

Background: Pantothenate kinase (PanK), an essential enzyme for Mycobacterium tuberculosis, catalyzes the rate-limiting step in the CoA pathway.
Results: Structures of M. tuberculosis PanK, complexed with new inhibitors, were determined, and their inhibitions were evaluated biochemically.
Conclusion: Inhibitor binding overlaps with the substrate/product sites; also, an alternative mode of ATP binding is proposed.
Significance: These are the first structures of a type I PanK complexed with inhibitors.
Keywords: Coenzyme A, Crystal Structure, Drug Design, Enzyme Inhibitors, Mycobacterium tuberculosis, CoaA, Pantothenate Kinase
Abstract
Mycobacterium tuberculosis, the bacterial causative agent of tuberculosis, currently affects millions of people. The emergence of drug-resistant strains makes development of new antibiotics targeting the bacterium a global health priority. Pantothenate kinase, a key enzyme in the universal biosynthesis of the essential cofactor CoA, was targeted in this study to find new tuberculosis drugs. The biochemical characterizations of two new classes of compounds that inhibit pantothenate kinase from M. tuberculosis are described, along with crystal structures of their enzyme-inhibitor complexes. These represent the first crystal structures of this enzyme with engineered inhibitors. Both classes of compounds bind in the active site of the enzyme, overlapping with the binding sites of the natural substrate and product, pantothenate and phosphopantothenate, respectively. One class of compounds also interferes with binding of the cofactor ATP. The complexes were crystallized in two crystal forms, one of which is in a new space group for this enzyme and diffracts to the highest resolution reported for any pantothenate kinase structure. These two crystal forms allowed, for the first time, modeling of the cofactor-binding loop in both open and closed conformations. The structures also show a binding mode of ATP different from that previously reported for the M. tuberculosis enzyme but similar to that in the pantothenate kinases of other organisms.
Introduction
Pantothenate kinase (PanK,2 EC 2.7.1.33) catalyzes the first and rate-limiting step in the universal CoA biosynthetic pathway, where pantothenate (vitamin B5) is converted to 4′-phosphopantothenate using ATP as a cofactor (1). Three types of PanK have been described, which differ in their biochemical and structural characteristics. Type I, encoded by the gene coaA (2), is found in a large number of bacterial species and is tightly feedback-regulated by CoA and its thioesters (3). It is exemplified by the extensively studied Escherichia coli PanK (EcPanK; Ref. 4, 5). The type II enzyme is mostly found in eukaryotes. Humans express four isoforms of the enzyme, named PanK1 to 4. Defects in the panK2 gene have been linked to neurodegenerative disease (6, 7). Based on sequence and structural homology some bacterial enzymes, such as Staphylococcus aureus PanK, are also classified as type II enzymes. Whereas the eukaryotic PanKs are feedback-regulated by CoA, the S. aureus enzyme is not (8). Type III, encoded by the gene coaX, is the most widespread type of the enzyme, with homologs present in 12 of the 13 major bacterial groups (9). It is not inhibited by CoA or its thioesters (10). Many bacteria have two PanK genes coding for different types of the enzyme.
Tuberculosis, caused by the pathogenic bacterium Mycobacterium tuberculosis, is estimated to infect one-third of the world population and is responsible for ∼1.4 million deaths annually (World Health Organization, 2011). High-burden countries for tuberculosis are found in sub-Saharan Africa, with high rates of the deadly combination of HIV and tuberculosis. Current treatment of tuberculosis relies on the frontline drugs isoniazid and rifampicin, developed in the 1950s and 1960s. Increasing incidence rates of multidrug-resistant tuberculosis, resistant to front-line drugs, and extensively drug-resistant tuberculosis, resistant to front-line and second-line drugs, make development of new antibiotics targeting M. tuberculosis a global health priority.
The M. tuberculosis genome contains both coaA and coaX genes, coding for a type I and type III PanK, respectively. However, it has been shown that coaA is the only PanK gene essential for bacterial growth in vitro and in vivo (11). The M. tuberculosis type I PanK (MtPanK) is a 312-amino acid (36 kDa) P-loop kinase with a central seven-stranded β-sheet surrounded by α-helices, containing a Walker A motif for nucleotide binding (12). It functions as a homodimer. MtPanK has been the target for extensive structural studies and has been crystallized in complex with its substrates, products, adenosine or guanosine cofactors, and feedback inhibitor CoA (13–16). The MtPanK structures in this study represent the first complexes with engineered inhibitory compounds and provide a starting point for development of novel antibiotics targeting pathogens that are dependent on type I PanK.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
The gene encoding MtPanK (coaA, Rv1092c) was PCR-amplified from genomic DNA, and an N-terminal His6 tag was added. The resulting construct was ligated into a pET15b expression vector (Novagen) using restriction enzymes NdeI and BamHI. Correct ligation of the sequence coding for a full-length His-tagged MtPanK was confirmed by sequencing. The resulting expression vector was used to transform E. coli BL21-AI competent cells (Invitrogen). Cells were incubated at 37 °C in Luria-Bertani growth medium, with 100 μg/ml ampicillin. Expression was induced by adding 0.2% (w/v) l-arabinose, at A600 ≈ 0.6, and cells were further incubated at 37 °C for 3 h. Cells were centrifuged at 4500 × g for 30 min, and the resulting cell pellet was resuspended in lysis buffer (50 mm Tris-HCl, pH 8.0, 300 mm NaCl, 20 mm imidazole) with 0.01 mg/ml RNase and 0.02 mg/ml DNase. Cells were lysed in a cell disruptor (Constant Systems, Ltd.) and the lysate was centrifuged at 18,000 × g for 30 min to remove cell debris. The His-tagged protein was then purified by binding it on a nickel-Sepharose (GE Healthcare) column. The column was washed with 10 column volumes of wash buffer (50 mm Tris-HCl, pH 8.0, 300 mm NaCl, 50 mm imidazole), and the protein was eluted with 4 column volumes of elution buffer (50 mm Tris-HCl, pH 8.0, 300 mm NaCl, and 500 mm imidazole). The elution fractions were pooled and then diluted at a 2:1 ratio with gel filtration buffer (50 mm Hepes, pH 7.5, 300 mm NaCl, 2% (v/v) glycerol, and 10 mm β-mercaptoethanol). The diluted protein sample was further purified using a Superdex 75 (GE Healthcare) size-exclusion column, pre-equilibrated with gel filtration buffer. A modified gel filtration buffer (50 mm Tris-HCl, pH 7.5, 300 mm NaCl, 10% (v/v) glycerol, 2 mm DTT, and 0.1 mm EDTA) was used in subsequent purifications of the enzyme for biochemical studies. Elution fractions were pooled, and analysis by SDS-PAGE showed it to contain ∼99% pure protein of correct size. Protein aliquots were stored at −70 °C. Only once-thawed aliquots were used for each experiment.
IC50, Minimum Inhibitory Concentration , and Thermal Stability Measurements
The biochemical and thermal shift assays have been described in detail by Venkatraman et al. (17). Briefly, MtPanK activity was measured in a pyruvate kinase-lactate dehydrogenase-coupled assay where the reaction rate was determined by spectrophotometric measurement of NADH consumption. IC50 measurements were performed with a suitable concentration range of inhibitory compound, while keeping the concentration of pantothenate and ATP at 122 and 395 μm, respectively, which is their respective Km (17). A second set of IC50 measurements were performed with the ATP concentration at 50× Km, to determine the effect of cofactor competition.
Thermal stability measurements were recorded with a real-time PCR thermal cycler where the fluorescence of SYPRO orange dye (Invitrogen) was used to monitor protein unfolding in the absence or presence of 25 μm inhibitory compound, at temperature intervals of 0.5 °C, from 25 °C to 85 °C. Measurements were repeated with the addition of 0.5 and 1.0 mm ATP.
The minimum inhibitory concentration assay was set up as per the Clinical and Laboratory Standards Institute guidelines. These assays were done in 96-well plate format, and the readout was by Microplate Alamar Blue Assay (18).
Crystallization
The purified protein was concentrated to 12 mg/ml in 20 mm Tris-HCl, pH 8.0, 0.3 m NaCl, 10 mm β-mercaptoethanol, and 10% (v/v) glycerol for initial crystallization trials. Trials were set up with the sitting-drop vapor diffusion method on an Oryx 6 robot (Douglas Instruments) in two-well MRC plates (Molecular Dimensions). Droplets composed of 0.3 μl of protein solution and 0.3 μl of reservoir solution were equilibrated against 80 μl of the latter. Inhibitors were dissolved in dimethyl sulfoxide. For cocrystallization experiments, a 10-molar excess of the inhibitor with and without 5 mm ATP and 1 mm MgCl2 was added to the protein immediately before crystallization trials. In addition, five non-hydrolyzable ATP analogues (Jena Bioscience, catalog no. NK-101), adenosine, adenosine ribose, and ADP were substituted for ATP.
Crystals of a binary complex (MtPanK-1a) in space group P3121 grew in 3–4 days at 20 °C from condition E11 of the JCSG+ screen (Qiagen) containing 14.4% (w/v) PEG 8000, 20% (v/v) glycerol, 0.08 m sodium cacodylate, pH 6.5, and 0.16 m calcium acetate. The optimized crystallization conditions were essentially the same except that magnesium chloride replaced the calcium. The cryosolution for these crystals consisted of 35% (w/v) PEG 3350, 0.2 m cacodylate, 10 mm MgCl2, and, where appropriate, 1 mm inhibitor. Crystals were placed in the cryosolution for 5 s before vitrification in liquid nitrogen. Soaking experiments on apo-crystals with this space group were unsuccessful.
Crystals of seven different ternary complexes (MtPanK-inhibitor-PO4) in space group P21212 were obtained with 1.8 m sodium/potassium phosphate, pH 8.2 (Quik screen, Hampton Research) at 20 °C in a 3–4 days. For optimization of these crystals, the protein concentration was reduced to 5 mg/ml, and the drops were seeded. These drops (0.5 μl each of protein and reservoir solution) were set up manually and equilibrated against 1 ml of the reservoir solution (1.8 m sodium/potassium phosphate, pH 8.2) in Nextal 24-well plates (Qiagen). The ternary complexes were obtained either by cocrystallization or soaking. The crystals (0.1 × 0.2 × 0.2 mm) were vitrified directly from the drops.
Data Collection and Processing
Diffraction data from MtPanK, co-crystallized or soaked with inhibitors, were collected from single crystals at the European Synchrotron Radiation Facility (Grenoble, France) or MAXlab (Lund, Sweden). Diffraction data were indexed and integrated using MOSFLM (19) and scaled with SCALA (20). These programs are part of the CCP4 program package (21). Data collection statistics are shown in Table 1.
TABLE 1.
Data collection and refinement statistics
| MtPanK-1a | MtPanK-1b-PO4 | MtPanK-1c-PO4 | MtPanK-1d-PO4 | MtPanK-1e-PO4 | MtPanK-1f-PO4 | MtPanK-2a-PO4 | MtPanK-2b-PO4 | |
|---|---|---|---|---|---|---|---|---|
| Data collection | ||||||||
| PDB code | 4BFS | 4BFT | 4BFU | 4BFV | 4BFW | 4BFX | 4BFY | 4BFZ |
| Beam line | I911-2, MAXlab | ID14-1, ESRFa | ID14-4, ESRF | ID14-1, ESRF | ID23-1, ESRF | ID14-4, ESRF | ID14-1, ESRF | ID14-4, ESRF |
| Detector | MAR 165 | ADSC Q210 | ADSC Q315r | ADSC Q210 | ADSC Q315r | ADSC Q315r | ADSC Q210 | ADSC Q315r |
| Wavelength (Å) | 1.0412 | 0.9340 | 0.9795 | 0.9334 | 1.0723 | 0.9795 | 0.9334 | 0.9795 |
| Space group | P3121 | P21212 | P21212 | P21212 | P21212 | P21212 | P21212 | P21212 |
| Cell axial lengths (Å) | 105.28, 105.28, 90.80 | 88.40, 149.69, 62.77 | 88.60, 150.69, 63.18 | 88.33, 151.77, 63.13 | 89.03, 150.71, 60.50 | 88.01, 149.51, 62.83 | 88.29, 149.28, 61.91 | 88.46, 148.28, 63.26 |
| Vm (Å3/Da)b | 4.07 | 2.91 | 2.96 | 2.97 | 2.85 | 2.90 | 2.86 | 2.91 |
| Resolution range (Å)c | 30.0–2.90 (3.06–2.90) | 40.0–2.29 (2.41–2.29) | 60.0–2.28 (2.40–2.28) | 40.0–2.29 (2.42–2.29) | 50.0–2.27 (2.39–2.27) | 60.0–2.70 (2.85–2.70) | 44.0–2.30 (2.42–2.30) | 58.0–2.10 (2.21–2.10) |
| No. of reflections measured | 78,322 (11,264) | 136,311 (19,054) | 266,498 (38,455) | 280,048 (40,356) | 125,447 (18,679) | 148,478 (21,019) | 168,008 (22,704) | 341,894 (50,728) |
| No. of unique reflections | 13,018 (1865) | 37,958 (5478) | 39,320 (5655) | 38,852 (5589) | 38,286 (5516) | 23,491 (3392) | 34,705 (4857) | 49,393 (7134) |
| Average multiplicity | 6.0 (6.0) | 3.6 (3.5) | 6.8 (6.8) | 7.2 (7.2) | 3.3 (3.4) | 6.3 (6.2) | 4.8 (4.7) | 6.9 (7.1) |
| Completeness (%) | 98.9 (99.5) | 99.4 (99.3) | 99.3 (99.3) | 100 (100) | 99.6 (99.7) | 99.9 (99.9) | 93.6 (91.6) | 99.9 (100.0) |
| Rmerged,e | 0.115 (0.491) | 0.111 (0.486) | 0.085 (0.430) | 0.095 (0.399) | 0.102 (0.500) | 0.114 (0.495) | 0.106 (0.431) | 0.107 (0.491) |
| Rmeasf | 0.127 (0.537) | 0.130 (0.571) | 0.093 (0.466) | 0.103 (0.430) | 0.122 (0.593) | 0.124 (0.541) | 0.118 (0.483) | 0.116 (0.529) |
| Rp.i.m.g | 0.051 (0.213) | 0.066 (0.291) | 0.036 (0.176) | 0.038 (0.159) | 0.066 (0.316) | 0.049 (0.215) | 0.051 (0.209) | 0.044 (0.197) |
| 〈I/σI〉 | 10.1 (3.3) | 10.1 (2.6) | 12.3 (3.7) | 19.2 (5.0) | 6.9 (2.3) | 10.6 (3.4) | 9.3 (3.2) | 12.0 (3.9) |
| Refinement statistics | ||||||||
| Resolution range (Å) | 30.0–2.90 (2.98–2.90) | 40.0–2.29 (2.35–2.29) | 60.0–2.28 (2.33–2.28) | 40.0–2.29 (2.35–2.29) | 50.0–2.27 (2.33–2.27) | 60.0–2.70 (2.77–2.70) | 44.0–2.30 (2.36–2.30) | 58.0–2.10 (2.16–2.10) |
| No. of reflections used in working set | 12,375 (807) | 36,047 (2420) | 37,299 (2497) | 36,854 (2466) | 36,326 (2446) | 22,238 (1455) | 32,917 (2166) | 46,841 (3162) |
| No. of reflections for Rfree calculation | 639 (41) | 1905 (125) | 1972 (135) | 1946 (138) | 1912 (135) | 1208 (82) | 1747 (105) | 2501 (170) |
| R (%)c | 19.9 | 22.5 | 22.1 | 20.6 | 21.6 | 20.9 | 20.3 | 20.7 |
| Rfree (%) | 24.5 | 28.6 | 26.3 | 26.2 | 27.3 | 28.5 | 25.7 | 24.8 |
| No. of non-hydrogen atoms | 2484 | 5109 | 5148 | 5229 | 5164 | 5075 | 5226 | 5307 |
| No. of solvent waters | 13 | 99 | 136 | 213 | 134 | 63 | 210 | 289 |
| Average B-factors (Å2) | ||||||||
| Protein atoms (A, B) | 52.0, −h | 45.4, 43.1 | 47.3, 48.8 | 32.0, 32.1 | 42.7, 46.3 | 44.6, 45.6 | 30.6, 33.9 | 30.5, 31.9 |
| Solvent atoms | 34.7 | 34.4 | 41.9 | 29.2 | 38.7 | 33.5 | 30.0 | 32.1 |
| Inhibitory compound (A, B) | 73.7, − | 51.6, 40.0 | 47.2, 51.3 | 34.9, 34.0 | 47.9, 50.5 | 64.8, 71.4 | 21.1, 24.4 | 22.8, 24.4 |
| Phosphate ion (A, B) | − | 31.5, 33.8 | 36.5, 34.9 | 22.2, 26.2 | 29.3, 31.3 | 37.5, 37.9 | 21.7, 24.9 | 26.4, 24.9 |
| Ramachandran plot outliers (A, B) (%)i | 3.5, − | 5.2, 5.5 | 6.5, 3.1 | 5.2, 4.1 | 4.1, 7.2 | 4.1, 4,5 | 3.8, 5.5 | 3.1, 2.7 |
| r.m.s.d. from ideal bond length (Å)j | 0.009 | 0.012 | 0.011 | 0.010 | 0.012 | 0.009 | 0.011 | 0.009 |
| r.m.s.d. from ideal bond angle | 1.479° | 1.795° | 1.650° | 1.612° | 1.897° | 1.564° | 1.725° | 1.501° |
a ESRF indicates the European Synchrotron Radiation Facility.
b Values were calculated using methods described by Matthews (Ref. 31).
c Values in parentheses refer to the outer resolution shell.
d Merging and crystallographic R-factors were generated by the computer programs described under “Experimental Procedures.”
e Rmerge = ΣhklΣi|Ii(hkl) − 〈I(hkl)〉|/ΣhklΣiIi(hkl).
f Rmeas = Σhkl[N/(N−1)]½ Σi|Ii(hkl) − 〈I(hkl)〉|/ΣhklΣiIi(hkl).
g Rp.i.m. = Σhkl[1/(N−1)]½ Σi|Ii(hkl) − 〈I(hkl)〉|/ΣhklΣiIi(hkl).
h −, value is not applicable.
i Calculated using a strict boundary Ramachandran plot definition (Ref. 32).
j Ideal values are from Engh & Huber (Ref. 33).
Structure Determination and Refinement
The MtPanK-1a binary complex (see Table 1) was crystallized in space group P3121, isomorphous with the published MtPanK-CoA structure (PDB code 2GEV; Ref. 13). These coordinates were stripped of ligands and water molecules and subjected to an initial round of rigid body crystallographic refinement with REFMAC5 (22). The seven structures of the MtPanK-inhibitor-PO4 ternary complexes (see Table 1) in space group P21212 were solved by molecular replacement with the program PHASER (23), using the refined MtPanK-1a structure, stripped of ligands and waters, as a search model.
All eight structures were improved by multiple, alternating cycles of crystallographic refinement with REFMAC5 and interactive rebuilding using the program O (24). Waters were added using the carbonyl oxygen profiling methods implemented in O. Coordinates and stereochemical restraints for inhibitory compounds were generated in O, and stereochemical dictionaries for refinement were generated by REFMAC5. Refinement statistics are shown in Table 1.
Other Methods
Structural figures were created in O and rendered with MOLRAY (25). Detailed structural comparisons were made in O with Cα matching pair cutoffs of 3.8 Å (26). Sequence alignments were made using ClustalW (27), and the corresponding figure was designed with ALINE (28).
RESULTS
Sequence Alignment
The sequence alignment of type I PanK sequences from 13 pathogenic bacterial species shows that the sequences are highly conserved, with a minimum of 41% pairwise sequence conservation (Fig. 1). The longest stretches of conserved residues are found around the P-loop, where the Walker A motif is identical in all 13 species, as are residues involved in substrate and cofactor binding. The Mt enzyme differs from the rest in the loop formed by residues 79–91 (MtPanK numbering), flanked by helix α4 and strand β2, which is extended by four residues. Some variation is seen in the length of the loop formed by residues 203–213 (MtPanK numbering), flanked by strand β6 and helix α10.
FIGURE 1.

Sequence alignment of type I PanKs from various pathogenic bacteria. MtPanK numbering and secondary structure are shown as defined by Das et al. (13). The blue triangle shows residue Arg-238, interacting with the phosphate bound in the P-loop, or inhibitory compounds. Red triangles show residues involved in pantothenate/phosphopantothenate binding. Green triangles show residues involved in adenine binding in the nucleoside binding site reported for Ec- and CbPanK. Sequences used in this alignment were as follows: MtPanK (UniProt ID P63810), Corynebacterium diphtheriae PanK (UniProt ID Q6NI48), EcPanK (UniProt ID P0A6I3), Salmonella typhi PanK (UniProt ID Q8Z318), Shigella dysenteriae PanK (UniProt ID Q32aF0), Klebsiella pneumoniae PanK (UniProt ID G0GSU7), Yersinia pestis PanK (UniProt ID Q8ZAN6), Haemophilus influenzae PanK (UniProt ID P44793), Vibrio cholerae PanK (UniProt ID Q9KV38), Coxiella burnetii PanK (UniProt ID Q83EV9), Bacillus subtilis PanK (UniProt ID P54556), Listeria monocytogenes PanK (UniProt ID Q8Y8I0), and Streptococcus pneumoniae PanK (UniProt ID Q97RH6).
The MtPanK-1a Binary Complex
The MtPanK-1a binary complex was solved in space group P3121, with one molecule in the asymmetric unit, and refined to a crystallographic R factor of 19.9% at a resolution of 2.9 Å (see Table 1 for crystallographic data). The crystal structure is in the same space group and has similar unit cell parameters as the previously reported MtPanK structures. It is isomorphous to what has been called crystal form II of the enzyme (13). The MtPanK-1a structure can be superimposed on all previously described MtPanK structures with root mean square deviations (r.m.s.d.) from 0.3 to 0.6 Å for 302 paired Cα atoms. The greatest differences between the MtPanK structures in this space group are located at the N-terminal and the loop formed by residues 79–91, which adopt different conformations in crystal forms I and II of the enzyme (13). The flexible part of this loop, residues 81–87, is disordered in our MtPanK-1a structure and has therefore been omitted from the model as well as the disordered first five residues and the N-terminal His tag.
The MtPanK-inhibitor-PO4 Ternary Complexes
Structures of seven ternary complexes of MtPanK with inhibitory compounds and phosphate ions were solved in space group P21212 (see Table 1 for crystallographic data). This is a new space group for this enzyme. One of these complexes, the MtPanK-2b-PO4 complex, diffracts to a resolution of 2.1 Å, which is the highest resolution reported for a type I PanK crystal structure. The structures in this space group have a complete homodimer in the asymmetric unit. This dimer is positioned such that the crystallographic 2-fold operator forms a tetramer with 222 point group symmetry. The active sites in both molecules contain a bound inhibitor and a phosphate ion. The first six residues and the N-terminal His tag are disordered in these structures and therefore omitted from the model. The A and B molecules of the homodimers can be superimposed with r.m.s.d. values ranging from 0.4 to 0.8 Å for all paired Cα atoms. The MtPanK-1a binary complex, as a representative of the structures in the trigonal space group, can be superimposed on the MtPanK-inhibitor-PO4 ternary complexes with an average r.m.s.d. of 1.1 Å for both A and B molecules, for all paired Cα atoms. The main differences are found in the second half of helix α4 at the dimerization interface. Here the end point, Leu-78, is shifted ∼6.5 Å as a result of helix bending, adopting a local conformation identical to that of the EcPanK (5, 29) and Coxiella burnetii PanK (CbPanK)3 structures (Fig. 2). Another difference is the cofactor-binding loop, or adenosine lid, which adopts an open conformation, in contrast to the closed conformation seen in all previous MtPanK structures (discussed in detail below). The open conformation of this loop forces a shift of helix α1 and the loop between helices α1 and α2.
FIGURE 2.

Cα superposition of A molecules of all MtPanK-inhibitor-PO4 complexes (red), MtPanK-1a (green), EcPanK-CoA (light blue, PDB code 1ESM), EcPanK-Pan-ADP (blue, PDB code 1SQ5), and CbPanK-ADP (yellow, PDB code 3TQC). Shown is the conserved conformation of the P-loop, the open and closed form of the adenosine lid (a-lid), the alternative conformation of helix α4, and the two loops (with MtPanK numbering) elongated in MtPanK, EcPanK, or CbPanK.
The MtPanK-inhibitor-PO4 ternary complex dimers superimpose on the various EcPanK or CbPanK dimers with average r.m.s.d. values of 1.3 and 1.2 Å, respectively, with more than ∼570 paired Cα atoms. Although the structures of the central β-sheet and the P-loop are highly conserved in all PanK structures, differences can be seen in the surrounding helices and loops. The main differences between the Mt-, Ec-, and CbPanKs are located to the loop formed by residues 203–213 (MtPanK numbering), flanked by strand β6 and helix α10 (Fig. 1). This loop is elongated by seven residues in the Ec and Cb enzymes and extends away from the center of the molecule in different conformations (Fig. 2). However, the loop formed by residues 79–91 (MtPanK numbering), flanked by helix α4 and strand β2, is elongated by four residues in the Mt enzyme (Figs. 1 and 2).
Binding of Inhibitors
We have determined crystal structures of eight protein-inhibitor complexes of MtPanK with inhibitors of two different classes. Compounds 1a to 1f are triazoles, whereas compounds 2a and 2b are biaryls (Table 2). Electron density is well defined for all of these inhibitors, except for the slightly less well defined electron density of the difluorophenyl ring in 1f (Fig. 3).
TABLE 2.
Structures of the inhibitory compounds and results from the biochemical assay, thermal shift, and MIC measurements
ND, not determined.
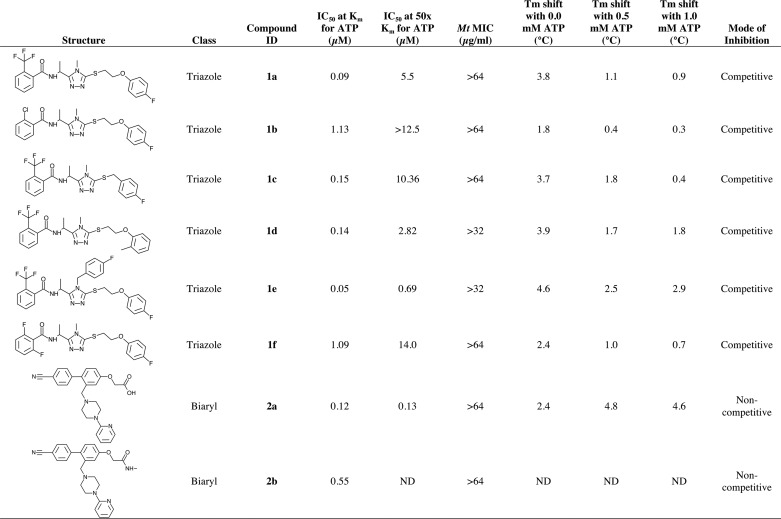
FIGURE 3.

Representative electron density for inhibitors in the A molecules of their respective complex structure. The σA-weighted (2m|Fo| − D|Fc|) electron density maps (34) are drawn at the level of the root mean square value of the respective map.
The binding mode of the triazole compounds is exemplified by triazole 1a in Fig. 4a. They overlap with pantothenate (MtPanK-Pan-AMPPCP, PDB code 2ZSE; Ref. 14), and the pantothenate moieties of phosphopantothenate (MtPanK-PPan-ADP, PDB code 2ZSA; Ref. 14) and CoA (MtPanK-CoA, PDB code 2ZSD) in the active site pocket (Fig. 5, a and b, respectively). These compounds fan out from the conserved triazole ring to form similar U-shaped conformations where the halogen-substituted phenyl rings are roughly co-planar, with ring edges abutting. In the 1b complex, for example, the vector normals to the phenyl rings form a 51° angle, with ring centers separated by 7.8 Å, and the pair of ring edge carbons separated by ∼5.5–6.5 Å. Each triazole ring forms a pair of conserved hydrogen bond interactions with side chains that are themselves highly conserved (Fig. 4a). Residues Tyr-235 and Asn-277 are involved in pantothenate and phosphopantothenate binding (14), respectively, but here, the hydroxyl group of Tyr-235 forms a hydrogen bond to one nitrogen in the triazole ring, whereas the amide group of the side chain of Asn-277 hydrogen bonds to another (Fig. 4a). Hydrogen bonding atoms in the peptide linkage make interactions with water molecules, where visible, except in the 1c complex where the carbonyl oxygen directly interacts with the hydroxyl of Tyr-177. No hydrogen bonding interactions are observed to the ether-linking oxygen. The removal of the ether-linked oxygen in 1c is easily accommodated by the inhibitor. Here, we see a ∼0.7 Å shift in the fluorophenyl group. This involves a small pivot of the triazole ring that maintains the protein-inhibitor hydrogen bonds but results in relatively large local changes in the enzyme. In particular, the side chain of Tyr-182 takes on a different rotamer conformation compared with all the other complexes. The tyrosyl ring comes closer to the space vacated by the truncated ether linkage, and there is a rigid-body shift in helix α12, one of the helices that define the binding site. A much larger change in structure has been made in 1e where a para-fluorobenzyl ring extends from the triazole ring. This change is easily accommodated by the enzyme, fitting into a preformed hydrophobic tunnel in the active site whose surface is created from four segments of the enzyme (helix α12 and α13, the loop between helix α7-α8 and sheets β4–β5).
FIGURE 4.

a, binding of a triazole compound (gray carbons) in the active site, exemplified by the MtPanK-1a complex. Also shown are the residues involved in compound interactions and the P-loop. b, binding of a biaryl compound (white carbons) in the A molecule active site, exemplified by the MtPanK-2b-PO4 complex. Also shown are the residues involved in compound interactions (yellow carbons) and the phosphate ion bound in the P-loop. c, superposition of all triazole compounds (gray carbons) and biaryl compounds (white carbons) in the MtPanK active site. The secondary structure is colored as in Fig. 1.
FIGURE 5.

a, superposition of MtPanK-1a, MtPanK-2b-PO4, MtPanK-Pan-AMPPCP (PDB code 2ZSE) and MtPanK-PPan-ADP (PDB code 2ZSA) showing the triazole compound 1a (green), the biaryl-compound 2b (red) with the phosphate ion (atom colors), pantothenate (blue), and phosphopantothenate (yellow). b, superposition of MtPanK-1a, MtPanK-2b-PO4, and MtPanK-CoA (PDB code 2ZSD) showing the triazole compound 1a (green), the biaryl compound 2b (red) with the phosphate ion (atom colors), and CoA (light blue). c, superposition of MtPanK-1a, MtPanK-2b-PO4, MtPanK-Pan-AMPPCP (PDB code 2ZSE) and EcPanK-AMPPNP (PDB code 1ESN) showing the triazole compound 1a (green), the biaryl compound 2b (red) with the phosphate ion (atom colors), AMPPCP bound in the Mt structure (gray), and AMPPNP bound in the Ec structure (magenta). The van der Waals radii of the fluorine in compound 1a and the γ-phosphate oxygen of AMPPCP and AMPPNP are shown as transparent spheres.
Many of the fluorine atoms are involved in the electrostatic interactions described by Müller et al. (30). One of the phenyl rings has a para-fluorine atom in five of the six triazole compounds, and this ring binds in a mostly hydrophobic pocket made up of side chains Val-99, Ala-100, Tyr-235, Phe-239, and Met-242. The side chain of Arg-238, however, stretches to the bound phosphate in the orthorhombic crystal forms (for five of the six triazoles), to form a salt link. The para-fluorophenyl ring is approximately parallel to the plane of the guanidinium group with the fluorine atom ∼3.3 Å above the plane. Another salt link exists between Asp-129 and His-179. The trifluoro-methyl at the ortho-phenyl substitution of 1e, for example, is 3.3 Å from a nitrogen of the imidazole ring but is more distant in the 1c complex (closest contact at 3.8 Å) as a consequence of the structural rearrangement in the enzyme. The relative orientation of the phenyl rings also means that the trifluoro-methyl group in one ring points toward the edge of the second ring in these structures.
The biaryl compounds, 2a and 2b (Fig. 4b), were both crystallized in ternary complexes with phosphate ions in the orthorhombic space group. These compounds also bind in the active site pocket, overlapping with the binding site of the triazole compounds (overlaid structure, Fig. 4c), and that of the pantothenate moieties in the previous MtPanK structures (Fig. 5, a and b). In the 2b complex, the Arg-238 side chain interacts with the phosphate ion as seen with the triazoles but changes conformation to form a salt link with the carboxylate of the inhibitor in the 2a complex. The Tyr-182 side chain has the same rotamer conformation observed in the 1c complex such that the hydroxyl group interacts with the ether linkage oxygen atom, as well as the carboxylate of one inhibitor and the amide nitrogen of the other (Fig. 4b). The para-nitrile phenyl group from each inhibitor packs into the hydrophobic tunnel used by the para-fluorobenzyl ring of 1e. The conserved residues Tyr-235 and Asn-277 that are important for triazole binding do not interact directly with the biaryl class of compounds. Instead, they form hydrogen bonds to a water molecule that, in turn, interacts with a nitrogen of the piperazine ring. The 2-pyridyl nitrogen also forms a hydrogen bond to another water molecule, structurally equivalent to the water forming the same interaction with the peptide oxygen of the triazoles (Fig. 4b). When the triazole and biaryl complexes are superimposed on the ATP analogue complexes of Ec- and MtPanK, the phosphate ion that we observe in the orthorhombic crystal form overlaps with the ATP β-phosphate group (Fig. 5c).
The Different Conformations of the Adenosine Lid
A structural feature of type I PanK enzymes is an adenosine-lid loop formed by residues 38–44 (MtPanK numbering) and flanked by helices α2 and α3 (Figs. 1 and 2). The loop is in an open form when the adenosine moiety of ADP or ATP is bound, exemplified by the EcPanK-Pan-ADP, EcPanK-AMPPNP, and CbPanK-ADP complex structures (Fig. 6a). In the absence of a bound adenosine, the loop adopts a closed conformation, exemplified by the EcPanK-CoA structure (Fig. 6b). When switching from an open to closed conformation the loop shifts ∼5 Å toward the adjacent helix α11. To accommodate this shift, the flanking helix α2 unwinds, contracting by one turn in the closed conformation. Helix α2 extends almost to helix α3 in the open form of the CbPank-ADP structure. The adenine ring of the cofactor is stacked between the loop and conserved residue His-302 (MtPanK numbering), which in turn stacks with the ring of conserved residue Trp-234 (MtPanK numbering) in the open form of the EcPanK and CbPanK structures (Fig. 6a). However, the MtPanK structures crystallized in complex with nucleoside cofactors exhibit an alternative binding-mode (Fig. 5c; Refs. 15 and 16), where the adenine or guanine moieties occupy roughly the same binding site as the adenine moiety of CoA (Fig. 5b). In these structures, the adenosine lid retains a closed conformation, unable to accommodate a nucleoside in this binding site. In the MtPanK-1a complex, crystallized in the previously reported trigonal space group, the adenosine lid adopts a closed conformation, identical to all previously reported MtPanK structures. However, in the MtPanK-inhibitor-PO4 structures crystallized in the orthorhombic space group, the loop adopts an open conformation. This conformation is similar to the open conformation of the EcPanK and CbPanK structures, although no adenosine is present in the adenosine-binding site (Fig. 6).
FIGURE 6.

a, superposition of MtPanK-2b-PO4 (red), EcPanK-AMPPNP (magenta, PDB code 1ESN), EcPanK-Pan-ADP (blue, PDB code 1SQ5), and CbPanK-ADP (yellow, PDB code 3TQC) showing the open form of the adenosine lid and conserved residues involved in adenosine binding. b, superposition of MtPanK-1a (green), EcPanK-CoA (light blue, PDB code 1ESM), MtPanK-2b-PO4 (red), EcPanK-AMPPNP (magenta, PDB code 1ESN), EcPanK-Pan-ADP (blue, PDB code 1SQ5), and CbPanK-ADP (yellow, PDB code 3TQC) showing the open and closed forms of the adenosine lid.
Attempts to obtain binary complexes of MtPanK with ATP or ternary complexes with inhibitory compounds and ATP, whether by co-crystallization or soaking in conditions for the orthorhombic space group, were unsuccessful. Non-hydrolyzable ATP analogues, adenosine, adenosine ribose, or ADP were also used in place of ATP but without success. Although some crystals were obtained and diffraction data were collected, the resolution and data quality did not allow modeling of the entities seen in the active site. The high phosphate concentration required for crystallization in this space group, resulting in a phosphate occupying the P-loop or potentially close contacts between our inhibitors and the γ-phosphate of ATP, may account for our inability to crystallize MtPanK with bound ATP analogues.
IC50, Minimum Inhibitory Concentration, and Thermal Stability Measurements
Detailed results for related inhibitory compounds of the classes presented here have been described (17). The loss of inhibition of the triazole compounds when the assay was run in excess of ATP (Table 2) indicates that these compounds compete with ATP. This is supported by the thermal stability measurements of these compounds where a decrease in the thermal shift is observed when the measurements are performed at higher ATP concentrations (Table 2). The biaryl compound 2a shows comparable levels of inhibition at low and high ATP concentrations, suggesting that this class of compounds is non-competitive with respect to ATP. The thermal stability measurements for this compound show an increase in thermal shift in the presence of ATP, also indicative of a non-competitive mode of inhibition. Unfortunately, inhibition in the presence of ATP and thermal stability measurements were not available for compound 2b. None of the compounds described here inhibited growth of Mt in the whole cell assay at their respective maximum concentration tested (Table 2).
DISCUSSION
We have determined the first enzyme-inhibitor complex structures of a type I PanK enzyme. The structures show that the two classes of inhibitory compounds, triazoles and biaryls, bind in roughly the same position, overlapping with binding sites for the substrate pantothenate and product phosphopantothenate.
The six triazole complexes provide 12 views of how this class of inhibitor interacts with MtPanK. The inhibitors share a common binding site with the same overall U-shaped structure and share many equivalent interactions with the enzyme and structurally conserved water molecules. In particular, the hydrogen bonding network of the triazole nitrogens with residues Tyr-235 and Asn-277 is a feature of this class of inhibitor. Modulation in IC50 values has been achieved by the introduction of halogen atoms and/or groups, by removal of the ether-linked oxygen, and by an extension from the central triazole ring. The benefits of introducing fluorine atoms on small molecule scaffolds is well documented, and structural data have been used to clarify the nature of fluorine-protein interactions. It has been suggested that C-F dipolar and multipolar interactions with protein side chain and main chain atoms can benefit the energetics of inhibitor binding (30). Favorable interactions with backbone and side chain amides as well as the guanidinium group of arginine residues appear to be common occurrences. The top four, most active triazoles (1a, 1c, 1d, and 1e) contain a trifluoromethyl substitution that is positioned close to the imidazole ring of His-179, with closest contacts in the range 3.3–3.7 Å. The trifluoromethyl group points toward the edge of the second phenyl ring of each inhibitor to form other, potentially stabilizing multipolar interactions. The combined effects may be sufficient to explain the 10-fold increase in IC50 values that are observed for 1b and 1f where the trifluoromethyl group has been substituted. In seven of the eight triazoles, the second phenyl ring is also fluorinated at the para-position. In these structures, we observe an interaction with the guanidinium group of Arg-238, but the removal of the fluorine atom does not reduce the activity of 1d. The replacement of the para-fluorophenyl ring by an ortho-methylphenyl ring in 1d results in a structural rearrangement such that the ring to ring centers are closer in 1d by 0.3 Å. This may benefit the interactions with the trifluoromethyl group sufficiently so that there is little change in the IC50 compared with 1a, for example.
IC50 values for the biaryl compounds are comparable with those for the triazole compounds, with 2a having the lowest IC50 (Table 2). The approximate 5-fold increase in IC50 for compound 2b can be due to the lack of a hydrogen bond to the guanido group of Arg-238 and the apparent weakening of the hydrogen bond to Tyr-182, which is absent in the B molecule due to side chain rotation.
Increasing the ATP concentration in the biochemical assay resulted in a large increase of the IC50 for all triazole compounds (Table 2). This suggests that these compounds exhibit a competitive mode of binding with respect to ATP. The same increase in ATP concentration does not affect the biaryl compounds (Table 2), suggesting that these compounds exhibit a non-competitive mode of binding. This observation is also supported by the thermal shift measurements where addition of ATP resulted in a decrease in Tm in the presence of triazole compounds and an increase with the biaryl compounds (Table 2). The superimposed ATP analogue complexes of the Mt and Ec enzymes (Fig. 5c) provide a likely explanation. In all triazole complexes, an electronegative fluorine or chlorine is in close contact with the γ-phosphate oxygen of an ATP analogue. The repulsive electrostatic force between these atoms results in the competitive binding mode of these compounds. In the biaryl complexes, these interactions are absent, making it possible for the non-competitive binding mode observed for this class of compounds.
In all previously reported MtPanK structures, solved in what has been described as crystal form I or II of the trigonal space group, the adenosine lid is in a closed conformation. The MtPanK-1a complex, crystallized in crystal form II of the trigonal space group, also exhibits a closed conformation of the adenosine lid. An examination of the crystal contacts in this crystal form reveals that the adenosine lid is in close contact with helix α7 of a symmetry-related molecule (Fig. 7). In particular, the guanidinium group of residue Arg-38 in the adenosine lid forms a hydrogen bond to the carbonyl oxygen of residue Arg-140 in helix α7 of the symmetry-related molecule. These crystal packing interactions are present in all MtPanK structures solved in crystal form II but not in the two structures solved in crystal form I (MtPanK-CoA, PDB codes 2GES and 2GET). Crystal packing interactions with the adenosine lid are also absent in our structures solved in the orthorhombic space group, where the adenosine lid adopts an open conformation. In contrast, the complex structures with nucleoside cofactors were crystallized in crystal form II of the trigonal space group, where the adenosine lid assumes a closed conformation. In these structures, the adenosine or guanosine moieties occupy roughly the same binding site as the adenosine moiety of CoA instead of binding in the adenosine binding site (Fig. 5, b and c). Crystal contacts with the adenosine lid in these structures may be responsible for keeping the adenosine lid in a closed conformation, forcing the nucleoside cofactors to adopt an alternative binding mode. To date, no crystal structure of a nucleoside-bound enzyme has been solved in the orthorhombic space group or crystal form I of the trigonal space group. However, the conserved nature of the adenosine binding site and the ability of MtPanK to adopt both open and closed forms of the adenosine-lid suggest that the same mode of nucleoside binding as seen in the Ec and Cb enzymes is biologically relevant.
FIGURE 7.

Crystal contacts between the adenosine lid (magenta) of MtPanK-1a (green) in the orthorhombic space group and a symmetry-related molecule (gray). Shown are interactions between Arg-38 in the adenosine lid (yellow carbons) and Arg-140 in helix α7 of the symmetry-related molecule (gray carbons).
Even though the compounds described in this study were inactive against Mt in the whole cell assay, they are potent inhibitors of MtPanK. The structural information from this study provides us with tools for structure-based optimization of these compounds and to design novel inhibitors that are active against this target in other pathogens.
Acknowledgments
We acknowledge the work of Kishore B. K. Reddy, Subramanyam J. Tantry, Sudhir Landge, Aishwarya Sundaram, Jagannathan Alagurajan, Harini Iyer, Jyothi Bhat, and N. Radha for compound synthesis and technical support.
This work was supported by the Foundation for Strategic Research, the Swedish Research Council, and Uppsala University.
The atomic coordinates and structure factors (codes 4BFS, 4BFT, 4BFU, 4BFV, 4BFW, 4BFX, 4BFY, and 4BFZ) have been deposited in the Protein Data Bank (http://wwpdb.org/).
M. C. Franklin, J. Cheung, M. Rudolph, M. Cassidy, E. Gary, F. Burshteyn, and J. Love, unpublished work.
- PanK
- pantothenate kinase
- MtPanK
- M. tuberculosis type I PanK
- EcPanK
- E. coli PanK
- PDB
- Protein Data Bank
- r.m.s.d.
- root mean square deviation
- CbPanK
- Coxiella burnetii PanK.
REFERENCES
- 1. Jackowski S., Rock C. O. (1981) Regulation of coenzyme A biosynthesis. J. Bacteriol. 148, 926–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dunn S. D., Snell E. E. (1979) Isolation of temperature-sensitive pantothenate kinase mutants of Salmonella typhimurium and mapping of the coaA gene. J. Bacteriol. 140, 805–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vallari D. S., Jackowski S., Rock C. O. (1987) Regulation of pantothenate kinase by coenzyme A and its thioesters. J. Biol. Chem. 262, 2468–2471 [PubMed] [Google Scholar]
- 4. Song W. J., Jackowski S. (1994) Kinetics and regulation of pantothenate kinase from Escherichia coli. J. Biol. Chem. 269, 27051–27058 [PubMed] [Google Scholar]
- 5. Yun M., Park C. G., Kim J. Y., Rock C. O., Jackowski S., Park H. W. (2000) Structural basis for the feedback regulation of Escherichia coli pantothenate kinase by coenzyme A. J. Biol. Chem. 275, 28093–28099 [DOI] [PubMed] [Google Scholar]
- 6. Zhou B., Westaway S. K., Levinson B., Johnson M. A., Gitschier J., Hayflick S. J. (2001) A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat. Genet. 28, 345–349 [DOI] [PubMed] [Google Scholar]
- 7. Rock C. O., Karim M. A., Zhang Y. M., Jackowski S. (2002) The murine pantothenate kinase (Pank1) gene encodes two differentially regulated pantothenate kinase isozymes. Gene 291, 35–43 [DOI] [PubMed] [Google Scholar]
- 8. Leonardi R., Chohnan S., Zhang Y. M., Virga K. G., Lee R. E., Rock C. O., Jackowski S. (2005) A pantothenate kinase from Staphylococcus aureus refractory to feedback regulation by coenzyme A. J. Biol. Chem. 280, 3314–3322 [DOI] [PubMed] [Google Scholar]
- 9. Yang K., Eyobo Y., Brand L. A., Martynowski D., Tomchick D., Strauss E., Zhang H. (2006) Crystal structure of a type III pantothenate kinase: insight into the mechanism of an essential coenzyme A biosynthetic enzyme universally distributed in bacteria. J. Bacteriol. 188, 5532–5540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brand L. A., Strauss E. (2005) Characterization of a new pantothenate kinase isoform from Helicobacter pylori. J. Biol. Chem. 280, 20185–20188 [DOI] [PubMed] [Google Scholar]
- 11. Awasthy D., Ambady A., Bhat J., Sheikh G., Ravishankar S., Subbulakshmi V., Mukherjee K., Sambandamurthy V., Sharma U. (2010) Essentiality and functional analysis of type I and type III pantothenate kinases of Mycobacterium tuberculosis. Microbiology 156, 2691–2701 [DOI] [PubMed] [Google Scholar]
- 12. Walker J. E., Saraste M., Runswick M. J., Gay N. J. (1982) Distantly related sequences in the α- and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1, 945–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Das S., Kumar P., Bhor V., Surolia A., Vijayan M. (2006) Invariance and variability in bacterial PanK: a study based on the crystal structure of Mycobacterium tuberculosis PanK. Acta Crystallogr. D Biol. Crystallogr. 62, 628–638 [DOI] [PubMed] [Google Scholar]
- 14. Chetnani B., Das S., Kumar P., Surolia A., Vijayan M. (2009) Mycobacterium tuberculosis pantothenate kinase: possible changes in location of ligands during enzyme action. Acta Crystallogr. D Biol. Crystallogr. 65, 312–325 [DOI] [PubMed] [Google Scholar]
- 15. Chetnani B., Kumar P., Surolia A., Vijayan M. (2010) M. tuberculosis pantothenate kinase: dual substrate specificity and unusual changes in ligand locations. J. Mol. Biol. 400, 171–185 [DOI] [PubMed] [Google Scholar]
- 16. Chetnani B., Kumar P., Abhinav K. V., Chhibber M., Surolia A., Vijayan M. (2011) Location and conformation of pantothenate and its derivatives in Mycobacterium tuberculosis pantothenate kinase: insights into enzyme action. Acta Crystallogr. D Biol. Crystallogr. 67, 774–783 [DOI] [PubMed] [Google Scholar]
- 17. Venkatraman J., Bhat J., Solapure S. M., Sandesh J., Sarkar D., Aishwarya S., Mukherjee K., Datta S., Malolanarasimhan K., Bandodkar B., Das K. S. (2012) Screening, identification, and characterization of mechanistically diverse inhibitors of the Mycobacterium tuberculosis enzyme, pantothenate kinase (CoaA). J. Biomol. Screen. 17, 293–302 [DOI] [PubMed] [Google Scholar]
- 18. Franzblau S. G., Witzig R. S., McLaughlin J. C., Torres P., Madico G., Hernandez A., Degnan M. T., Cook M. B., Quenzer V. K., Ferguson R. M., Gilman R. H. (1998) Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. J. Clin. Microbiol. 36, 362–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Leslie A. G. (2006) The integration of macromolecular diffraction data. Acta Crystallogr. D Biol. Crystallogr. 62, 48–57 [DOI] [PubMed] [Google Scholar]
- 20. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 21. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jones T. A., Zou J. Y., Cowan S. W., Kjeldgaard M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 (Pt 2), 110–119 [DOI] [PubMed] [Google Scholar]
- 25. Harris M., Jones T. A. (2001) Molray–a web interface between O and the POV-Ray ray tracer. Acta Crystallogr. D Biol. Crystallogr. 57, 1201–1203 [DOI] [PubMed] [Google Scholar]
- 26. Kleywegt G. J., Jones T. A. (1997) Detecting folding motifs and similarities in protein structures. Methods Enzymol. 277, 525–545 [DOI] [PubMed] [Google Scholar]
- 27. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 28. Bond C. S., Schüttelkopf A. W. (2009) ALINE: a WYSIWYG protein-sequence alignment editor for publication-quality alignments. Acta Crystallogr. D Biol. Crystallogr. 65, 510–512 [DOI] [PubMed] [Google Scholar]
- 29. Ivey R. A., Zhang Y. M., Virga K. G., Hevener K., Lee R. E., Rock C. O., Jackowski S., Park H. W. (2004) The structure of the pantothenate kinase.ADP.pantothenate ternary complex reveals the relationship between the binding sites for substrate, allosteric regulator, and antimetabolites. J. Biol. Chem. 279, 35622–35629 [DOI] [PubMed] [Google Scholar]
- 30. Müller K., Faeh C., Diederich F. (2007) Fluorine in pharmaceuticals: looking beyond intuition. Science 317, 1881–1886 [DOI] [PubMed] [Google Scholar]
- 31. Matthews B. W. (1968) Solvent content of protein crystals. J. Mol. Biol. 33, 491–497 [DOI] [PubMed] [Google Scholar]
- 32. Kleywegt G. J., Jones T. A. (1996) Phi/psi-chology: Ramachandran revisited. Structure 4, 1395–1400 [DOI] [PubMed] [Google Scholar]
- 33. Engh R. A., Huber R. (1991) Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallogr. A 47, 392–400 [Google Scholar]
- 34. Read R. J. (1986) Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Crystallogr. D Biol. Crystallogr. 42, 140–149 [Google Scholar]