

Background: Nrf2 is up-regulated in response to reoxygenation after hypoxia.
Results: Hypoxia suppressed Nrf2 expression and induced Siah2 expression. Knockdown of Siah2 rescued hypoxic suppression of an Nrf2 mutant that mimicked phosphorylation at serine 40 or lacked this phosphorylation site.
Conclusion: Siah2 serves as a novel regulator of Nrf2.
Significance: Association of Siah2 with Nrf2 causes the degradation of Nrf2 irrespective of its phosphorylation status at serine 40.
Keywords: E3 Ubiquitin Ligase, Hypoxia, Ischemia, Nrf2, Reactive Oxygen Species (ROS), Siah2
Abstract
Under pathological conditions such as ischemia-reperfusion, Nrf2 acts as a key regulator of cellular oxidative response. Provided that Nrf2 is sensitive to hypoxia during ischemia, Nrf2 may affect reactive oxygen species metabolism during reoxygenation. In this study, hypoxia suppressed Nrf2 protein, and its hypoxic suppression was not recovered with knockdown of the Nrf2 repressor Keap1. Moreover, an Nrf2 mutant lacking the Keap1 binding domain was suppressed under hypoxia, suggesting that Keap1 does not contribute to hypoxic Nrf2 suppression. HIF-1α and Siah2 are both key regulators of hypoxic responses. Hypoxia induced the Siah2 protein. Although inhibition or knockdown of Siah2 prevented the suppression of Nrf2, knockdown of HIF-1α did not. Moreover, Siah2 interacted with Nrf2 through a binding motif, suggesting that Siah2 contributes to the suppression of Nrf2. Some cytosolic kinases also play important roles in Nrf2 regulation. In this study, PKC phosphorylates serine residues of Nrf2 during hypoxia. Knockdown of Siah2 rescued hypoxic decreases in an Nrf2 mutant that mimicked phosphorylation at serine 40 or lacked this phosphorylation site, suggesting that Siah2 contributes to the degradation of Nrf2 irrespective of its phosphorylation status. Moreover, knockdown of Siah2 attenuated ubiquitination of the Nrf2 mutant, suggesting that association of Siah2 with Nrf2 causes proteasome-mediated degradation of Nrf2.
Introduction
Ischemia-reperfusion occurs when blood flow to tissues and organs is disrupted and subsequently reintroduced. Rapid reintroduction of oxygen generates reactive oxygen species (ROS),2 leading to the oxidative damage (1). Oxidative damage is implicated in a variety of pathophysiological processes, including myocardial infarction, stroke, acute renal failure, and post-transplantation injury (2). Antioxidant proteins such as heme oxygenase play important roles in protecting cells from such damage and are induced at the transcriptional level in response to electrophiles and oxidative stress (3). The antioxidant-response element (ARE) is a cis-regulatory element critical to the expression of cytoprotective genes encoding antioxidant proteins (4). NF-E2-related factor 2 (Nrf2) is a key transcriptional activator of AREs and a central regulator of the induction of antioxidant responsive enzymes such as HO-1 and NAD(P)H-quinone oxidoreductase-1 (NQO-1) (5, 6).
Under oxidative stress, the liberation of Nrf2 from Kelch-like ECH-associated protein 1 (Keap1) is required for the activation of AREs (7). Oxidative stress and electrophilic agents modify key sulfhydryl groups of Keap1, leading to a conformational change that does not allow binding of Keap1 to Nrf2 (8). Under normal conditions, Nrf2 is constantly degraded through the ubiquitin-proteasome pathway in a Keap1-mediated manner. Two molecules of Keap1 form a cherry-bob structure to which one molecule of Nrf2 binds via two binding motifs localized in the N-terminal region of Nrf2, the Neh2 domain (9). Association of Keap1 with Neh2 causes ubiquitination of Nrf2 and subsequent degradation through the proteasome pathway (10). Thus, Keap1 maintains steady-state levels of Nrf2 and blocks Nrf2-mediated transcription.
Several studies indicate that phosphorylation of Nrf2 is an important mechanism for the activation of AREs. Stress-mediated cytosolic kinases such as protein kinase C (PKC), phosphoinositide 3-kinase (PI3K), and p38 mitogen-activated protein kinase (MAPK) have been shown to modify Nrf2 and thereby affect transcription from AREs (11–13). In particular, phosphorylation of Nrf2 at serine 40 by PKC liberates Nrf2 from Keap1 (11). It has recently been reported that Keap1- and cytosolic kinase-dependent mechanisms work in concert (14).
It is well known that ischemic hypoxia is accompanied by significant increases in cytoplasmic ROS, which are produced by cytosolic enzymes such as xanthine oxygenase and by mitochondrial complex III (15). Hence, oxygen deprivation (hypoxia) before reoxygenation is considered an important factor in the generation of ROS.
Hypoxia-inducible factor-1α (HIF-1α) is a central transcription factor that plays a major role in protecting cells against hypoxic stress (16). Under normal conditions, HIF-1α is regulated by the hydroxylation of its proline residues. Prolyl hydroxylases (PHDs) are central to this mechanism (17), resulting in the association of HIF-1α with von Hippel-Lindau protein and subsequent proteasomal degradation of HIF-1α (18). Nakayama et al. (19) have reported that although PHDs are inactive under hypoxia, they are active under mild hypoxia and that seven in absentia homolog 2 (Siah2), which is involved in the degradation of PHDs through the proteasome pathway, concomitantly affects HIF-1α accumulation under mild hypoxia.
Siah2 is a potent RING finger E3 ubiquitin ligase that limits its own availability through self-ubiquitination and is a known regulator of hypoxia-activated pathways (19, 20). Under stress and hypoxia, p38 MAPK and Akt pathways regulate stabilization and induction of Siah2 (21, 22). Siah2 binds directly to its substrates, such as the netrin membrane receptor (deleted in colorectal cancer; DCC) via AXVXP substrate motifs, and initiates their proteasomal degradation (23).
As an inducer of cytoprotective genes, Nrf2 plays an important role in ROS metabolism. Indeed, its regulation under oxidative conditions is well characterized. However, despite the involvement of ROS in cell damage during hypoxia of ischemia-reperfusion injury (24), the mechanisms of Nrf2 regulation under hypoxia remain unclear. A better understanding of Nrf2 regulation under hypoxia may lead to improvements in therapy for ischemia-reperfusion injury. Thus, in this study, we have investigated mechanisms of Nrf2 regulation under hypoxia.
EXPERIMENTAL PROCEDURES
Materials and Antibodies
Fetal bovine serum (FBS), MG132, menadione, and geneticin (G418) were purchased from Sigma; SB203580, LY294002, calphostin C, Dulbecco's modified Eagle's medium (DMEM), DMEM/Ham's F-12, and enhanced chemiluminescence (ECL) system were from Wako (Tokyo, Japan); KOD FX DNA polymerase was from TOYOBO (Tokyo, Japan); and Gene Porter 2 was from Gene Therapy Systems (San Diego). Anti-c-Myc, anti-DYKDDDDK (FLAG) tag, and anti-HA monoclonal antibodies were purchased from Wako; anti-phosphoserine antibody was from BD Biosciences; anti-human ubiquitin antibody was from Enzo Life Sciences (Farmingdale, NY); anti-human Siah2 antibody was from Santa Cruz Biotechnology (Santa Cruz, CA); anti-human β-actin antibody was from Sigma; and horseradish peroxidase-conjugated anti-rabbit and anti-mouse IgG antibodies were from Bio-Rad. The antibodies against human HIF-1α, human Keap1, and human Nrf2 were prepared as follows. The full lengths of HIF-1α, Keap1, and Nrf2 were ligated into pQE80L vectors (Qiagen, Hilden, Germany), which allow protein expression in Escherichia coli strains. HIF-1α, Keap1, and Nrf2 proteins were expressed in E. coli DH5α and then purified using nickel-nitrilotriacetic acid-agarose (Qiagen). Antibodies were raised against HIF-1α, Keap1, and Nrf2 in rabbits by a previously described method (25). The cross-reactivity of these antibodies was confirmed by each purified HIF-1α, Keap1, and Nrf2 protein.
Isolation of RNA and Quantitative Real Time-PCR (qRT-PCR)
Total RNA was extracted from Hep3B cells with Isogen (Nippon Gene, Toyama, Japan) according to the manufacturer's instructions. Total RNA was converted to cDNA by reverse transcription as follows. A reaction mixture containing 1 μg of RNA and 200 units of reverse transcriptase (Thermo Scientific, Waltham, MA) was incubated according to the manufacturer's instructions as follows: 10 min at 25 °C followed by 60 min at 42 °C and then 10 min at 70 °C to stop the reaction. Quantitative real time PCR was performed using a Thermal Cycler Dice Real Time System Single TP850 (Takara Bio Inc., Shiga, Japan). SYBR Primer Ex TagII, 10 pmol of forward and reverse primers, and 1 μg of cDNA were mixed, and qRT-PCR was then performed according to the manufacturer's instructions. The PCR was carried out at 95 °C for 10 s, followed by 40 cycles of 95 °C for 5 s and 60 °C for 20 s. Primers for human histone H4 (GenBankTM accession number NM_003548), human Nrf2 (GenBankTM accession number NM_006164), human Siah2 (GenBankTM accession number NM_005067), human HO-1 (GenBankTM accession number NM_002123), and human NQO-1 (GenBankTM accession number NM_000903) are shown in Table 1. Quantification was performed using the second derivative maximum method according to the manufacturer's instructions. Values are expressed relative to the transcription of the housekeeping gene histone H4.
TABLE 1.
Primers for quantitative real time PCR
| Factor | Forward/reverse | Sequence |
|---|---|---|
| Histone H4 | Forward | CCG CAA GGT CTT GAG AGA CAA A |
| Reverse | CTA GCC TCC GAA GCC GTA CA | |
| Nrf2 | Forward | CGG TAT GCA ACA GGA CAT TG |
| Reverse | AGG ATG CTG CTG AAG GAA TC | |
| Siah2 | Forward | ACC TGG CTA TGG AGA AGG TG |
| Reverse | CAC ACC GTC ATG AAT CGA AC | |
| HO-1 | Forward | CCA GCC ATG CAG CAC TAT GT |
| Reverse | AGC CCT ACA GCA ACT GTC GC | |
| NQO-1 | Forward | TGA TCG TAC TGG CTC ACT CA |
| Reverse | GTC AGT TGA GGT TCT AAG AC |
Isolation of Human Nrf2, Keap1, and Siah2
cDNAs of human Nrf2, human Keap1 (GenBankTM accession number NM_203500), and human Siah2 were amplified by PCR. Forty cycles of PCR (98 °C for 10 s, 57 °C for 30 s, and 68 °C for 2 min) were performed using the cDNA obtained from reverse transcription of total RNA from Hep3B cells as the template, using the KOD FX DNA polymerase and corresponding primer pairs. The primer pair shown in Table 2 include primers 1 and 2 for pQE80L Nrf2, primers 3 and 4 for pCMV-myc Nrf2, primers 5 and 6 for pQE80L Keap1, primers 7 and 8 for pCMV-HA Keap1, and primers 9 and 10 for Siah2. The cDNA of Nrf2 was digested with the restriction enzymes BamHI and KpnI or with SalI and XhoI; it was then ligated into pQE80L (Qiagen, Valencia, CA) or pCMV-Myc (Clontech), respectively. The cDNA of Keap1 was digested with BamHI and HindIII and then ligated into pQE80L or pCMV-HA (Clontech), respectively. The cDNA of Siah2 was digested with BamHI and EcoRI and then ligated into pcDNA4 TO-3×FLAG (Invitrogen). The cDNA of Myc-Nrf2 was amplified by PCR using primers 4 and 11. The plasmid pCMV-Myc containing full-length wild type (WT) Nrf2 was used as a template. The cDNA of Myc-Nrf2 was digested with BamHI and XhoI and then ligated into pcDNA3.1(+) (Invitrogen).
TABLE 2.
Primers for cloning human Nrf2, Keap1, and Siah2 and for construction of Nrf2 mutants
| Factor | Primer no. | Sequence |
|---|---|---|
| Nrf2 | 1 | AAG GAT CCA TCA TGA TGG ACT TGG AGC T |
| 2 | AAG GTA CCA TTC AAC ATA CTG ACA CTC C | |
| 3 | ATA GTC GAC CAT GAT GGA CTT GGA GC | |
| 4 | AGA CTC GAG AAC ATA CTG ACA CTC CAA TG | |
| Keap1 | 5 | ATG GAT CCC CCA TGC AGC CAG ATC CCA G |
| 6 | TTA AGC TTA ATC AAC AGG TAC AGT TCT GCT | |
| 7 | ATTA AGC TTC CCA TGC AGC CAG ATC C | |
| 8 | CTG GAT CCT CAA CAG GTA CAG TTC TG | |
| Siah2 | 9 | ATG GAT CCG CGA TGA GCC GC |
| 10 | GCG GAA TTC GAA AGT CAC ATC ATG GAC | |
| pCMV | 11 | CAG GAT CCA TGG CAT CAA TGC AGA AG |
| Nrf2(93–604) | 12 | ATA GTC GAC CCA GTC AGA AAC CAG TG |
| V172A | 13 | TCT GTT GCT CAG GCA GCC CCT GTT GAT TTA |
| 14 | TAA ATC AAC AGG GGC TGC CTG AGC AAC AGA | |
| V172A/P174A | 15 | GTT GCT CAG GCA GCC GCT GTT GAT TTA GAC |
| 16 | GTC TAA ATC AAC AGC GGC TGC CTG AGC AAC | |
| S40N | 17 | GAG AAG TAT TTG ACT TCA ATC AGC GAC GGA |
| 18 | TCC GTC GCT GAT TGA AGT CAA ATA CTT CTC | |
| S40D | 19 | GAG AAG TAT TTG ACT TCG ATC AGC GAC GGA |
| 20 | TCC GTC GCT GAT CGA AGT CAA ATA CTT CTC |
Preparation of Nrf2 Mutants and shRNAs and Transfection
The Nrf2 mutants Nrf2(93–604), V172A, V172A-P174A, S40N, and S40D were prepared. Nrf2(93–604) lacks the first 92 amino acid residues of Nrf2. In the Nrf2 mutants V172A and V172A-P174A, Val and/or Pro residues at the Siah2-binding site (Val-172 and Pro-174) were substituted with Ala residues. In the mutants S40N or S40D, the Ser residue in the PKC phosphorylation site (Ser-40) was substituted with Asn or Asp, respectively. The cDNA of Nrf2(93–604) was constructed by amplification of full-length WT Nrf2 with primers 4 and 12, digestion with restriction enzymes SalI and XhoI, and then ligation into pCMV-Myc. The cDNA of Myc-Nrf2(93–604) was then amplified by PCR using primers 4 and 11. The pCMV-Myc plasmid containing Nrf2(93–604) was used as a template. The cDNAs of V172A, V172A/P174A, S40N, and S40D were obtained after two PCR steps. In the first PCR, the nucleotide fragment 1 of V172A was amplified using primers 11 and 14, nucleotide fragment 2 of V172A/P174A with primers 11 and 16, nucleotide fragment 3 of S40N with primers 11 and 18, and nucleotide fragment 4 of S40D with primers 11 and 20. Furthermore, the nucleotide fragment 5 of V172A was amplified with primers 4 and 13, fragment 6 of V172A/P174A with primers 4 and 15, fragment 7 of S40N with primers 4 and 17, and fragment 8 of S40D with primers 4 and 19. The second round of PCR was performed using primers 4 and 11 with fragments 1 and 5 (V172A), fragments 2 and 6 (V172A/P174A), fragments 3 and 7 (S40N), and fragments 4 and 8 (S40D). The cDNAs of Myc-Nrf2(93–604), V172A, V172A/P174A, S40N, and S40D were digested with BamHI and XhoI and ligated into pcDNA3.1(+). The primers 11–20 are shown in Table 2. For the knockdown of Keap1, HIF-1α, and Siah2 using shRNA and the MOCK control with GFP shRNA, specific target regions of Keap1, HIF-1α, Siah2, and GFP were designated according to a previously described procedure (26–29). The sense, antisense, and hairpin loop sequences of these were inserted stepwise into the pBAsi-hU6 Neo Vector (Takara Bio Inc.) according to the manufacturer's instructions. The sequences of the inserted nucleotides are shown in Table 3. The DNA sequences of all constructs were confirmed by DNA sequence analysis. Plasmids were transfected into cells using Gene PORTERTM2 according to the manufacturer's instructions. Transfectants were selected using G418.
TABLE 3.
Nucleotides for Keap1, HIF-1α, Siah2, and GFP shRNAs
| Factor | Sequence | Ref. |
|---|---|---|
| Keap1 | GCA GGC CTT TGG CAT CAT GAA CGT GTG CTG TCC GTT CAT GAT GCC AAA GGC CTG CTT TTT T | 26 |
| HIF-1α | GCA ACT GTC ATA TAT AAC ACC CCC CTG TGA AGC CAC AGA TGG GGG GGG TGT TAT ATA TGA CAG TTG CTT TTT T | 27 |
| Siah2 | ACC CGG AGT GCT TAT CTT AAA CTG TGA AGC CAC AGA TGG GTT TAA GAT AAG CAC TCC GGG TTT TTT T | 28 |
| GFP | GTA CAG GAA CGC ACT ATA TCT CTG TGA AGC CAC AGA TGG GAG ATA TAG TGC GTT CCT GTA CTT TTT T | 29 |
Cell Cultures and Hypoxic Stimulation
Human hepatic carcinoma Hep3B cells were obtained from the Cell Resource Center for Biomedical Research at the Institute of Development, Aging, and Cancer of Tohoku University (Miyagi, Japan). Human cervix carcinoma HeLa cells and human embryonic kidney HEK293 cells were generous gifts from Prof. Yamasaki and Prof. Hirai of Kwansei Gakuin University, respectively. Hep3B and HeLa cells were grown in DMEM supplemented with 10% FBS, penicillin (100 units/ml), and streptomycin (100 μg/ml). HEK293 cells were grown in DMEM/Ham's F-12 supplemented with 10% FBS, penicillin (100 units/ml), and streptomycin (100 μg/ml). These cells were incubated at 37 °C in 95% air and 5% CO2. To simulate hypoxic conditions, the cells were cultured in a multi-gas incubator, APM-30D (Astec, Shizuoka, Japan), set to 1% O2 and 5% CO2.
Immunoprecipitation and Western Blotting
Immunoprecipitation experiments and Western blotting analysis were performed as described previously (30). In the immunoprecipitation experiments, the following were used: lysis buffer containing 50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1% Triton X-100, and 0.1 μg/ml PMSF; wash buffer containing 50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 0.1% Triton X-100; protein A- and protein G-Sepharose (GE Healthcare); and negative control mouse or rabbit IgG, anti-c-Myc, anti-FLAG, anti-Siah2, anti-Keap1, or anti-Nrf2 antibodies. For Western blotting analysis, anti-β-actin, anti-Nrf2, anti-Keap1, anti-Siah2, anti-HIF-1α, anti-ubiquitin, anti-phosphoserine, anti-c-Myc, anti-HA, or anti-FLAG antibodies were used. In this study, β-actin was used as a loading control.
RESULTS
Suppression of Nrf2 Protein under Hypoxia
Hep3B, HEK293, and HeLa cells were subjected to hypoxia for 3, 6, or 9 h, and changes in Nrf2 protein levels were examined (Fig. 1A). Nrf2 protein was suppressed by hypoxia in the Hep3B, HEK293, and HeLa cells. Similarly, changes in Nrf2 mRNA expression were examined (Fig. 1B) and were found to be unchanged under hypoxia. These results suggest that hypoxia affects the stabilization of Nrf2 protein.
FIGURE 1.
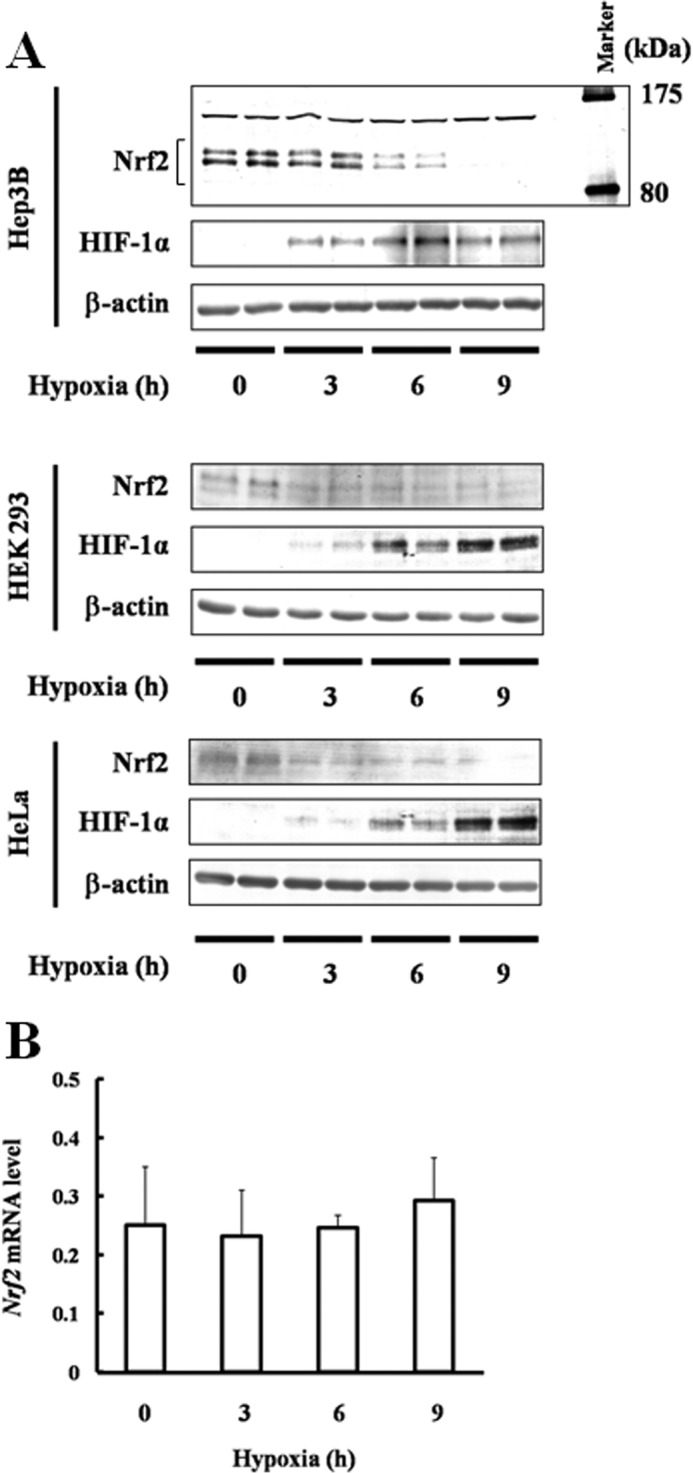
Effect of hypoxia on Nrf2 accumulation and transcription. A, Hep3B, HEK293, and HeLa cells were exposed to hypoxia for 3, 6, or 9 h. Twenty, 40, and 5 μg of Hep3B lysates prepared were immunoblotted with anti-Nrf2, anti-HIF-1α, or anti-β-actin antibodies (upper panel). Forty, 50, and 10 μg of lysates prepared from HEK293 and HeLa cells were immunoblotted with anti-Nrf2, anti-HIF-1α, or anti-β-actin antibodies (middle and lower panels). B, Hep3B cells were exposed to hypoxia for 3, 6, or 9 h. Total RNA was isolated, and Nrf2 mRNA was determined using qRT-PCR. Values are expressed as the mean ± S.D. of three replicates.
Contributions of HIF-1α or Keap1 to Suppression of Nrf2
HIF-1α is known to be a central factor in the regulation of hypoxic responses (16). We knocked down HIF-1α using shRNA in Hep3B cells and then measured changes in the levels of Nrf2 protein (Fig. 2A). The presence of Nrf2 protein was reduced under hypoxia and was not recovered by knockdown of HIF-1α, suggesting that HIF-1α does not contribute to the suppression of Nrf2 under hypoxia.
FIGURE 2.

Effect of HIF-1α knockdown or Keap1 overexpression or knockdown on Nrf2 accumulation. A, WT Hep3B, MOCK (GFP shRNA control), and HIF-1α knockdown Hep3B cells were grown under normoxia or hypoxia for 6 h in the presence or absence of MG132 (5 μm). Two independent HIF-1α knockdown clones were used. Total cell lysates were immunoblotted with anti-Nrf2, anti-HIF-1α, or anti-β-actin antibodies. B, Hep3B cells were transfected with empty pcDNA4 vector (MOCK) or pcDNA3.1 containing HA-Keap1. Forty eight hours after transfection, the transfected and WT Hep3B cells were grown under normoxia or hypoxia for 6 h. Two independent clones were used. Total lysates were immunoblotted using anti-Nrf2, anti-HA, anti-Keap1, anti-HIF-1α, or anti-β-actin antibodies. C, WT Hep3B, MOCK (GFP shRNA control), and Keap1 knockdown Hep3B, HEK293, and HeLa cells were grown under normoxia or hypoxia for 6 h in the presence or absence of MG132 (5 μm). Two independent Keap1 knockdown clones were used in each cell line. Total cell lysates were immunoblotted with anti-Nrf2, anti-Keap1, anti-HIF-1α, or anti-β-actin antibodies.
As Nrf2 is a well known substrate for Keap1 (9), its suppression under hypoxia may be related to the induction and/or activation of Keap1 following hypoxia. However, in Western blotting experiments, Keap1 protein levels were not changed by hypoxia (Fig. 2, B and C), suggesting that hypoxia does not induce Keap1 expression. In Hep3B cells overexpressing Keap1, Nrf2 protein levels were reduced under hypoxia (Fig. 2B), suggesting that Keap1 is active even under hypoxia. To investigate the effects of active Keap1 on the stabilization of Nrf2, Keap1 was knocked down by shRNA in Hep3B, HEK293, and HeLa cells (Fig. 2C). As a result, Nrf2 protein expression increased under normoxia; however, its suppression under hypoxia was not completely recovered by knockdown of Keap1 in any cell line. Nrf2 binds to its repressor Keap1 via the Neh2 domain (9), which is absent in the Myc-Nrf2(93–604) construct. Interestingly, under hypoxia, Myc-Nrf2(93–604) as well as WT Nrf2 were suppressed (Fig. 3). These results suggest that Keap1 does not contribute to the suppression of Nrf2 under hypoxia.
FIGURE 3.
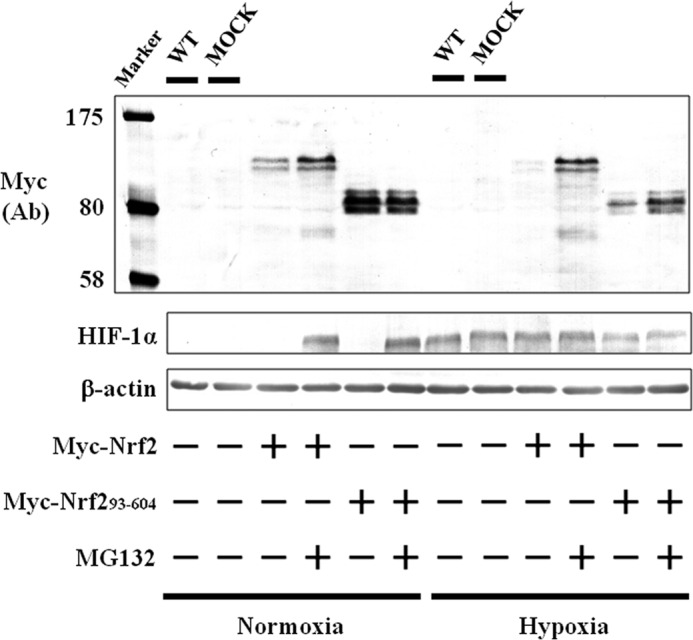
Effect of hypoxia and/or MG132 on accumulation of Nrf2(93–604). Hep3B cells were transfected with the empty pcDNA3.1 vector (MOCK), pcDNA3.1 containing Myc-Nrf2, or Myc-Nrf2(93–604). Forty eight hours after transfection, the transfected and WT Hep3B cells were grown under normoxia or hypoxia for 6 h in the presence or absence of MG132 (5 μm). Total cell lysates were immunoblotted with anti-c-Myc, anti-HIF-1α, or anti-β-actin antibodies. Ab, antibody.
Using the proteasome inhibitor MG132, we also showed that suppression of Nrf2 (Fig. 2, A and C) and Myc-Nrf2(93–604) (Fig. 3) is dependent on the proteasome under hypoxia as well as normoxia. Hence, we predicted that some unidentified hypoxia-activated E3 ubiquitin ligase may be involved in the degradation of Nrf2.
Contribution of Siah2 to Nrf2 Degradation
Siah2 is known to be an important regulator of hypoxia-activated pathways and is also involved in the degradation of substrates such as PHD3 through the E3 ubiquitin ligase and proteasome pathways (19). In this study, hypoxia induced Siah2 expression (Fig. 4). Thus, we investigated the effect of menadione, an inhibitor of Siah2 ubiquitin ligase activity (31), on the stabilization of Nrf2. As menadione inhibits both Siah2 and Keap1 activities (26), specific inhibition of Siah2 by menadione in Hep3B cells was achieved either by suppression of Keap1 (Fig. 5A) or expression of Myc-Nrf2(93–604) (Fig. 5B). In the presence of menadione, expression of Nrf2 and Myc-Nrf2(93–604) proteins were recovered, indicating that Siah2 serves as a novel regulator of Nrf2.
FIGURE 4.
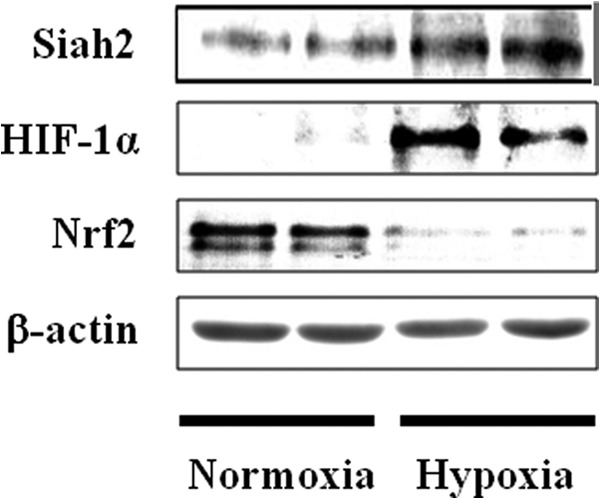
Effect of hypoxia on Siah2 accumulation. Hep3B cells were exposed to hypoxia for 6 h. Sixty, 40, 20, and 5 μg of lysates were immunoblotted with anti-Siah2, anti-HIF-1α, anti-Nrf2, or anti-β-actin antibodies.
FIGURE 5.
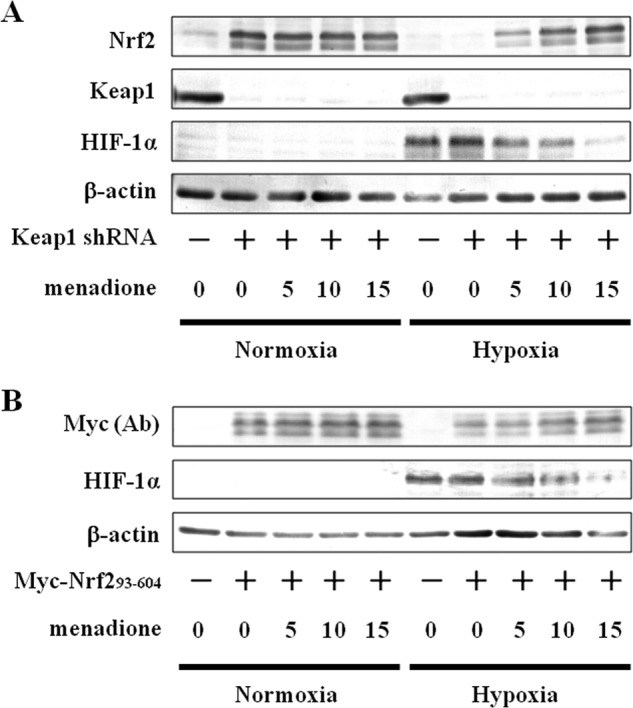
Effect of menadione on Nrf2 accumulation in Keap1 knockdown cells and on accumulation of Nrf2(93–604). A, MOCK (GFP shRNA control) or Keap1 knockdown Hep3B cells were grown under normoxia or hypoxia for 6 h in the presence of DMSO (vehicle control) or menadione (5, 10, or 15 μm). Total cell lysates were immunoblotted with anti-Nrf2, anti-Keap1, anti-HIF-1α, or anti-β-actin antibodies. B, Hep3B cells were transfected with empty pcDNA3.1 vector (MOCK) or pcDNA3.1 containing Myc-Nrf2(93–604). Forty eight hours after transfection, the cells were grown under normoxia or hypoxia for 6 h in the presence of DMSO (vehicle control) or menadione (5, 10, or 15 μm). Total cell lysates were immunoblotted using anti-c-Myc, anti-HIF-1α, or anti-β-actin antibodies. Ab, antibody.
In subsequent experiments, knockdown of Siah2 significantly stabilized endogenous Nrf2 in the Hep3B, HEK293, and HeLa cells (Fig. 6A) and induced the Nrf2-responsive genes HO-1 and NQO-1 in Hep3B cells (Fig. 6B). In contrast, when Siah2 was overexpressed in Hep3B cells (Fig. 7), Nrf2 protein levels were significantly decreased. These results provide further evidence that Siah2 contributes to the suppression of Nrf2 and suggest that Siah2 blocks Nrf2-mediated transcription.
FIGURE 6.

Effect of Siah2 knockdown on Nrf2 accumulation and transcription of Nrf2-responsive genes. A, WT Hep3B, MOCK (GFP shRNA control), or Siah2 knockdown Hep3B, HEK293 and HeLa cells were grown under normoxia or hypoxia for 6 h. Two independent Siah2 knockdown clones were used in each cell line. Total cell lysates were immunoblotted using anti-Nrf2, anti-HIF-1α, or anti-β-actin antibodies. Four dots with cross, effect of transfection with the Siah2 pBAsi plasmid are indicated by HIF-1α blots. B, MOCK (GFP shRNA control) or Siah2 knockdown Hep3B cells were exposed to hypoxia for 6 h. Total RNA was isolated and Siah2, HO-1, or NQO-1 mRNAs were determined using qRT-PCR. Values are expressed as the mean ± S.D. of three replicates. **, p < 0.01; *, p < 0.05 significantly different from MOCK.
FIGURE 7.
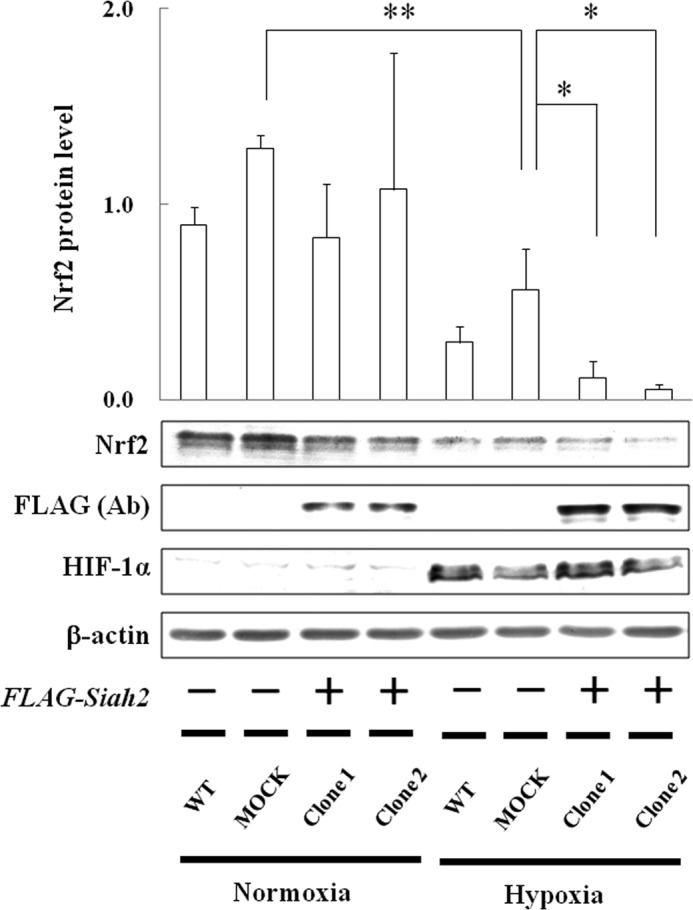
Effect of Siah2 overexpression on Nrf2 accumulation. Hep3B cells were transfected with empty pcDNA4 vector (MOCK) or pcDNA4 containing FLAG-Siah2. Forty eight hours after transfection, the transfected and WT Hep3B cells were grown under normoxia or hypoxia for 6 h. Two independent clones were used. Total cell lysates were immunoblotted using anti-Nrf2, anti-FLAG, anti-HIF-1α, or anti-β-actin antibodies. Values are expressed as the mean ± S.D. of three replicates. **, p < 0.01 significantly different from MOCK under normoxia, *, p < 0.05 significantly different from MOCK under hypoxia. Ab, antibody.
Siah2 directly binds to substrates carrying the AXVXP-binding motif (23). Interestingly, we found a putative Siah2-binding motif, AQVAP, at positions 170–174 in Nrf2. To identify the importance of this Siah2-binding motif, Nrf2 mutants V172A and V172A/P174A, which contain single or double mutations in this putative Siah2-binding motif, respectively, were expressed in the Hep3B cells (Fig. 8). Neither V172A nor V172A/P174A was degraded under hypoxia, suggesting that this binding motif is essential for the degradation of Nrf2 by Siah2 and confirming that Siah2 directly regulates Nrf2.
FIGURE 8.
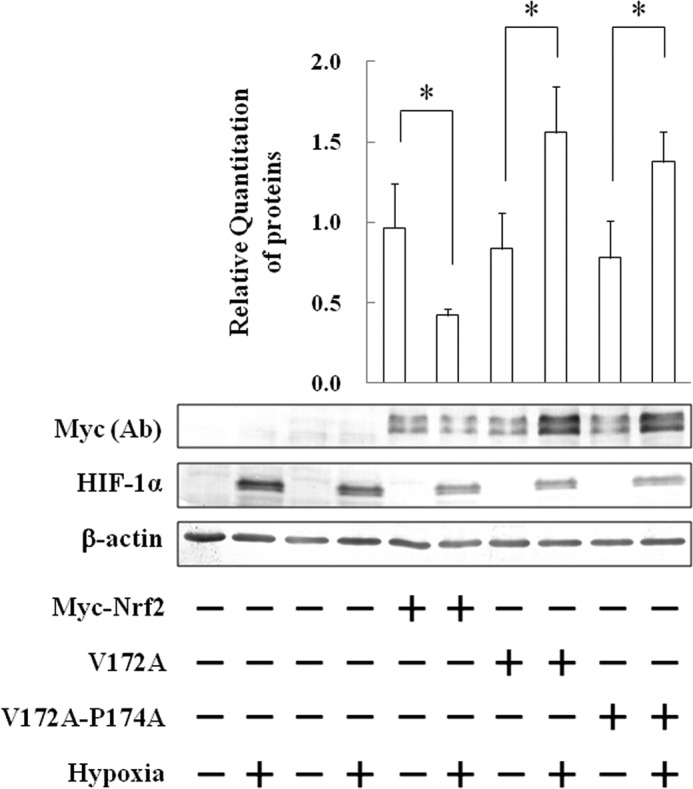
Determination of Siah2-binding site in Nrf2 amino acid residues. Hep3B cells were transfected with empty pcDNA3.1 vector (MOCK) or pcDNA3.1 containing Myc-Nrf2, V172A, or V172A/P174A. Forty eight hours after transfection, the cells were grown under normoxia or hypoxia for 6 h. Total cell lysates were immunoblotted using anti-c-Myc, anti-HIF-1α, or anti-β-actin antibodies. Values are expressed as the mean ± S.D. of three replicates. *, p < 0.05 significantly different from normoxia. Ab, antibody.
Direct Interaction of Siah2 with Nrf2
Next, we investigated the direct binding interactions of Siah2 and Nrf2 using immunoprecipitation and Myc-Nrf2-, V172A-, or V172A-P174A-expressing Hep3B cells (Fig. 9A). In these experiments, both V172A and Myc-Nrf2 interacted with Siah2, whereas V172A/P174A did not. A subsequent reciprocal immunoprecipitation assay also confirmed that Siah2 binds to Nrf2 through the abovementioned binding motif. Moreover, in both immunoprecipitation and reciprocal immunoprecipitation assays, endogenous Siah2 interacted with Nrf2 in WT cells (Fig. 9B). These data offer further evidence of the Siah2-Nrf2 interaction.
FIGURE 9.
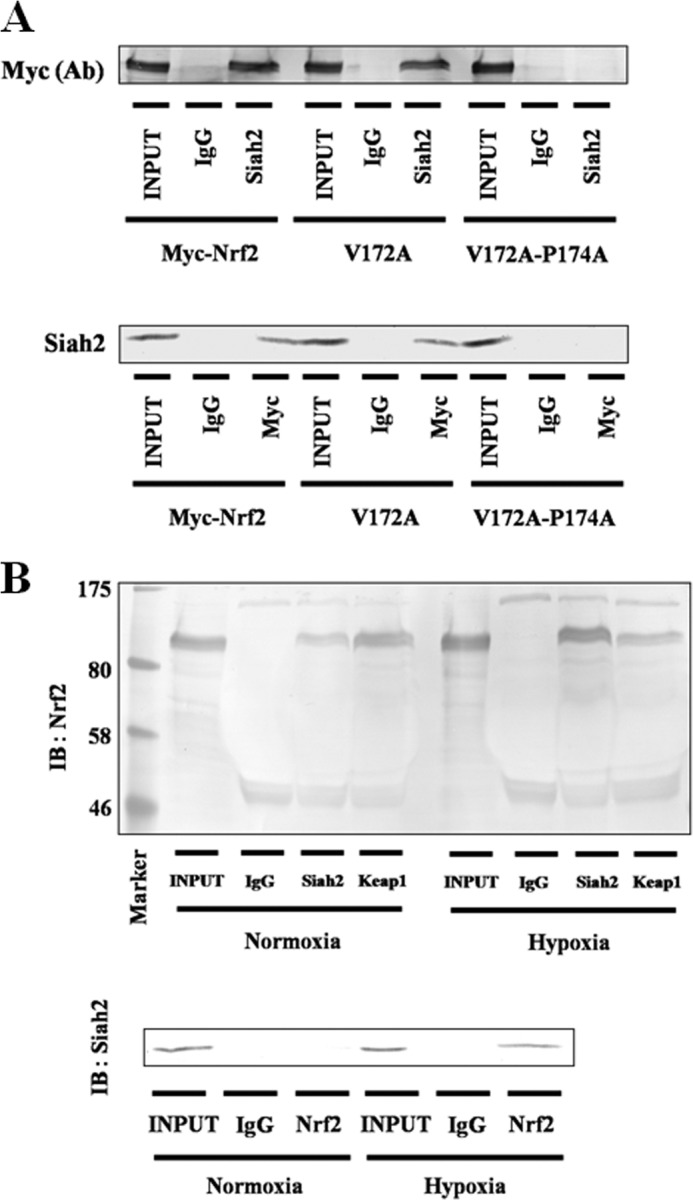
Association of Siah2 with Nrf2. A, Hep3B cells were transfected with pcDNA3.1 containing Myc-Nrf2, V172A, or V172A/P174A. Forty eight hours after transfection, the cells were exposed to hypoxia for 6 h in the presence of MG132 (5 μm). Total cell lysates were immunoprecipitated with anti-IgG or anti-Siah2 antibodies and then immunoblotted (IB) using anti-c-Myc antibody (upper panel). Lysates were immunoprecipitated with anti-IgG or anti-c-Myc antibodies and then immunoblotted using anti-Siah2 antibody (lower panel). Ab, antibody. B, Hep3B cells were grown under normoxia or hypoxia in the presence of MG132 (5 μm). Total cell lysates were immunoprecipitated with anti-IgG, anti-Siah2, or anti-Keap1 antibodies and then immunoblotted using anti-Nrf2 antibody (upper panel). Lysates were immunoprecipitated with anti-IgG or anti-Nrf2 antibodies and then immunoblotted using anti-Siah2 antibody (lower panel).
Contribution of the Phosphorylation of Nrf2 by PKC to Nrf2 Suppression by Siah2
Hypoxia is involved in the activation of several cytosolic kinases (32), and the phosphorylation of Nrf2 at serine residues by stress-related cytosolic kinases is an important stabilizer of Nrf2 (11–13). Thus, we investigated if serine residues of Nrf2 are phosphorylated under hypoxia (Fig. 10A). As p38 MAPK, PI3K, and PKC are involved in phosphorylation of Nrf2, p38 MAPK inhibitor (SB203580), PI3K inhibitor (LY294002), and PKC inhibitor (calphostin C) were used. Although treatment with calphostin C thoroughly inhibited phosphorylation at serine residues of Nrf2 under hypoxia, neither SB203580 nor LY294002 had any effect under hypoxia. In addition, hypoxia enhanced the phosphorylation of serine residues of Nrf2 by PKC (Fig. 10B).
FIGURE 10.
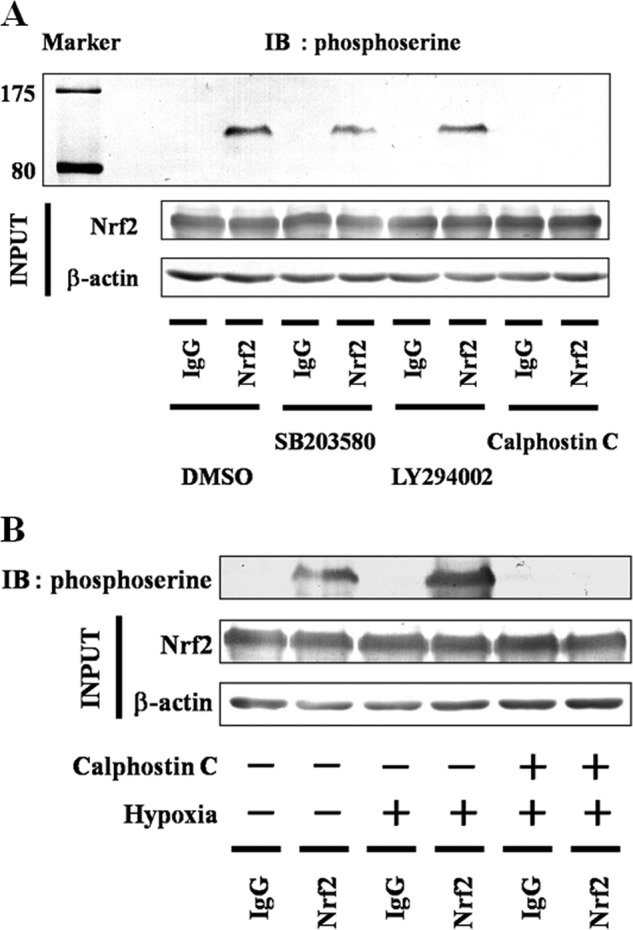
Effect of hypoxia on Nrf2 phosphorylation. A, Hep3B cells were exposed to hypoxia for 6 h in the presence of MG132 (5 μm), SB203580 (5 μm) plus MG132 (5 μm), LY294002 (50 μm) plus MG132 (5 μm), or calphostin C (0.2 μm) plus MG132 (5 μm). Total cells lysates were immunoblotted (IB) using anti-Nrf2 or anti-β-actin antibodies. Lysates were immunoprecipitated using anti-IgG or anti-Nrf2 antibodies and then immunoblotted using anti-phosphoserine antibody. B, Hep3B cells were grown under normoxia or hypoxia for 6 h in the presence of MG132 (5 μm) with and without calphostin C (0.2 μm). Total cell lysates were immunoblotted using anti-Nrf2 or anti-β-actin antibodies. Lysates were immunoprecipitated using anti-IgG or anti-Nrf2 antibodies and then immunoblotted using anti-phosphoserine antibody.
PKC phosphorylates the serine residue of Nrf2 at position 40, leading to the liberation of Nrf2 from Keap1 (11). To investigate the relationship between phosphorylation of Nrf2 at serine 40 and the stabilization of Nrf2 under hypoxia, Asn and Asp mutants, Myc-S40N and Myc-S40D, were prepared to mimic the nonphosphorylated and phosphorylated states, respectively (Fig. 11, A and B). In these experiments, knockdown of Keap1 stabilized Myc-S40N protein under normoxia, although it did not aid the recovery of either Myc-S40N or Myc-S40D proteins under hypoxia (Fig. 11A), suggesting that nonphosphorylated Nrf2 at serine 40 is a target of Keap1 under normoxia; however, it is not a target of only Keap1 under hypoxia. Similarly, although knockdown of Siah2 had little effect under normoxia, it rescued the hypoxic decrease in Myc-S40D protein (Fig. 11B), suggesting that phosphorylated Nrf2 at serine 40 is a target of Siah2 under hypoxia. To further examine the effect of PKC-mediated phosphorylation of Nrf2 on the degradation of Nrf2 by Keap1, Keap1 knockdown of Hep3B cells were grown in the presence of calphostin C (Fig. 11C). Under hypoxia, Nrf2 protein, which was not subject to the phosphorylation by PKC, was not completely recovered by calphostin C in these cells, further suggesting that nonphosphorylated Nrf2 is degraded by additional factors to Keap1 under hypoxia. Similarly, to evaluate if PKC-mediated phosphorylation of Nrf2 contributes to the degradation of Nrf2 by Siah2, Siah2 knockdown Hep3B cells were grown in the presence of calphostin C (Fig. 11D). In these cells, Nrf2 protein levels were decreased, suggesting that after phosphorylation by PKC, Nrf2 is subject to Siah2-mediated degradation.
FIGURE 11.

Targeted degradation of Nrf2 after phosphorylation by PKC. A, MOCK (GFP shRNA control) or Keap1 knockdown Hep3B cells were transfected with empty pcDNA3.1 vector or pcDNA3.1 containing Myc-S40N or Myc-S40D. Forty eight hours after transfection, the cells were grown under normoxia (Nor) or hypoxia (Hyp) for 6 h. Total cell lysates were immunoblotted using anti-c-Myc, anti-Keap1, anti-HIF-1α, or anti-β-actin antibodies. Values are expressed as the mean ± S.D. of three replicates. *, p < 0.05 significantly different from Myc-S40N alone transfected cells; #, p < 0.05 significantly different from Myc-S40D alone transfected cells. B, MOCK (GFP shRNA control) or Siah2 knockdown Hep3B cells were transfected with empty pcDNA3.1 vector or pcDNA3.1 containing Myc-S40N or Myc-S40D. Forty eight hours after transfection, the cells were grown under normoxia or hypoxia for 6 h. Total cell lysates were immunoblotted using anti-c-Myc, anti-HIF-1α, or anti-β-actin antibodies. Values are expressed as the mean ± S.D. of three replicates; *, p < 0.05 significantly different from Myc-S40D alone transfected cells under normoxia; #, p < 0.05 significantly different from Myc-S40D alone transfected cells under hypoxia. C, MOCK (GFP shRNA control) or Keap1 knockdown Hep3B cells were grown under normoxia or hypoxia for 6 h in the presence of DMSO (vehicle control) or calphostin C (0.1 or 0.2 μm). Total cell lysates were immunoblotted with anti-Nrf2, anti-Keap1, anti-HIF-1α or anti-β-actin antibodies. *, p < 0.05; **, p < 0.01 significantly different from untreated MOCK under normoxia; #, p < 0.05 significantly different from untreated Keap1 knockdown cells under hypoxia. D, MOCK (GFP shRNA control) or Siah2 knockdown Hep3B cells were grown under normoxia or hypoxia for 6 h in the presence of DMSO (vehicle control) or calphostin C (0.1 or 0.2 μm). Total cell lysates were immunoblotted with anti-Nrf2, anti-HIF-1α, or anti-β-actin antibodies. **, p < 0.01 significantly different from untreated MOCK under normoxia; ##, p < 0.01 significantly different from untreated MOCK under hypoxia; *, p < 0.05 significantly different from untreated Siah2 knockdown cells under hypoxia.
Contribution of Siah2 to the Degradation and Ubiquitination of Nonphosphorylated Nrf2
Myc-Nrf2(93–604) lacks the Neh2 domain, which includes the phosphorylation site at serine 40; however, the treatment with either MG132 or menadione led to recovery of its expression (Figs. 3 and 5B), as described above. Moreover, our results proposed that nonphosphorylated Nrf2 is degraded by additional factors to Keap1 under hypoxia (Fig. 11, A and C). Hence, we predicted that Siah2 may be involved in the degradation of not only phosphorylated Nrf2 but also nonphosphorylated Nrf2. To identify the importance of the phosphorylation site in Neh2 domain under hypoxia, Myc-Nrf2 or Myc-Nrf2(93–604) was expressed in Siah2 knockdown Hep3B cells (Fig. 12A). Hypoxic decreases in Myc-Nrf2(93–604) and Myc-Nrf2 were prevented in the Siah2 knockdown cells. Moreover, in both immunoprecipitation and reciprocal immunoprecipitation assays, Myc-Nrf2(93–604) as well as Myc-Nrf2 interacted with Siah2 (Fig. 12B), suggesting that the phosphorylation site is not required for the degradation of Nrf2 by Siah2 or the interaction between Siah2 and Nrf2. Importantly, although Nrf2 phosphorylated by PKC is subject to Siah2-mediated degradation, Siah2 as well as Keap1 contributes to the degradation of nonphosphorylated Nrf2.
FIGURE 12.

Effect of Siah2 knockdown on accumulation and ubiquitination of Nrf2(93–604) and association of Siah2 with Nrf2(93–604). A, empty pcDNA3.1 vector or pcDNA3.1 containing Myc-Nrf2, or Myc-Nrf2(93–604) was transfected into MOCK (GFP shRNA control) or Siah2 knockdown Hep3B cells, respectively. Forty eight hours after transfection, the cells were grown under normoxia or hypoxia for 6 h. Total cell lysates were immunoblotted using anti-c-Myc, anti-HIF-1α, or anti-β-actin antibodies. Ab, antibody. B, Hep3B cells were transfected with pcDNA3.1 containing Myc-Nrf2 or Myc-Nrf2(93–604). Forty eight hours after transfection, the cells were grown under normoxia or hypoxia for 6 h in the presence of MG132 (5 μm). Total cell lysates were immunoprecipitated with anti-IgG or anti-Siah2 antibodies and then immunoblotted using anti-c-Myc antibody (upper panel). Lysates were immunoprecipitated with anti-IgG or anti-c-Myc antibodies and then immunoblotted using anti-Siah2 antibody (lower panel). C, MOCK (GFP shRNA control) or Siah2 knockdown Hep3B cells were transfected with pcDNA3.1 containing Myc-Nrf2(93–604). Forty eight hours after transfection, the cells were grown under normoxia or hypoxia for 6 h in the presence or absence of MG132 (5 μm). Total cell lysates were immunoblotted using anti-c-Myc, anti-HIF-1α, or anti-β-actin antibodies. Lysates were immunoprecipitated using anti-c-Myc antibodies and then immunoblotted using anti-ubiquitin antibody. Four dots with cross, effect of transfection with the Siah2 pBAsi plasmid is indicated by HIF-1α blots.
In a Keap1-mediated manner, Neh2 is ubiquitinated, and then Nrf2 is degraded through the proteasome pathway (10). We analyzed the ubiquitination and degradation of Myc-Nrf2(93–604) in the presence and absence of MG132 (Fig. 12C). Most of Myc-Nrf2(93–604) was ubiquitinated, and knockdown of Siah2 attenuated the ubiquitination of Myc-Nrf2(93–604), suggesting that association of Siah2 with Nrf2 caused ubiquitination of Nrf2 and subsequent degradation through the proteasome pathway.
Significance of the Phosphorylation of Nrf2
To investigate interactions of phosphorylated Nrf2 with Keap1 and Siah2, Hep3B cells were grown in the presence of calphostin C, and immunoprecipitation was performed (Fig. 13, A and B). The interaction between Siah2 and Nrf2 was attenuated by treatment of calphostin C, whereas the interaction between Keap1 and Nrf2 was enhanced (Fig. 13A). In addition, the interaction between Siah2 and phosphorylated Nrf2 was attenuated by treatments with calphostin C (Fig. 13B). These results suggest that PKC-mediated phosphorylation of Nrf2 was required for the escape of Nrf2 from Keap1, resulting in the association of Nrf2 with Siah2.
FIGURE 13.
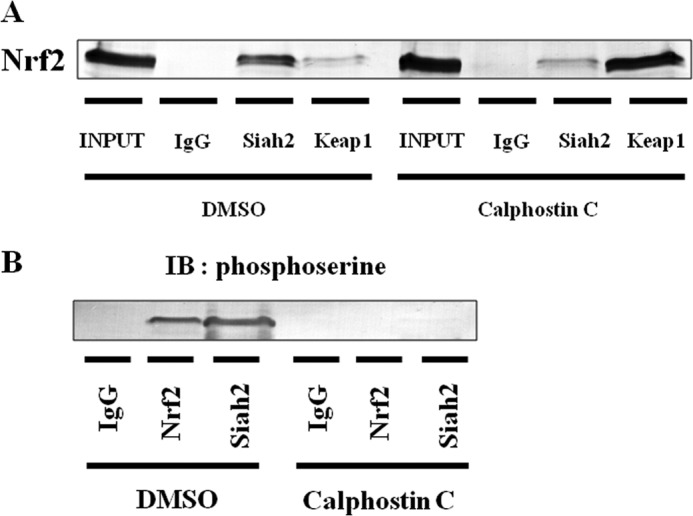
Significance of Nrf2 phosphorylation by PKC. A, total cell lysates were immunoprecipitated using anti-IgG, anti-Siah2, or anti-Keap1 antibodies and then immunoblotted using anti-Nrf2 antibody. B, lysates were immunoprecipitated using anti-IgG, anti-Nrf2, or anti-Siah2 antibodies and then immunoblotted with anti-phosphoserine antibody. Four dots with cross, effect of transfection with the Siah2 pBAsi plasmid is indicated by HIF-1α blots. Ab, antibody.
DISCUSSION
Ischemia reperfusion generates ROS and leads to cellular damage and oxidative stress in hypoxia and subsequent reoxygenation (1). Although this mechanism is accepted for reoxygenation, the association of cellular damage with hypoxia-ischemia is not as well known. Three major sources of ROS, including mitochondria, xanthine oxygenase, and Ca2+ flux, are activated during ischemic hypoxia, representing an important mechanism of cytotoxicity (15). ROS causes lipid peroxidation as well as protein and DNA oxidation (1), and it also stimulates ischemic cells to secrete inflammatory cytokines and chemokines that induce cell damage (33). Cellular oxidative stress defense systems are tightly regulated through synthesis and degradation of many transcription factors such as Nrf2 and HIF-1α. Although the generation of ROS under hypoxia causes cellular injury, the regulation of Nrf2 in hypoxia has been investigated.
Hypoxia suppresses the stabilization of Nrf2 and enhances that of HIF-1α. HIF-1α plays a central role in the regulation of hypoxic responses (16). Our previous study showed that a partner of Nrf2, MafG, binds to HIF-1α and leads to transcription from hypoxia-responsive elements (HREs) (34). Indeed, the induction of HIF-1α might prevent interactions of MafG with Nrf2, leading to the hypoxic suppression of Nrf2. However, knockdown of HIF-1α did not recover hypoxic suppression of Nrf2, suggesting that HIF-1α does not suppress Nrf2.
As a transcription factor, Nrf2 binds to AREs and induces a variety of genes that encode cytoprotective enzymes such as HO-1 and NQO-1 (5, 6). These defense enzymes play critical roles in protecting cells from cellular degeneration. Under stressful conditions, Nrf2 is up-regulated and activates AREs. In these conditions, a conformational change in Keap1 and/or phosphorylation of Nrf2 by stress-related cytosolic kinases prevents binding of Keap1 to Nrf2, wherein Nrf2 is available for ARE binding and induces cytoprotective genes. Similarly, under hypoxia, HREs are activated, and cytoprotective genes are induced (35). In this study, hypoxia significantly suppressed accumulation of Nrf2. Conversely, overexpression of Nrf2 suppressed the activation of HREs by hypoxia, suggesting that suppression of Nrf2 is required for full activation of HREs.
In this study, we have defined a novel Nrf2 regulatory pathway. Our results show that hypoxia-activated E3 ubiquitin ligase Siah2 regulates both nonphosphorylated Nrf2 and Nrf2 phosphorylated by stress-related kinase PKC, which is activated by hypoxia. The biological significance of our findings is best illustrated by the abundance of Nrf2 in hypoxia in Siah2 knockdown cells compared with that in Keap1 knockdown cells.
Importantly, although Myc-Nrf2(93–604) lacks the phosphorylation site at serine 40, it was recovered by inhibition or knockdown of Siah2. Moreover, under normoxia, phosphorylated Nrf2 was barely observed, and Nrf2 accumulation was decreased by overexpressing of Siah2 even under normoxia. Furthermore, Myc-S40N was still degraded in Keap1 knockdown cells, confirming that Siah2 binds and degrades both phosphorylated and nonphosphorylated Nrf2.
In a Keap1-mediated manner, the Neh2 domain is required for the ubiquitination of Nrf2, because the Neh2 domain includes the Keap1-binding site and lysines for ubiquitin attachment; however, our results showed that the Neh2 domain is not required for the ubiquitination of Nrf2, suggesting that in the degradation of Nrf2, the Siah2-mediated manner is different from that of Keap1. It means that Neh domains except the Neh2 domain include a novel site for ubiquitination. Indeed, the domains include the Siah2-binding site.
Previous investigations have established that Nrf2 is degraded in response to stress by the Keap1-Cullin3 complexes (10) and that phosphorylation of Nrf2 leads to dissociation from Keap1 (11). Under hypoxia and other stress conditions, p38 MAPK and Akt pathways regulate stabilization and induction of Siah2 (21, 22). In agreement, the present data suggest that although the Keap1 complex limits the expression of Nrf2, Siah2 limits the expression of Nrf2 before recruitment and/or after liberation from Keap1.
Given that the activity of Siah2 is inhibited in proportion to increasing oxygen concentrations (36), we investigated its significance to the degradation of Nrf2 under hypoxia. Under mild hypoxia, Siah2-mediated degradation of Nrf2 is attenuated, leading to an abundance of Nrf2. Under these conditions, ROS are likely to be generated, and activation of Nrf2 is required for protection from oxidative stress. In contrast, severe hypoxia is not associated with ROS because too few oxygen molecules are available to generate hydrogen peroxide or superoxide. These conditions lead to the maintenance of active Keap1, although PKC continues to phosphorylate Nrf2 and cause dissociation from Keap1. We propose that Siah2-mediated degradation of phosphorylated Nrf2 is of physiological importance as a mechanism that prevents excess activation of AREs by Nrf2 and blocks Nrf2-mediated transcription of target genes.
In this study, experiments were predominantly performed in Hep3B, HEK293, and HeLa cells. Importantly, the interaction of Siah2 with Nrf2 was observed in all these cell lines, suggesting that Siah2 contributes to the accumulation of Nrf2 ubiquitously. However, the effects of Keap1 knockdown on the accumulation of Nrf2 under hypoxia differed between cells types. Indeed, knockdown of Keap1 facilitated Nrf2 recovery in the Hep3B and HEK293 cells but only partially in the HeLa cells. Hence, we predicted that the sensitivity, accumulation, and activities of cytosolic kinase-related pathways, especially PKC, may also differ between these cell types.
Given that Siah2 regulates the abundance of HIF-1α by degrading PHD3 (19), it may be involved in the regulation of hypoxia-induced ROS. Our results demonstrate that inhibition or knockdown of Siah2 causes suppression of HIF-1α and increases Nrf2. Hence, we proposed that the inhibition of Siah2 may be followed by inactivation of HIF-1α-mediated pathways and activation of Nrf2-mediated pathways, leading to attenuated ROS production. Therefore, Siah2 may be a novel target for ROS metabolites. This study provides clues for understanding the therapeutic approaches to pathological states caused by ischemia-reperfusion injury.
In summary, our study demonstrates that hypoxia induces the phosphorylation of Nrf2 by PKC and thus decreases Keap1-mediated degradation compared with that under normoxia. Moreover, concomitant stabilization of Siah2 protein provides a dominating Nrf2 degradation pathway over that of Keap1 under hypoxia, leading to hypoxic suppression of Nrf2 by Siah2.
This work was supported in part by a grant-in-aid for scientific research from the Japan Society for the Promotion of Science, a grant-in aid from the Hyogo Science and Technology Association, the Support Project to Assist Private Universities in Developing Bases from the Ministry of Education, Culture, Sports, Science and Technology, and by a grant-in-aid from Kwansei Gakuin University.
This article was selected as a Paper of the Week.
- ROS
- reactive oxygen species
- PHD
- prolyl hydroxylase
- qRT
- quantitative real time
- ARE
- antioxidant-response element
- HRE
- hypoxia-responsive element.
REFERENCES
- 1. Chan P. H. (1996) Role of oxidants in ischemic brain damage. Stroke 27, 1124–1129 [DOI] [PubMed] [Google Scholar]
- 2. Bauer C. E., Elsen S., Bird T. H. (1999) Mechanisms for redox control of gene expression. Annu. Rev. Microbiol. 53, 495–523 [DOI] [PubMed] [Google Scholar]
- 3. Ishii T., Itoh K., Takahashi S., Sato H., Yanagawa T., Katoh Y., Bannai S., Yamamoto M. (2000) Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 275, 16023–16029 [DOI] [PubMed] [Google Scholar]
- 4. Rushmore T. H., Pickett C. B. (1990) Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene. Characterization of a xenobiotic-responsive element controlling inducible expression by phenolic antioxidants. J. Biol. Chem. 265, 14648–14653 [PubMed] [Google Scholar]
- 5. Dalton T. P., Shertzer H. G., Puga A. (1999) Regulation of gene expression by reactive oxygen. Annu. Rev. Pharmacol. Toxicol. 39, 67–101 [DOI] [PubMed] [Google Scholar]
- 6. Jaiswal A. K. (2000) Regulation of genes encoding NAD(P)H:quinone oxidoreductases. Free Radic. Biol. Med. 29, 254–262 [DOI] [PubMed] [Google Scholar]
- 7. Cullinan S. B., Gordan J. D., Jin J., Harper J. W., Diehl J. A. (2004) The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 24, 8477–8486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wakabayashi N., Dinkova-Kostova A. T., Holtzclaw W. D., Kang M. I., Kobayashi A., Yamamoto M., Kensler T. W., Talalay P. (2004) Protection against electrophile and oxidative stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. U.S.A. 101, 2040–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McMahon M., Itoh K., Yamamoto M., Hayes J. D. (2003) Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element driven gene expression. J. Biol. Chem. 278, 21592–21600 [DOI] [PubMed] [Google Scholar]
- 10. Zhang D. D., Lo S. C., Cross J. V., Templeton D. J., Hannink M. (2004) Keap1 is a redox-regulated substrate adapter protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 24, 10941–10953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang H. C., Nguyen T., Pickett C. B. (2002) Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 277, 42769–42774 [DOI] [PubMed] [Google Scholar]
- 12. Kang K. W., Choi S. H., Kim S. G. (2002) Peroxynitrite activates NF-E2-related factor 2/antioxidant response element through the pathway of phosphatidylinositol 3-kinase: the role of nitric oxide synthase in rat glutathione S-transferase A2 induction. Nitric Oxide 7, 244–253 [DOI] [PubMed] [Google Scholar]
- 13. Yu R., Lei W., Mandlekar S., Weber M. J., Der C. J., Wu J., Kong A.-N. (1999) Role of a mitogen-activated protein kinase pathway in the induction of phase II detoxifying enzymes by chemicals. J. Biol. Chem. 274, 27545–27552 [DOI] [PubMed] [Google Scholar]
- 14. Niture S. K., Jain A. K., Jaiswal A. K. (2009) Antioxidant-induced modification of INrf2 cysteine 151 and PKC-δ-mediated phosphorylation of Nrf2 serine 40 are both required for stabilization and nuclear translocation of Nrf2 and increased drug. J. Cell Sci. 122, 4452–4464 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15. Abramov A. Y., Scorziello A., Duchen M. R. (2007) Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J. Neurosci. 27, 1129–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Semenza G. L. (1999) Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 15, 551–578 [DOI] [PubMed] [Google Scholar]
- 17. Huang J., Zhao Q., Mooney S. M., Lee F. S. (2002) Sequence determinants in hypoxia-inducible factor-1α for hydroxylation by the prolyl hydroxylases PHD1, PHD2, and PHD3. J. Biol. Chem. 277, 39792–39800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maxwell P. H., Pugh C. W., Ratcliffe P. J. (2001) The pVHL-hIF-1 system. A key mediator of oxygen homeostasis. Adv. Exp. Med. Biol. 502, 365–376 [PubMed] [Google Scholar]
- 19. Nakayama K., Frew I. J., Hagensen M., Skals M., Habelhah H., Bhoumik A., Kadoya T., Erdjument-Bromage H., Tempst P., Frappell P. B., Bowtell D. D., Ronai Z. (2004) Siah2 regulates stability of prolyl-hydroxylases, controls HIFα abundance, and modulates physiological response to hypoxia. Cell 117, 941–952 [DOI] [PubMed] [Google Scholar]
- 20. Nakayama K., Gazdoiu S., Abraham R., Pan Z. Q., Ronai Z. (2007) Hypoxia-induced assembly of prolyl hydroxylase PHD3 into complexes: implications for its activity and susceptibility for degradation by the E3 ligase Siah2. Biochem. J. 401, 217–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mo C., Dai Y., Kang N., Cui L., He W. (2012) Ectopic expression of human MutS homologue 2 on renal carcinoma cells is induced by oxidative stress with interleukin-18 promotion via p38 mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK) signaling pathways. J. Biol. Chem. 287, 19242–19254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jing Y., Liu L. Z., Jiang Y., Zhu Y., Guo N. L., Barnett J., Rojanasakul Y., Agani F., Jiang B. H. (2012) Cadmium increases HIF-1 and VEGF expression through ROS, ERK, and AKT signaling pathways and induces malignant transformation of human bronchial epithelial cells. Toxicol. Sci. 125, 10–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. House C. M., Frew I. J., Huang H. L., Wiche G., Traficante N., Nice E., Catimel B., Bowtell D. D. (2003) A binding motif for Siah2 ubiquitin ligase. Proc. Natl. Acad. Sci. U.S.A. 100, 3101–3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kaur C., Foulds W. S., Ling E.-A. (2008) Hypoxia-ischemia and retinal ganglion cell damage. Clin. Ophthalmol. 2, 879–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Osada M., Imaoka S., Sugimoto T., Hiroi T., Funae Y. (2002) NADPH-cytochrome P-450 reductase in the plasma membrane modulates the activation of hypoxia-inducible factor 1. J. Biol. Chem. 277, 23367–23373 [DOI] [PubMed] [Google Scholar]
- 26. Devling T. W., Lindsay C. D., McLellan L. I., McMahon M., Hayes J. D. (2005) Utility of siRNA against Keap1 as a strategy to stimulate a cancer chemopreventive phenotype. Proc. Natl. Acad. Sci. U.S.A. 102, 7280–7285A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jansen M. P., Ruigrok-Ritstier K., Dorssers L. C., van Staveren I. L., Look M. P., Meijer-van Gelder M. E., Sieuwerts A. M., Helleman J., Sleijfer S., Klijn J. G., Foekens J. A., Berns E. M. (2009) Down-regulation of SIAH2, an ubiquitin E3 ligase, is associated with resistance to endocrine therapy in breast cancer. Breast Cancer Res. Treat. 116, 263–271 [DOI] [PubMed] [Google Scholar]
- 28. Chen C., Yu Z. (2009) siRNA targeting HIF-1α induces apoptosis of pancreatic cancer cells through NF-κB-independent and -dependent pathways under hypoxic conditions. Anticancer Res. 29, 1367–1372 [PubMed] [Google Scholar]
- 29. Tschuch C., Schulz A., Pscherer A., Werft W., Benner A., Hotz-Wagenblatt A., Barrionuevo L. S., Lichter P., Mertens D. (2008) Off-target effects of siRNA specific for GFP. BMC Mol. Biol. 9, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kubo T., Maezawa N., Osada M., Katsumura S., Funae Y., Imaoka S. (2004) Bisphenol A, an environmental endocrine-disrupting chemical, inhibits hypoxic response via degradation of hypoxia-inducible factor 1α (HIF-1α): structural requirement of bisphenol A for degradation of HIF-1α. Biochem. Biophys. Res. Commun. 318, 1006–1011 [DOI] [PubMed] [Google Scholar]
- 31. Shah M., Stebbins J. L., Dewing A., Qi J., Pellecchia M., Ronai Z. A. (2009) Inhibition of Siah2 ubiquitin ligase by vitamin K3 (menadione) attenuates hypoxia and MAPK signaling and blocks melanoma tumorigenesis. Pigment Cell Melanoma Res. 22, 799–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ristoiu V., Shibasaki K., Uchida K., Zhou Y., Ton B. H., Flonta M. L., Tominaga M. (2011) Inhibition of Siah2 ubiquitin ligase by vitamin K3 (menadione) attenuates hypoxia and MAPK signaling and blocks melanoma tumorigenesis. Pain 152, 936–94521376466 [Google Scholar]
- 33. Wang Q., Tang X. N., Yenari M. A. (2007) The inflammatory response in stroke. J. Neuroimmunol. 184, 53–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ueda K., Xu J., Morimoto H., Kawabe A., Imaoka S. (2008) MafG controls the hypoxic response of cells by accumulating HIF-1α in the nuclei. FEBS Lett. 582, 2357–2364 [DOI] [PubMed] [Google Scholar]
- 35. Kimura H., Weisz A., Ogura T., Hitomi Y., Kurashima Y., Hashimoto K., D'Acquisto F., Makuuchi M., Esumi H. (2001) Identification of hypoxia-inducible factor 1 ancillary sequence and its function in vascular endothelial growth factor gene induction by hypoxia and nitric oxide. J. Biol. Chem. 276, 2292–2298 [DOI] [PubMed] [Google Scholar]
- 36. Khurana A., Nakayama K., Williams S., Davis R. J., Mustelin T., Ronai Z. (2006) Regulation of the ring finger E3 ligase Siah2 by p38 MAPK. J. Biol. Chem. 281, 35316–35326 [DOI] [PMC free article] [PubMed] [Google Scholar]