could be enriched in either CD8+ cKit+ or CD8+ cKit− cells (supplemental Fig. S5). This is not surprising, as a similar phenotypic heterogeneity of tumor-initiating cells has been reported in gliomas, in which both CD133+ and CD133− cells have tumorigenic activity (26).
Up-regulation of NOTCH1 Signaling Is Involved in Both T-ALL Initiation and Progression
We found that NOTCH1 signaling can be up-regulated by overexpression of surface NOTCH1 and/or Notch1 mutations. Although Notch1 mutations appear to promote T-ALL development only at a later stage, we believe that up-regulation of NOTCH1 signaling by overexpression of surface NOTCH1 is involved in T-ALL initiation at the pre-leukemia stage (Fig. 4B). In addition, overexpression of CD44 (13) and likely other unknown epigenetic and genetic events contribute to T-ALL development at the pre-leukemia stage. The majority of tumor samples have two different types of Notch1 mutations. It is not clear whether this is due to incremental NOTCH1 activation in the same cells and/or the presence of multiple clones in these tumor samples. In either case, the heterogeneous Notch1 mutations might at least partially explain the variation we observed in T-LIC frequency and immunophenotypes.
Different Notch1 mutations arise from distinct mechanisms. For example, the Notch1 type 1 deletion is mediated by RAG1/2 recombinase (19). Therefore, these mutations likely occur spontaneously during T-cell development and are subsequently selected during T-ALL progression. In contrast, the type 2 deletion is associated with genome instability (27). Our prior comparative genomic hybridization analysis of T-ALL samples indicated low-level or no genome instability (14). Consistently, the frequency of the Notch1 type 2 deletion is very low in our T-ALL model (Fig. 3).
Compared with mouse T-ALL, overexpression of intracellular NOTCH1 is achieved through similar but not identical mechanisms in human T-ALL. For example, RAG1/2-mediated Notch1 deletion is predominant in mouse T-ALL models. In contrast, in a small fraction of human T-ALL cases, the chromosomal translocation t(7;9) results in deregulated expression of a truncated activated form of NOTCH1 driven by the TCR-β promoter (28). Nonetheless, consistent with our finding, NOTCH1 mutations are secondary events in some T-ALL patients at diagnosis or at relapse (29). Some mutant-positive patients at diagnosis relapsed with the same mutation(s) at the same high level, whereas the others showed a change in mutations at relapse.
Consistent with our hypothesis, we found that Notch1 mutations occur in CD8+ T-cells (including both CD8+ CD4+ and CD8+ CD4− cells) but not in HSCs (Fig. 5). Similarly, Li et al. (30) reported that, in bone marrow cells overexpressing intracellular NOTCH1, the first tumorigenic cells were detected among more immature CD4− CD8+ TCR-αβ− cells. In addition, the malignant CD4+ CD8+ TCR-αβ+ and CD4− CD8+ TCR-αβ+ cells derived from the immature CD4− CD8+ TCR-αβ− cells were able to cause T-ALL in recipient mice as well.
Notch1 mutations are also present in 100% of T-ALL tumors induced by Kras G12D: CD44−/−, Nras G12D/+, or Nras G12D/G12D (supplemental Fig. S2) (13, 17, 31). Furthermore, Kras G12D thymocytes isolated at the pre-leukemia stage (without detectable Notch1 mutations) did not induce T-ALL in secondary recipient mice (Fig. 5). This result is not surprising because we expect that, without Notch1 mutations, these pre-leukemia cells are not transformed and thus are non-leukemogenic. Taken together, our data suggest that gain-of-function mutations in Notch1 may represent a common mechanism contributing to T-LIC transformation in endogenous Ras G12D-induced T-ALL models.
We also examined the Wnt/β-catenin pathway in our model and did not observe up-regulation of this pathway (Fig. 6). This result is probably not surprising because in Pten deficiency-induced T-ALL, Notch1 mutations are not identified, but tumor cells show overexpression of c-MYC and up-regulation of the Wnt/β-catenin pathway (11). These results suggest that T-ALLs induced from different genetic alterations use different genes and pathways to achieve T-LIC transformation. Up-regulation of NOTCH1 signaling through gain-of-function mutations and up-regulation of Notch1 surface expression in the Kras G12D model may functionally substitute for other abnormalities observed in the Pten−/− model. Consistent with this possibility, loss of PTEN is associated with resistance to NOTCH1 inhibition in human T-ALL cell lines (32).
In summary, we propose that in the Kras G12D-induced T-ALL model, Kras G12D targets HSCs to initiate T-ALL, whereas secondary genetic hits (e.g. Notch1 mutations) occur in T-lineage-committed CD8+ cells to promote T-ALL progression (Fig. 7). Notch1 mutations and Kras G12D contribute cooperatively to leukemogenic transformation of normal T-cells and thus provide a further rationale for targeting NOTCH1 and oncogenic RAS signaling pathways in T-ALL.
FIGURE 7.
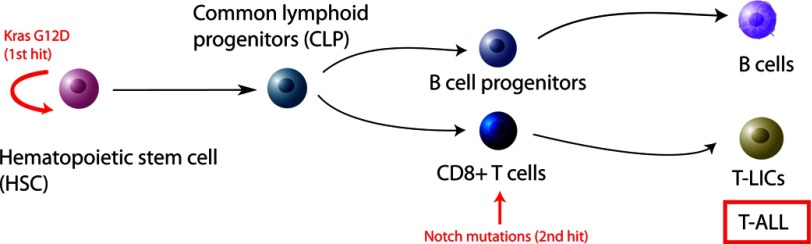
Model of Kras G12D-induced T-ALL.
Supplementary Material
Acknowledgment
We thank the University of Wisconsin Carbone Cancer Center for use of the shared services to complete this research.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 CA152108 (to J. Z.) and P30 CA014520 (to the University of Wisconsin Comprehensive Cancer Center) from NCI. This work was also supported by a research grant from the Elsa Pardee Foundation, a Shaw Scientist Award from the Greater Milwaukee Foundation, an ASH Scholar Award from the American Society of Hematology, a V Scholar Award from the V Foundation for Cancer Research, and an Investigator Initiated Grant from the University of Wisconsin Carbone Cancer Center (to J. Z.).

This article contains supplemental Figs. S1–S5.
- T-ALL
- acute T-cell lymphoblastic leukemia/lymphoma
- TCR
- T-cell receptor
- T-LIC
- T-cell leukemia/lymphoma-initiating cell
- HSC
- hematopoietic stem cell
- B6
- C57BL/6.
REFERENCES
- 1. Ferrando A. A., Neuberg D. S., Staunton J., Loh M. L., Huard C., Raimondi S. C., Behm F. G., Pui C. H., Downing J. R., Gilliland D. G., Lander E. S., Golub T. R., Look A. T. (2002) Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 1, 75–87 [DOI] [PubMed] [Google Scholar]
- 2. Teitell M. A., Pandolfi P. P. (2009) Molecular genetics of acute lymphoblastic leukemia. Annu. Rev. Pathol. 4, 175–198 [DOI] [PubMed] [Google Scholar]
- 3. Uckun F. M., Gaynon P. S., Sensel M. G., Nachman J., Trigg M. E., Steinherz P. G., Hutchinson R., Bostrom B. C., Sather H. N., Reaman G. H. (1997) Clinical features and treatment outcome of childhood T-lineage acute lymphoblastic leukemia according to the apparent maturational stage of T-lineage leukemic blasts: a Children's Cancer Group study. J. Clin. Oncol. 15, 2214–2221 [DOI] [PubMed] [Google Scholar]
- 4. Brunning R. D., Flandrin G., Barowitz M., Swerdlow S. H., Matutes E., et al. , (2001) WHO histological classification of precursor B-cell and T-cell neoplasms. in Pathology & Genetics: Tumours of Haematopoietic and Lymphoid Tissues (Jaffe E. S., Harris N. L., Stein H., Vardiman J. W., eds.) pp. 110–117, IARC Press, Lyon, France [Google Scholar]
- 5. Aifantis I., Raetz E., Buonamici S. (2008) Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat. Rev. Immunol. 8, 380–390 [DOI] [PubMed] [Google Scholar]
- 6. Weng A. P., Ferrando A. A., Lee W., Morris J. P., 4th, Silverman L. B., Sanchez-Irizarry C., Blacklow S. C., Look A. T., Aster J. C. (2004) Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269–271 [DOI] [PubMed] [Google Scholar]
- 7. van Grotel M., Meijerink J. P., van Wering E. R., Langerak A. W., Beverloo H. B., Buijs-Gladdines J. G., Burger N. B., Passier M., van Lieshout E. M., Kamps W. A., Veerman A. J., van Noesel M. M., Pieters R. (2008) Prognostic significance of molecular-cytogenetic abnormalities in pediatric T-ALL is not explained by immunophenotypic differences. Leukemia 22, 124–131 [DOI] [PubMed] [Google Scholar]
- 8. Genot E., Cantrell D. A. (2000) Ras regulation and function in lymphocytes. Curr. Opin. Immunol. 12, 289–294 [DOI] [PubMed] [Google Scholar]
- 9. von Lintig F. C., Huvar I., Law P., Diccianni M. B., Yu A. L., Boss G. R. (2000) Ras activation in normal white blood cells and childhood acute lymphoblastic leukemia. Clin. Cancer Res. 6, 1804–1810 [PubMed] [Google Scholar]
- 10. Zhang J., Ding L., Holmfeldt L., Wu G., Heatley S. L., Payne-Turner D., Easton J., Chen X., Wang J., Rusch M., Lu C., Chen S. C., Wei L., Collins-Underwood J. R., Ma J., Roberts K. G., Pounds S. B., Ulyanov A., Becksfort J., Gupta P., Huether R., Kriwacki R. W., Parker M., McGoldrick D. J., Zhao D., Alford D., Espy S., Bobba K. C., Song G., Pei D., Cheng C., Roberts S., Barbato M. I., Campana D., Coustan-Smith E., Shurtleff S. A., Raimondi S. C., Kleppe M., Cools J., Shimano K. A., Hermiston M. L., Doulatov S., Eppert K., Laurenti E., Notta F., Dick J. E., Basso G., Hunger S. P., Loh M. L., Devidas M., Wood B., Winter S., Dunsmore K. P., Fulton R. S., Fulton L. L., Hong X., Harris C. C., Dooling D. J., Ochoa K., Johnson K. J., Obenauer J. C., Evans W. E., Pui C. H., Naeve C. W., Ley T. J., Mardis E. R., Wilson R. K., Downing J. R., Mullighan C. G. (2012) The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481, 157–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guo W., Lasky J. L., Chang C. J., Mosessian S., Lewis X., Xiao Y., Yeh J. E., Chen J. Y., Iruela-Arispe M. L., Varella-Garcia M., Wu H. (2008) Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature 453, 529–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kelly P. N., Dakic A., Adams J. M., Nutt S. L., Strasser A. (2007) Tumor growth need not be driven by rare cancer stem cells. Science 317, 337. [DOI] [PubMed] [Google Scholar]
- 13. Du J., Liu Y., Meline B., Kong G., Tan L. X., Lo J. C., Wang J., Ranheim E., Zhang L., Chang Y. I., Ryu M. J., Zhang J. F., Zhang J. (2013) Loss of CD44 attenuates aberrant GM-CSF signaling in Kras G12D hematopoietic progenitor/precursor cells and prolongs the survival of diseased animals. Leukemia 27, 754–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang J., Wang J., Liu Y., Sidik H., Young K. H., Lodish H. F., Fleming M. D. (2009) Oncogenic Kras-induced leukemogeneis: hematopoietic stem cells as the initial target and lineage-specific progenitors as the potential targets for final leukemic transformation. Blood 113, 1304–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kindler T., Cornejo M. G., Scholl C., Liu J., Leeman D. S., Haydu J. E., Fröhling S., Lee B. H., Gilliland D. G. (2008) K-RasG12D-induced T-cell lymphoblastic lymphoma/leukemias harbor Notch1 mutations and are sensitive to γ-secretase inhibitors. Blood 112, 3373–3382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sabnis A. J., Cheung L. S., Dail M., Kang H. C., Santaguida M., Hermiston M. L., Passegué E., Shannon K., Braun B. S. (2009) Oncogenic Kras initiates leukemia in hematopoietic stem cells. PLoS Biol. 7, e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang J., Liu Y., Li Z., Wang Z., Tan L. X., Ryu M. J., Meline B., Du J., Young K. H., Ranheim E., Chang Q., Zhang J. (2011) Endogenous oncogenic Nras mutation initiates hematopoietic malignancies in a dose- and cell type-dependent manner. Blood 118, 368–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chiang M. Y., Xu L., Shestova O., Histen G., L'Heureux S., Romany C., Childs M. E., Gimotty P. A., Aster J. C., Pear W. S. (2008) Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras-initiated leukemia. J. Clin. Invest. 118, 3181–3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ashworth T. D., Pear W. S., Chiang M. Y., Blacklow S. C., Mastio J., Xu L., Kelliher M., Kastner P., Chan S., Aster J. C. (2010) Deletion-based mechanisms of Notch1 activation in T-ALL: key roles for RAG recombinase and a conserved internal translational start site in Notch1. Blood 116, 5455–5464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Van Meter M. E., Díaz-Flores E., Archard J. A., Passegué E., Irish J. M., Kotecha N., Nolan G. P., Shannon K., Braun B. S. (2007) K-RasG12D expression induces hyperproliferation and aberrant signaling in primary hematopoietic stem/progenitor cells. Blood 109, 3945–3952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guo Z., Dose M., Kovalovsky D., Chang R., O'Neil J., Look A. T., von Boehmer H., Khazaie K., Gounari F. (2007) β-Catenin stabilization stalls the transition from double-positive to single-positive stage and predisposes thymocytes to malignant transformation. Blood 109, 5463–5472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dietrich W. F., Lander E. S., Smith J. S., Moser A. R., Gould K. A., Luongo C., Borenstein N., Dove W. (1993) Genetic identification of Mom-1, a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell 75, 631–639 [DOI] [PubMed] [Google Scholar]
- 23. Zhang X., Ren R. (1998) Bcr-Abl efficiently induces a myeloproliferative disease and production of excess interleukin-3 and granulocyte-macrophage colony-stimulating factor in mice: a novel model for chronic myelogenous leukemia. Blood 92, 3829–3840 [PubMed] [Google Scholar]
- 24. Parikh C., Subrahmanyam R., Ren R. (2007) Oncogenic NRAS, KRAS, and HRAS exhibit different leukemogenic potentials in mice. Cancer Res. 67, 7139–7146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Perentesis J. P., Bhatia S., Boyle E., Shao Y., Shu X. O., Steinbuch M., Sather H. N., Gaynon P., Kiffmeyer W., Envall-Fox J., Robison L. L. (2004) RAS oncogene mutations and outcome of therapy for childhood acute lymphoblastic leukemia. Leukemia 18, 685–692 [DOI] [PubMed] [Google Scholar]
- 26. Beier D., Hau P., Proescholdt M., Lohmeier A., Wischhusen J., Oefner P. J., Aigner L., Brawanski A., Bogdahn U., Beier C. P. (2007) CD133+ and CD133− glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 67, 4010–4015 [DOI] [PubMed] [Google Scholar]
- 27. Bagley B. N., Keane T. M., Maklakova V. I., Marshall J. G., Lester R. A., Cancel M. M., Paulsen A. R., Bendzick L. E., Been R. A., Kogan S. C., Cormier R. T., Kendziorski C., Adams D. J., Collier L. S. (2012) A dominantly acting murine allele of Mcm4 causes chromosomal abnormalities and promotes tumorigenesis. PLoS Genet. 8, e1003034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grabher C., von Boehmer H., Look A. T. (2006) Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 6, 347–359 [DOI] [PubMed] [Google Scholar]
- 29. Mansour M. R., Duke V., Foroni L., Patel B., Allen C. G., Ancliff P. J., Gale R. E., Linch D. C. (2007) Notch-1 mutations are secondary events in some patients with T-cell acute lymphoblastic leukemia. Clin. Cancer Res. 13, 6964–6969 [DOI] [PubMed] [Google Scholar]
- 30. Li X., Gounari F., Protopopov A., Khazaie K., von Boehmer H. (2008) Oncogenesis of T-ALL and nonmalignant consequences of overexpressing intracellular NOTCH1. J. Exp. Med. 205, 2851–2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang J., Liu Y., Li Z., Du J., Ryu M. J., Taylor P. R., Fleming M. D., Young K. H., Pitot H., Zhang J. (2010) Endogenous oncogenic Nras mutation promotes aberrant GM-CSF signaling in granulocytic/monocytic precursors in a murine model of chronic myelomonocytic leukemia. Blood 116, 5991–6002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gutierrez A., Look A. T. (2007) NOTCH and PI3K-AKT pathways intertwined. Cancer Cell 12, 411–413 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.