

Background: Mycobacterium tuberculosis FadD10 transfers fatty acyl chains onto an acyl carrier protein via adenylated intermediates.
Results: The structures of M. tuberculosis FadD10 reveal a ligand-bound conformation previously unseen in the adenylate-forming superfamily.
Conclusion: The structures of M. tuberculosis FadD10 provide a novel structural basis for the enzyme's function compared with the homologues.
Significance: Fatty acyl transfer is an important biological process involved in the synthesis of bioactive lipopeptides.
Abstract
Mycobacterium tuberculosis has a group of 34 FadD proteins that belong to the adenylate-forming superfamily. They are classified as either fatty acyl-AMP ligases (FAALs) or fatty acyl-CoA ligases based on sequence analysis. FadD10, involved in the synthesis of a virulence-related lipopeptide, was mis-annotated as a fatty acyl-CoA ligase; however, it is in fact a FAAL that transfers fatty acids to an acyl carrier protein (Rv0100). In this study, we have determined the structures of FadD10 in both the apo-form and the complexed form with dodecanoyl-AMP, where we see for the first time an adenylate-forming enzyme that does not adopt a closed conformation for catalysis. Indeed, this novel conformation of FadD10, facilitated by its unique inter-domain and intermolecular interactions, is critical for the enzyme to carry out the acyl transfer onto Rv0100 rather than coenzyme A. This contradicts the existing model of FAALs that rely on an insertion motif for the acyltransferase specificity and thus makes FadD10 a new type of FAAL. We have also characterized the fatty acid preference of FadD10 through biological and structural analyses, and the data indicate long chain saturated fatty acids as the biological substrates of the enzyme.
Introduction
Adenylate-forming enzymes are found in a variety of important biological processes in both eukaryotes and prokaryotes (1). They show wide diversity in their catalytic activities and biological functions. The adenylate-forming superfamily of enzymes is composed of luciferases, amino acid adenylation domains of nonribosomal peptide synthetases (NRPSs),2 acyl-coenzyme A (acyl-CoA) synthetases, and acyl-acyl carrier protein (acyl-ACP) synthetases. These enzymes first use ATP to adenylate the carboxylate group of the cognate substrate, which is then followed by the transfer of the acyl moieties to the corresponding acceptors, for example, luciferyl to oxygen, aminoacyl to peptidyl carrier protein domains, and fatty acyl to CoA or ACPs. The fatty acyl-CoA synthetases (EC 6.2.1.3) and fatty acyl-ACP synthetases (EC 6.2.1.20) have also been referred to as fatty acyl-CoA ligases (FACLs) and fatty acyl-AMP ligases (FAALs), respectively (2–5). Both enzymes have acyl-AMP ligase activity. However, FACLs transfer fatty acyl chains to coenzyme A, whereas FAALs transfer fatty acyl chains to ACPs.
Within the genome of Mycobacterium tuberculosis, there are 34 fadD genes that have been annotated to be members of the adenylate-forming superfamily (6). Although the precise functions of most are unclear, several have been shown to be involved in the synthesis of bioactive lipids that are essential for bacterial survival or virulence (7–10) and thus represent potential targets for drug discovery. Multisequence alignment of the 34 M. tuberculosis FadDs revealed two subclasses, 12 FAALs and 22 FACLs (2). The 12 FAALs share a much higher sequence homology (70–80% identity) than the FACLs (20–30% identity). Interestingly, 10 of the 12 M. tuberculosis FAAL-encoding genes are located in close proximity to multienzyme polyketide synthases. Biochemical studies have shown that three of the FAALs (FAAL26, FAAL30, and FAAL32) transfer long chain fatty acyl moieties to the polyketide synthases, PpsA, PKS6, and PKS13, respectively (2, 11, 12). Therefore, it has been suggested that FAAL enzymes activate fatty acids as adenylate derivatives and sequentially transfer acyl moieties to cognate modular enzymes, particularly their ACP domains, to be further processed into complex lipids (2, 3). Recent structural and biochemical studies of M. tuberculosis FAAL28 (3) and FAALs from Escherichia coli and Legionella pneumophila (4) have provided a molecular basis for their catalytic mechanism. These studies have identified a signature sequence motif, consisting of an insertion of about 20 amino acids, that defines the function of this newly recognized subclass of the adenylate-forming superfamily and distinguishes them from FACLs (3, 13).
FadD10 (Rv0099) is present on an operon consisting of Rv0096–Rv0101. The operon has been the subject of many studies (14–16) because it is involved in making a virulence-essential lipopeptide using an NRPS (Rv0101, nrp). Mycobacterium smegmatis has a similar NPRS, called mps, which synthesizes a glycopeptidolipid (17). We have shown that Rv0098 (14) is a fatty acyl-CoA thioesterase. Although the identity of the lipopeptide produced by M. tuberculosis Rv0096–Rv0101 operon is not known, its biological significance was indicated in several studies. For example, Rv0096 was demonstrated to be required for M. tuberculosis survival in mouse macrophages (18), and Rv0098 to Rv0101 were predicted, using transposon site hybridization, to be required for M. tuberculosis survival in a mouse model of infection (19). It has been suggested that the products of the operon repressed the SigM factor that further regulates a series of metabolic pathways and modulates the host-bacteria interactions (20). Despite the biological significance of the lipopeptide produced by the Rv0096–Rv0101 operon, neither the isolation nor the identity of this lipopeptide have been reported; therefore, an alternative approach to define its chemical structure is to characterize the function and mechanism of the individual proteins involved in its synthesis.
Rv0098–Rv0101 are essential for M. tuberculosis survival in the mouse model of infection (19). Hence, they are likely to have direct enzymatic roles in the synthesis of the virulence-related lipopeptide product of the operon. As mentioned above, our biochemical and structural characterization of Rv0098 has shown that it encodes a thioesterase capable of hydrolyzing a long chain fatty acyl-CoA to release a fatty acid and coenzyme A (14). Rv0100 is clearly an ACP according to sequence analysis, and it shares 24.4% sequence identity to the primary M. tuberculosis ACP (Rv0033) involved in type II fatty acid biosynthesis. Rv0101 is a two-module NRPS, with the first module of an undefined amino acid specificity and the second module to incorporate phenylalanine (21). M. tuberculosis FadD10 (Rv0099) was designated as an FACL based on sequence analysis (2). However, the presence of a thioesterase (Rv0098) immediately upstream suggests that fadD10 is unlikely to function as an acyl-CoA synthetase because this would put two enzymes with opposing functions next to each other in the operon. Indeed, the fact that fadD10 is located in close proximity with a multifunction enzyme (Rv0101) is analogous to the M. tuberculosis FAALs. These findings suggest that M. tuberculosis FadD10 activates and transfers the fatty acyl chain to the cognate ACP-Rv0100. Chhabraa et al. (16) recently showed that FadD10 does not acylate CoA but is able to transfer a radioactively labeled dodecanoyl moiety to Rv0100 in vitro. This suggests that FadD10 has FAAL activity even though it has a primary sequence more similar to FACLs. FadD10 has much lower sequence similarity to the members of the M. tuberculosis FAALs cluster and moreover lacks the signature motif critical for FAAL activity (Fig. 1); hence, it is intriguing to delineate the molecular basis underlying this discrepancy.
FIGURE 1.

Multisequence alignment of M. tuberculosis FadDs reveals a signature insertion (boxed) for FAALs cluster.
In this study, we have solved the structures of both apo- and dodecanoyl-AMP-bound M. tuberculosis FadD10, leading to the characterization of a novel ligand-bound conformation for the adenylate-forming superfamily. Instead of undergoing conformational rearrangement between the N- and C-terminal domains, as observed in all the other reported adenylate-forming homologues (22), FadD10 retains a single open conformational state for both the apo- and ligand-bound forms. We find that FadD10's FAAL activity can be explained by its unique inter-domain and intermolecular interactions, a mechanism distinct from that known for the other FAALs. Our modeling studies identify the binding site of the ACP (Rv0100) onto FadD10 in the acyl transfer reaction. Also, our studies provide a structural basis for the fatty acid preference of M. tuberculosis FadD10, which may eventually aid in elucidating the chemical structure of the virulence-related lipopeptide produced by the Rv0096–Rv0101 operon.
EXPERIMENTAL PROCEDURES
Cloning, Protein Expression, and Purification
The M. tuberculosis fadD10 (Rv0099) gene was amplified by PCR, incorporated into the pDEST17 vector by gateway cloning (Invitrogen), and then transformed into either Novagen BL21(DE3)pLys E. coli cells for expression of native protein or Novagen B834(DE3)pLys cells for selenomethionine-incorporated protein expression.
The cells were cultured at 37 °C until an A600 of 0.8 was reached. For native protein expression, 1 mm isopropyl 1-thio-β-d-galactopyranoside was added to induce expression, and the cells were grown overnight at 20 °C. For selenomethionine-incorporated protein expression, the cells were collected at an A600 of 0.8, centrifuged, and then resuspended and transferred into minimal media with selenomethionine. Induction of expression and growth were the same as for the native protein.
After harvesting, the cells were resuspended in 25 mm Tris (pH 8.0), 500 mm NaCl, and 2 mm β-mercaptoethanol and lysed by French press. Recombinant FadD10 with an N-terminal poly-His tag was purified by nickel affinity chromatography followed by gel filtration. The His6 tag was then cleaved by tobacco etch virus protease, and the untagged protein was passed through another nickel column in 25 mm Tris (pH 8.0), 100 mm NaCl, and 2 mm β-mercaptoethanol. It was then concentrated to 15 mg ml−1, flash-frozen, and stored at −80 °C.
The M. tuberculosis Rv0100 gene was amplified by PCR and ligated into the Novagen pET28b vector. The E. coli phosphopantetheine transferase Sfp was cloned into Novagen pETduet-1 vector. The plasmids containing Rv0100 and Sfp, respectively, were co-transformed into Novagen BL21(DE3) E. coli cells and then expressed as described for the native FadD10. Recombinant holo-Rv0100 with a His6 tag was purified by nickel affinity chromatography in 25 mm Tris (pH 8.0), 100 mm NaCl, and 2 mm β-mercaptoethanol.
Crystallization
Crystals of Se-FadD10 (selenomethionine-incorporated) were grown at 18 °C by hanging drop vapor diffusion. Each drop contained an equal volume of the protein solution and reservoir solution (0.32–0.36 mm LiSO4 and 15–30% polyethylene glycol 6000).
FadD10 in complex with dodecanoyl-AMP was obtained by incubating the protein solution for 1 h with the reaction mixture in a 10:1 volume ratio. The reaction mixture was made by incubating 10 μm FadD10 with 2.5 mm ATP, 10 mm MgCl2, and 1 mm dodecanoic acid for 1 h at 37 °C, then filtering out the protein, and concentrating the mixture 10-fold. Crystals formed in hanging drops after 4 days in 4 m potassium formate.
Data Collection and Processing
Diffraction data from a single apo-Se-FadD10 crystal was collected at 120 K using a cryo-protection solution consisting of the crystallization condition with the addition of 30% glycerol. Crystals diffracted to 2.20 Å at beam line 5.0.2 at the Advanced Light Source. A total of 180° diffraction data were collected at the wavelength of 0.9795 Å, which is the absorption peak of Se-FadD10 crystals. Diffraction data of FadD10 in complex with dodecanoyl-AMP were collected to 2.44 Å at the wavelength of 1.542 Å using Rigaku R-AXIS IV++ at home source. All the data were processed and reduced using HKL2000 (23). The crystals of apo-Se-FadD10 are in the space group P21, with two molecules in each asymmetric unit. The crystals of FadD10 binding dodecanoyl-AMP are in the space group P3121, with one molecule in each asymmetric unit (Table 1).
TABLE 1.
Data collection and refinement statistics
| PDB ID | 4ISB | 4IR7 |
|---|---|---|
| Crystal | Se-FadD10 | C12-FadD10 |
| Ligands | SO42 − | Dodecanoyl-AMP Mg2+ |
| Data collection | ||
| Space group | P21 | P3121 |
| Unit cell dimensions | a = 57.32Å, b = 107.91Å, c = 85.69 Å | a = b = 138.16 Å, c = 82.47 Å |
| α = 90.0°, β = 106.9°, γ = 90.0° | α = β = 90.0°, γ = 120.0° | |
| Molecules/asymmetric unit | 2 | 1 |
| Wavelength | 0.9795 Å | 1.542 Å |
| Resolution | 48.57 to 2.20 Å | 39.84 to 2.80 Å |
| Completenessa | 99.3% (98.9%) | 100.0% (99.7%) |
| No. of reflections | 49,204 | 24,707 |
| I/σIa | 9.50 (1.94) | 14.01 (2.15) |
| Rsyma | 0.103 (0.703) | 0.0567 (0.504) |
| Refinement statistics | ||
| Resolution | 48.50 to 2.20 Å | 39.03 to 2.80 Å |
| No. of reflection work | 49,014 | 22,632 |
| No. of protein atoms | 7422 | 3734 |
| No. of water molecules | 59 | 32 |
| No. of heteroatoms | 20 | 36 |
| Rcryst | 22.69% | 22.61% |
| Rfree | 27.28% | 27.95% |
| r.m.s.d.b bond length | 0.01 Å | 0.002 Å |
| r.m.s.d. angle | 1.26° | 0.60° |
| Mean temperature factor | 25.5 Å2 | 62.0 Å2 |
a Numbers in parentheses indicate data for highest resolution shell.
b r.m.s.d., root mean square deviation.
Structure Determination and Model Refinement
The phase of Se-FadD10 was determine by single-wavelength anomalous dispersion using Autosol in PHENIX (24). Twenty two selenium atoms were located and refined per asymmetric unit until the overall figure of merit reaches 0.32. An initial model was built by Autobuild in PHENIX (24). Manual rebuilding was then performed to improve the model using Coot (25). The final model was obtained after further cycles of model building and PHENIX refinement yielding Rcryst and Rfree of 22.69 and 27.28%, respectively. There are two subunits, designated as A and B, as well as four sulfate ions and 59 water molecules per asymmetric unit in the refined model. A total of 508 and 502 out of 540 residues for chain A and B, respectively, were visible and built into the electron density. The missing residues are due to the absence of interpretable electron density. They include the N-terminal residues 1–8, the C-terminal residues 532–540, and the loop residues 145–154, 424, and 477–480 of chain A and the N-terminal residues 1–3, the C-terminal residues 532–540, and the loop residues 125–131, 145–154, 161–164, and 179–183 of chain B. Residues Ser-178, Thr-181, Glu-183, and Lys-185 of chain A and residues Glu-426, Arg-451, Ser-474, Glu-476, and Leu-477 of chain B were built as alanine due to ambiguous side chain electron density.
The structure of FadD10 in complex with dodecanoyl-AMP was solved by molecular replacement using MOLREP (26) in CCP4. A single solution for the molecular replacement was obtained using the chain A of apo-FadD10 as the search model. Dodecanoyl-AMP was manually built in the model by examining the Fo − Fc map in Coot (25). The final model was obtained after further cycles of model building and PHENIX refinement, to 2.80 Å, yielding Rcryst and Rfree of 22.61 and 27.95%, respectively. There is one subunit of FadD10, one dodecanoyl-AMP, and 32 water molecules in each asymmetric unit. A total of 508 residues were built into the refined model. The N-terminal residues 1–10, the C-terminal residues 532–540, and the loop residues 124–128, 147–152, and 179–180 are missing from the model due to the absence of interpretable electron density. The complete refinement statistics are given in Table 1.
Enzymatic Assays to Detect Acylation of Rv0100
FadD10 (1.5 μm) and Rv0100 (20 μm) were incubated with 2 mm ATP, 5 mm MgCl2 in the presence of 200 μm different fatty acids, in 25 mm ammonium bicarbonate buffer at pH 7.8, for 1 h. The salts were removed by diluting and concentrating the solutions by 10-fold in 25 mm ammonium bicarbonate buffer for three cycles. The resulted samples were mixed with acetonitrile and formic acid in a ratio of 1:1:0.002 and then analyzed by Bruker microQTOF-QII.
HPLC-MS Analysis of Dodecanoyl Amino Acids
FadD10 (10 μm) was incubated with 2 mm ATP, 8 mm MgCl2, 200 μm dodecanoic acid in the presence of 2 mm of different amino acids, in 50 mm phosphate buffer at pH 6.5, for 1 h. The macromolecules were removed by passing the solutions through 5-kDa cutoff filters. 100 μl of each reaction product were injected in to HPLC coupled with ESI-MS analysis. The HPLC was performed with an Atlantis T3 5-μm column (4.6 × 250 mm), using a 5% acetonitrile, 0.1% TFA wash for 10 min followed by a 5–80% acetonitrile, 0.1% TFA gradient over 1 h.
RESULTS AND DISCUSSION
Crystal Structures of M. tuberculosis FadD10 Subunit
To understand the molecular basis of the FAAL activity of FadD10 arising from a primary sequence more similar to FACLs, we have determined the structures of FadD10 in both apo- and complexed forms. The full-length apo M. tuberculosis FadD10 crystallized in space group P21 and its structure was solved using selenium single wavelength anomalous dispersion. The asymmetric unit contains a dimer (Fig. 2A), consistent with the gel filtration analysis (data not shown). The two subunits are very similar with an r.m.s.d. of 0.68 Å for 491 α-carbons after superimposition (Fig. 2C). For the chains designated as A and B, a total of 508 and 502 out of 540 residues, respectively, were visible and built into the electron density. Residues Ser-178, Thr-181, Glu-183, and Lys-185 of chain A and residues Glu-426, Arg-451, Ser-474, Glu-476, and Leu-477 of chain B were built as alanine due to ambiguous side chain electron density. The structure was refined to 2.2 Å resolution with a final R-factor of 22.7% and an R-free of 27.3%. Statistics of the refined structures are listed in Table 1.
FIGURE 2.

A, ribbon representation of the dimeric FadD10. The N-terminal domains of the two subunits, designated as A and B, are colored yellow and green, and the corresponding C-terminal domains are colored orange and cyan, respectively. B, Fo − Fc map is calculated when dodecanoyl-AMP and magnesium ion are omitted from the model and are contoured at the 2σ level. C, FadD10 (gray) in complex with dodecanoyl-AMP (pink) is superimposed with the two subunits (colored as in A) from the apo structure. D, cylinder representation of FadD10 that demonstrates the two-domain organization and the secondary structural elements of each subunit. The inter-domain loop is colored in black. E, inter-domain hydrogen bonds shown as dotted lines. The residues involved in the interactions are shown as sticks. The identity of the helices involved in forming the hydrogen bonds is shown in color.
FadD10 complexed with the half-reaction product dodecanoyl-AMP crystallized in space group P3121. Its structure was solved by molecular replacement using chain A of our refined apo structure as the search model. Dodecanoyl-AMP was built into a clear electron density (Fo − Fc map) located at the active site (Fig. 2B). In this crystal, M. tuberculosis FadD10 packed with one molecule per asymmetric unit, and the two subunits of the dimer are related to each other by a crystallographic 2-fold symmetry. The structure, with 508 residues built into the electron density, was refined to 2.8 Å resolution with an R-factor of 22.6% and an R-free of 27.9%. Superposition of the complexed M. tuberculosis FadD10 subunit with the chain A and chain B from the apo-crystal yields an r.m.s.d. of 0.596 and 0.637 Å, respectively, for the α-carbons (Fig. 2C).
Each subunit of M. tuberculosis FadD10 is composed of two domains (Fig. 2D). The N-terminal domain consists of 420 amino acids (residue 1–420), forming a central α/β structure surrounded by a distorted β-sheet on one side and three β-strands with intervening α-helices on the other side. The loop containing the residues 421KGRSS425 extends from the β-strand B15 and continues into the C-terminal domain, consisting of 115 amino acids (residues 426–540). This domain is composed of three β-strands surrounded by two α-helices and a pair of short anti-parallel β-strands at the beginning of the domain.
Analysis of the complexed FadD10 subunit structure using Vector Alignment Search Tool (27) revealed that the highest structural similarity was with adenylate-forming enzymes, namely Alcaligenes sp. 4-chlorobenzoate-CoA synthetase, the M. tuberculosis very-long-chain fatty acyl-CoA synthetase FACL13, and the Neotyphodium lolii NRPS activation domain SidNA3. Superposition of the full-length M. tuberculosis FadD10 with these structures yielded fairly poor alignments. For instance, superposition between M. tuberculosis FadD10 and FACL13 (PDB code 3R44) showed an alignment with only part (301 out of 420 α-carbons) of the N-terminal domain (r.m.s.d. 2.61 Å). When the N-terminal domains were aligned, the C-terminal domains were found to be in a completely different orientation, even though the secondary structural elements of their C-terminal domains were quite similar. Indeed, dividing FadD10 into N- and C-terminal domains allowed 80% of its N-terminal structure and 70% of the C-terminal structure to align with FACL13, yielding r.m.s.d. of 2.80 Å for 345 N-terminal domain α-carbons and 4.24 Å for 101 C-terminal domain α-carbons. A similar result was observed when M. tuberculosis FadD10 was compared with the other adenylate-forming homologues identified by Vector Alignment Search Tool, i.e. structural superposition could only be obtained when the N- and C-terminal domains were overlaid independently. Although all of the adenylate-forming enzymes complexed with adenylate (including analogues) share a similar inter-domain orientation, M. tuberculosis FadD10 adopts a distinctive inter-domain conformation (Fig. 3A). As expected, similar alignments were obtained with the apo structure of FadD10.
FIGURE 3.

A, FadD10 (yellow) complexed with dodecanoyl-AMP (ball and stick) is aligned with human medium chain acyl-CoA synthetase (gray, PDB code 3DAY) at their N-terminal domains. The latter is shown only at the C-terminal domain, and the N-terminal domain of FadD10 is shown in carbon trace for clarity. B, variation of the secondary structure equivalent to FadD10 helix A11, among adenylate-forming enzymes (panel i), is exemplified by M. tuberculosis FACL13 (PDB code 3R44) (panel ii), M. tuberculosis FAAL28 (PDB code 3E53) (panel iii), and Bacillus brevis PheA (PDB code 1AMU) (panel iv). Glu-340 in FadD10 is shown as ball and stick. A11 and its equivalents are colored red.
The adenylate-forming superfamily of proteins that have been structurally characterized revealed remarkable conformational flexibility between the N- and C-terminal domains (28–34). The inter-domain orientations of these proteins vary noticeably among the apo structures as well as between the apo and the respective ligand-bound structures of the individual proteins. Such variations are not correlated to the functions of different subclasses. They likely arise because the two domains form no significant protein-protein inter-domain contacts and are connected by only a flexible loop. This feature allows the two domains to rearrange upon substrates binding to desolvate the active site, a process that has been referred to as the “domain alternation” mechanism for the adenylate-forming superfamily (22). This mode of ligand binding is exemplified by the Thermus thermophilus long chain fatty acyl-CoA synthetase (32). When this protein binds AMPPNP or myristoyl-AMP, its C-terminal domain rotates almost 180° relative to the inter-domain linkage loop (432DRLK435) and moves toward the N-terminal domain forming a lid over the active site. A similar rearrangement, with varying extents of rotation, has also been observed for Photinus pyralis luciferase (28), the aryl acid adenylation domain of bacillibactin synthetase (DhbE) (30), Alcaligenes sp. 4-chlorobenzoate-CoA synthetase (31), and Homo sapiens medium chain acyl-CoA synthetase (33), for which both apo and ligand-bound structures are available. Thus, the closure of the C-terminal domain toward the N-terminal domain is believed to be a common substrate binding event for the adenylate-forming superfamily. In this respect, M. tuberculosis FadD10 is clearly unique in comparison with other homologous adenylate-forming proteins, as it maintains an “open” conformation for the apo structure, as well as for FadD10 complexed to the half-reaction product dodecanoyl-AMP.
Novel Ligand-bound Conformation of M. tuberculosis FadD10
The apo and complexed structures of M. tuberculosis FadD10 have provided several insights into the structural determinants for its unique open ligand-bound conformation. The first factor that contributes to the maintenance of the open conformation in the M. tuberculosis FadD10 structures is the inter-domain interactions between the α-helix A15 from the C-terminal domain and the three-helix (A9, A10, A11) cluster from the N-terminal domain (Fig. 2E). Specifically, the side-chain oxygen of Ser-524 forms hydrogen bonds with both the side-chain oxygen of Glu-340 (2.5 Å) and the backbone nitrogen of Ala-301 (3.3 Å). More inter-domain hydrogen bonds are formed by the backbone nitrogens of Ala-523 (3.1 Å) and Ser-524 (3.3 Å) with the side-chain carboxylate of Glu-340, as well as between the side-chain amine of Lys-279 and the side-chain oxygen atoms of Asp-511 (3.1 and 3.3 Å). These interactions are maintained in both the apo-M. tuberculosis FadD10 structure and the complexed structure with dodecanoyl-AMP, which greatly limits the inter-domain movement of the protein. A similar inter-domain contact has not been observed in the other adenylate-forming proteins. The reason is likely due to the absence of the structural elements found in M. tuberculosis FadD10 accounting for this interaction. Although A9, A10, and A15 are structurally conserved, the structural equivalent to helix A11 of FadD10 (Fig. 3B, panel i) notably varies among adenylate-forming proteins. A short helix equivalent to A11 is found in only four members, including the adenylation domains DhbE (30) and DltA (35), as well as the acyl-CoA synthetases, long chain fatty acyl-CoA synthetase (32), and M. tuberculosis FACL13 (29). However, the glutamate residue (Glu-340) essential to forming inter-domain hydrogen bonds in M. tuberculosis FadD10 is replaced by nonpolar residues in these four homologous proteins (Fig. 3B, panel ii). A significant structural alteration of A11 is observed in FAALs, including the N-terminal domain of M. tuberculosis FAAL28 (3), the full-length E. coli, and L. pneumophila FAALs (4), where the helix A11 is replaced by a β-strand followed by an α-helix or a loop (Fig. 3B, panel iii). This is the location where the ∼20-amino acid insertion motif occurs in FAALs, which has been implicated in determining FAAL activity. In all of the other structures that have been determined for the adenylate-forming superfamily, the region equivalent to the helix A11 is disordered (Fig. 3B, panel iv).
The second factor contributing to the rigidity of M. tuberculosis FadD10 open conformation arises from the quaternary organization of the dimer. According to the current understanding of adenylate-forming proteins, most of them, including FAALs (4), are functional as monomers. The only subclass that can form dimers are the acyl-CoA synthetases, and for these proteins the dimerization interface is always limited to the N-terminal domains. This observation is consistent with the acyl-CoA synthetic mechanism, which requires the C-terminal domain to move freely relative to the N-terminal domain to bind ATP and then coenzyme A in the stepwise reactions. M. tuberculosis FadD10 is also shown to be a dimer in solution based on both gel filtration analysis (data not shown), and PISA (Protein Interfaces, Surfaces, and Assemblies) calculation (36). In both the apo structure of M. tuberculosis FadD10 and its complexed structure with dodecanoyl-AMP, the two subunits of the dimer form an extensive network of intermolecular interactions, including eight hydrogen bonds and many van der Waals interactions. All of the hydrogen bonds and most of the van der Waals interactions are between residues from the C-terminal domain of one subunit and residues from the N-terminal domain of the other subunit. This confers a contact area of about 1600 Å2 per 22,000 Å2 for each subunit. FadD10 is the first adenylate-forming enzyme other than acyl-CoA synthetases to be identified as a dimer. More importantly, the two subunits of the dimer interact in a manner that was previously unseen in the other homologues, and we propose this dimerization mode has played a role in maintaining the open conformation of M. tuberculosis FadD10 upon binding dodecanoyl-AMP.
Conformation of M. tuberculosis FadD10 Prevents Coenzyme A Binding
The adenylate-forming proteins, as a superfamily, share 10 conserved sequence motifs (37), a similar structural scaffold (22), and a common half-reaction, the adenylation of the carboxylate group of a substrate. Moreover, structural characterization of the superfamily revealed that a phosphopantetheine binding cavity is structurally conserved in the N-terminal domain. This is consistent with the notion of an evolutionary relationship within the adenylate-forming superfamily, and it strongly argues that the ubiquitous acyl-CoA synthetases are likely to be ancestral (13, 38, 39). Functional conservation of acyl-CoA synthesis activity has also been reported in other sub-classes of the adenylate-forming superfamily. For example, firefly luciferase (38) as well as five different adenylation domains of NRPSs (39) were shown to synthesize luciferyl-CoA and aminoacyl-CoAs, respectively, when their genuine acyl acceptors were absent, and coenzyme A was added as substrate. In contrast, M. tuberculosis FAALs (acyl-ACP synthetases) that have been studied to date, which have also been proposed to be descendants of acyl-CoA synthetases, lack acyl-CoA synthesis activity (3, 13), even though they retain the binding elements of phosphopantetheine. This was demonstrated as well for FadD10.
Structural comparison and analyses of relative domain orientation of FAALs and FadD10 with acyl-CoA synthetases provide a plausible explanation to their inability to turn over acyl-COA. The structures of three acyl-CoA synthetases, Alcaligenes sp. 4-chlorobenzoate-CoA synthetase, human medium chain fatty acyl-CoA synthetase, and E. coli acetyl-CoA synthetase, have all been reported in complex with coenzyme A or derivatives (31, 33, 40). When these proteins bind to coenzyme A, their C-terminal domains all rotate about 140° relative to the position in the adenylate-bound form. Also, for all of these proteins, the C-terminal domain is explicitly engaged in the binding of coenzyme A through hydrophobic interactions with its nucleotide moiety. This domain reorganization is believed to be requisite for coenzyme A binding by the adenylate-forming enzymes, and hence for the formation of acyl-CoA.
Sequence analysis of M. tuberculosis FadD proteins reveals an N-terminal domain insertion of 10–24 amino acids only present in the FAAL cluster (Fig. 1). Using the sequence insertion as an indicator, a number of putative FAALs can be identified in various organisms. Structural studies of E. coli FAAL, L. pneumophila FAAL (4), and the N-terminal domain of M. tuberculosis FAAL28 (3) indicate that the inserted sequence flanks the interface of the two domains. In M. tuberculosis FadD28 and E. coli FAAL, the insertion folds into a β-strand followed by a short α-helix and a short extension of β-strand; in L. pneumophila FAAL, the insertion folds into a β-strand and then a loop. The structural insertion does not appear to interfere with the closure of the C-terminal domain to form the adenylate-bound conformation (4). However, it prevents the FAALs from undergoing the large scale inter-domain rotation associated with coenzyme A binding. The functional role of the insertion was further established by showing that its deletion and addition manipulated the gain and loss of acyl-CoA synthesis activity in M. tuberculosis FAALs and FACLs, respectively (3, 13).
M. tuberculosis FadD10 does not have the FAALs insertion; therefore, the lack of acyl-CoA synthesis activity for M. tuberculosis FadD10 must be based on a different mechanism. We propose that it is the unique inter-domain and intermolecular interactions of FadD10, as described above, that prevent the required inter-domain rearrangement for coenzyme A binding. The introduction of an acyl moiety into a natural product made by a PKS or NRPS is a well recognized function of the acyl-CoA synthetases. Although the acyl-CoA synthetases have been extensively studied, the CoA-independent acyl transfer function has only been recently discovered, and the understanding of its mechanism is limited to FAALs (41). Functionally analogous to FAALs, M. tuberculosis FadD10 has revealed a distinct strategy to prevent acyl-CoA synthesis. It contradicts the existing model for the catalysis of FAALs dependent on an insertion motif, and therefore it establishes M. tuberculosis FadD10 as a new type of FAAL.
Modeling Studies of the Interactions between M. tuberculosis FadD10 and Rv0100
We have identified the likely binding interface for ACP (Rv0100) on FadD10. Because the transfer of a fatty acyl chain to the ACP by FadD10 is analogous to the transfer of aminoacyl groups to peptidyl carrier proteins by NRPS adenylation domains, we have compared FadD10 with the structure of Pseudomonas aeruginosa PA1221, which is a didomain construct containing an adenylation domain and a peptidyl carrier protein (42). When the N-terminal domains of the FadD10 and PA1221 adenylation domains (21% identity) are superimposed, their C-terminal domains are in different orientations as expected. Interestingly, the peptidyl carrier protein of the didomain structure is positioned in an orientation poised for the FadD10 substrate to bind. Specifically, the side chain of the active site serine residue of the peptidyl carrier protein is directed toward and is approximately the correct distance to the phosphopantetheine-binding cavity of FadD10 (Fig. 4A). When the interaction between FadD10 and the PA1221 peptidyl carrier protein is examined over the surface of the FadD10 dimer, we observe a relatively good fit of the carrier protein onto the intermolecular space of the dimer. Because ACPs are highly functional and structurally homologous to the peptidyl carrier proteins, we have generated a homology model of Rv0100 based on the structure of PA1221 peptidyl carrier protein (19% identity) (43). When the ACP model is overlaid on top of the superposition of FadD10 and PA1221, the interaction of ACP to FadD10 is similar to that of the PA1221 peptidyl carrier protein to FadD10 (Fig. 4B), which suggests that the open conformation of FadD10 has room for the ACP (Rv0100) to bind for acyl transfer.
FIGURE 4.

A, ribbon representation of the superposition of FadD10 subunit A with the didomain structure of PA1221 (PDB code 4DG9). The N- and C-terminal domains of FadD10 are colored in yellow and orange, respectively. The N-terminal domain of the PA1221 adenylation domain is colored in white, and its C-terminal domain is hidden for clarity. The PA1221 peptidyl carrier protein is colored in blue, and its active site residue Ser-533 is shown in stick. Phosphopantetheine, shown in green stick, is modeled based on comparison with human medium chain fatty acyl synthetase in complex with butyl-CoA (PDB code 3EQ6). B, surface representation of the interaction between the ACP (Rv0100) and FadD10 dimer. The subunit A of FadD10 dimer and PA1221 peptidyl carrier protein are colored as in A. The N- and C-terminal domains of the FadD10 subunit B are colored in green and cyan, respectively. The Rv0100 model is shown as pink ribbon.
Substrate-binding Site of M. tuberculosis FadD10
Several structures of adenylate-forming proteins in complex with adenylated derivatives (including three proteins bound to different long chain acyl-AMPs (4, 32)) have been reported, wherein multiple residues from both the N- and C-terminal domains are involved in the binding of adenylate moiety. However, the interactions between the amino acids of the C-terminal domain to the adenylate moiety are missing in M. tuberculosis FadD10, due to its open conformation. Conserved between FadD10 and the other homologous structures are primarily the interactions between the residues from the N-terminal domain of the enzyme and the adenylate group. The adenosine moiety, which is coordinated by a network of both hydrophobic interactions and hydrogen bonding (Fig. 5A), sits at the entrance to the catalytic cavity. The planar adenine is sandwiched by the hydrophobic side chains of Tyr-317 and Val-344 on one side, and the backbone atoms of 294GGSR297 on the other side. The exocyclic nitrogen forms hydrogen bonds with the side-chain oxygen of Gln-315 (3.2 Å) and the backbone oxygen of Val-316 (3.2 Å). The major binding determinants of ribose are Gly-295 and Asp-408. Specifically, the backbone oxygen of the former forms hydrogen bond (3.3 Å) with the ribose ring oxygen, and a side chain oxygen of the latter interacts with the 3′ (3.2 Å) ribosyl hydroxyl via hydrogen bond. The α-phosphate of AMP interacts with Ser-320 by forming hydrogen bonds between the O2 of the phosphate and the backbone nitrogen (3.3 Å) of this residue.
FIGURE 5.
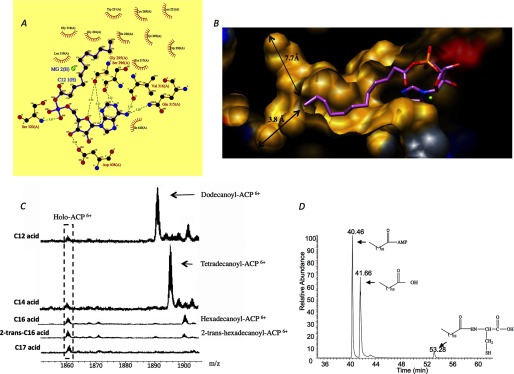
A, schematic representation of the hydrogen bonds (dotted lines) and hydrophobic interactions (bows) between dodecanoyl-AMP and FadD10. B, fatty acid binding tunnel of M. tuberculosis FadD10. The hydrophobic, positive, negative, and neutral surfaces of FadD10 are colored yellow, blue, red, and gray, respectively. Dodecanoyl-AMP is shown in stick and colored in pink. The arrows indicate the distance from the terminal carbon of dodecanoyl chain to the molecular surface of the binding tunnel, taking into account the atomic van der Waals radii. C, ESI-QTOF mass spectrometry of the holo-ACP (Rv0100) acylated by M. tuberculosis FadD10, in addition to fatty acids with varying chain length and degree of saturation. The ratio of abundance between the unreacted holo-ACP and the acylated ACP is approximately 1:5.5, 1:6.9, 1:1.5, 1:0.6, and 1:0, respectively, for C12, C14, C16, 2-trans-C16, and C17 fatty acid. D, FadD10 was incubated with dodecanoic acid, ATP, and cysteine and then analyzed by LC-MS. The result is simplified for clarity by only showing the elution of dodecanoic acid, dodecanoyl-AMP, and dodecanoyl-cysteine.
The dodecanoyl aliphatic chain is buried inside a preformed narrow closed-end tunnel by β-strands B9, B10, B11, and α-helix A7 (Fig. 5B). In the apo structure of FadD10, this fatty acyl binding cavity is vacant. The tunnel that contains the aliphatic chain is lined primarily by the backbone atoms of B9, B10, and B11 (i.e. 264TCLV267, 291VGYGG295, and 317YG318), although only three hydrophobic side chains, Val-316, Ile-226, and Trp-231, are within van der Waal's distance to the aliphatic chain. Approximately 11 Å from the portal of the fatty acyl binding cavity, which is near the C12 of the dodecanoyl chain, the tunnel splits into two directions. In one direction, it extends along strand B9 to Leu-290, whose dimethyl carbons are 5.5 Å from C12 of the dodecanoyl chain. In the other direction, it bends into a highly hydrophobic groove made by the side chains of Leu-201, Val-209, Trp-211, Trp-230, and Tyr-348. At the distal end, it is terminated by the side chains of Val-197 and Pro-198. The bottom of this groove is about 9.4 Å from C12 of the dodecanoyl chain, raising the possibility that the enzyme could accommodate a longer fatty acid substrate. We compared the fatty acid binding tunnels of the very long chain fatty acyl-CoA synthetase M. tuberculosis FACL13 (active on fatty acid with up to 26 carbons) (29), FadD10, and the human medium chain fatty acyl-CoA synthetase (active on fatty acid with up to 10 carbons) (33). The linear distances between the distal end and the portal of their fatty acyl binding tunnels are ∼17, 14, and 10 Å, respectively, which indicates that the biological substrate of FadD10 is probably a long chain fatty acid. Based on the geometric modeling and taking into account of the contact distance associated with atomic van der Waals radii, the fatty acid binding tunnel in FadD10 could accommodate at most 16 carbons (i.e. hexadecanoic acid).
To further determine the fatty acid specificity of FadD10, we incubated M. tuberculosis FadD10 and the holo-ACP (Rv0100), with fatty acids of varying chain length and degree of saturation, and we then analyzed the results for the acylation of ACP (Rv0100) using ESI-QTOF mass spectrometry. We observed acylated ACP even when hexadecanoic acid was used as a substrate (Fig. 5C). This is consistent with the modeling study that shows a hexadecyl chain can be accommodated in the fatty acid binding tunnel of FadD10 and the study by Chhabraa et al. (16) showing that FadD10 could utilize fatty acids with up to 16 carbons in the adenylation reaction. The lower product yield for hexadecanoic acid could reflect differences in solubility or affinity. We have also observed a higher activity of FadD10 with hexadecanoic acid than 2-trans-hexadecanoic acid, which suggests that FadD10 prefers saturated fatty acid substrates. We previously identified Rv0098 as a long chain (C12–C18) fatty acyl-CoA thioesterase. Taken together with the structural analyses and enzymatic characterization of FadD10 (Rv0099), it suggests that the lipopeptide produced by the M. tuberculosis Rv0096–Rv0101 operon incorporates a long chain fatty acid, particularly tetradecanoic acid that demonstrated the highest activity to acylate the ACP (Rv0100).
ATP-dependent amide bond synthesis activity has been previously reported for the short chain acyl-CoA synthetases as well as for firefly luciferase (44). Specifically, this was observed when cysteine analogues, including d-cysteine, homocysteine, and l-cysteine, were used as substrates to react with the preferred acid substrate of each enzyme. N-Acylated cysteine derivatives were detected (44). We have observed a similar activity in FadD10 using cysteine as a substrate (Fig. 5D). Unlike the side reactions observed in short chain acyl-CoA synthetases and firefly luciferase, whose amide forming activity is limited to cysteine analogues (44), FadD10 is promiscuous with respect to other amino acids, specifically histidine, aspartate, glycine, and phenylalanine as we have tested (data not shown). Other amide synthetases belonging to the adenylate-forming superfamily have been reported. NovL, CloL, CouL, and SimL involved in novobiocin, clorobiocin, coumermycin A1, and simocyclinone D8 synthesis, respectively, can catalyze the formation of an amide bond between the amino group on the aminocoumarin ring and a carboxylate moiety through forming an adenylated intermediate (45–48). Similar to fadD10, these four enzymes are in proximity to and cooperate with NRPS-encoding genes. This suggests that amide formation, a side reaction to thioesterification of adenylate-forming enzymes, may evolve into a major biological function.
Superposition of FadD10 with acyl-CoA synthetase complexed with coenzyme A derivatives shows that the phosphopantetheine binding cavity is in the N-terminal domain of FadD10. In our two structures, the continuity between the phosphopantetheine cavity and the fatty acyl binding tunnel is blocked by the side chain of His-225. A conserved aromatic residue (either histidine, phenylalanine, or tryptophan), equivalent to His-225 in FadD10, is found in the sequences of all of the adenylate-forming enzymes. It is thought to function in the proper positioning of a fatty acid substrate into its cognate binding tunnel instead of extending into the phosphopantetheine-binding site. More clearly described in the human medium chain fatty acyl CoA synthase (33), the indole ring of Trp-265 (equivalent to His-225 in FadD10) rotates about 180° relative to the main chain in the ATP- or coenzyme A-binding state. This action switches off and on the connection between the fatty acid binding tunnel and the phosphopantetheine binding cavity. FadD10 should adopt a similar scheme to reorient His-225 in the context of the overall reaction to allow for the extension of the phosphopantetheine terminus of the ACP (Rv0100) into the fatty acid-binding site.
Conclusion
We have determined the apo structure and dodecanoyl-AMP-bound structures of M. tuberculosis FadD10, leading to the characterization of a new type of adenylate-forming enzyme. FadD10, independent of the presence of ligand, adopts and maintains an open conformation wherein the inter-domain orientation prevents the binding of coenzyme A. Although M. tuberculosis FadD10 has a primary sequence similar to FACLs, we have clearly shown that it is indeed an FAAL that is only able to acylate an ACP (Rv0100) rather than coenzyme A. This activity is consistent with the structural features of FadD10 and is in agreement with the operon organization of M. tuberculosis Rv0099 (FadD10)–Rv0101 (nrp), an assembly line for the production of lipopeptide(s). Because many similar gene clusters, involving an FAAL, an ACP, and an NRPS, have been observed in the synthetic pathways for bioactive lipopeptides (49–54), it is therefore very likely that enzymes with similar mechanisms and structures to M. tuberculosis FadD10 will be discovered in nature.
Acknowledgments
We thank Dr. Li-Wei Hung at the Advanced Light Source for collecting the diffraction data of the apo-FadD10. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the United States Department of Energy under Contract DE-AC02–05CH11231. We thank Amir Safi and Tracey Musa for editing the manuscript and Dr. Inna Krieger, Anup Aggarwal, and Mallikarjun Lalgondar for helpful comments on the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant P01AIO68135. This work was also supported by Robert A. Welch Foundation Grant A-0015.
The atomic coordinates and structure factors (codes 4ISB and 4IR7) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- NRPS
- non-ribosomal peptide synthetase
- FAAL
- fatty acyl-AMP ligase
- FACL
- fatty acyl-CoA ligase
- ACP
- acyl carrier protein
- PDB
- Protein Data Bank
- r.m.s.d.
- root mean square deviation
- AMPPNP
- adenosine 5′-(β,γ-imino)triphosphate.
REFERENCES
- 1. Schmelz S., Naismith J. (2009) Adenylate-forming enzymes. Curr. Opin. Struct. Biol. 19, 666–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Trivedi O. A., Arora P., Sridharan V., Tickoo R., Mohanty D., Gokhale R. S. (2004) Enzymic activation and transfer of fatty acids as acyl-adenylates in mycobacteria. Nature 428, 441–445 [DOI] [PubMed] [Google Scholar]
- 3. Arora P., Goyal A., Natarajan V. T., Rajakumara E., Verma P., Gupta R., Yousuf M., Trivedi O. A., Mohanty D., Tyagi A., Sankaranarayanan R., Gokhale R. S. (2009) Mechanistic and functional insights into fatty acid activation in Mycobacterium tuberculosis. Nature Chem. Biol. 5, 166–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang Z., Zhou R., Sauder J. M., Tonge P. J., Burley S. K., Swaminathan S. (2011) Structural and functional studies of fatty acyl adenylate ligases from E. coli and L. pneumophila. J. Mol. Biol. 406, 313–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Duckworth B. P., Nelson K. M., Aldrich C. C. (2012) Adenylating enzymes in Mycobacterium tuberculosis as drug targets. Curr. Top. Med. Chem. 12, 766–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Camus J. C., Pryor M. J., Médigue C., Cole S. T. (2002) Re-annotation of the genome sequence of Mycobacterium tuberculosis H37Rv. Microbiology 148, 2967–2973 [DOI] [PubMed] [Google Scholar]
- 7. Rindi L., Bonanni D., Lari N., Garzelli C. (2004) Requirement of gene fadD33 for the growth of Mycobacterium tuberculosis in a hepatocyte cell line. New Microbiol. 27, 125–131 [PubMed] [Google Scholar]
- 8. Siméone R., Léger M., Constant P., Malaga W., Marrakchi H., Daffé M., Guilhot C., Chalut C. (2010) Delineation of the roles of FadD22, FadD26, and FadD29 in the biosynthesis of phthiocerol dimycocerosates and related compounds in Mycobacterium tuberculosis. FEBS J. 277, 2715–2725 [DOI] [PubMed] [Google Scholar]
- 9. Dunphy K. Y., Senaratne R. H., Masuzawa M., Kendall L. V., Riley L. W. (2010) Attenuation of Mycobacterium tuberculosis functionally disrupted in a fatty acyl-coenzyme A synthetase gene fadD5. J. Infect. Dis. 201, 1232–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Casabon I., Crowe A. M., Liu J., Eltis L. D. (2012) FadD3 is an acyl-CoA synthetase that initiates catabolism of cholesterol rings C and D in actinobacteria. Mol. Microbiol. 87, 263–283 [DOI] [PubMed] [Google Scholar]
- 11. Léger M., Gavalda S., Guillet V., van der Rest B., Slama N., Montrozier H., Mourey L., Quémard A., Daffé M., Marrakchi H. (2009) The dual function of the Mycobacterium tuberculosis FadD32 required for mycolic acid biosynthesis. Chem. Biol. 16, 510–519 [DOI] [PubMed] [Google Scholar]
- 12. Gavalda S., Léger M., van der Rest B., Stella A., Bardou F., Montrozier H., Chalut C., Burlet-Schiltz O., Marrakchi H., Daffé M., Quémard A. (2009) The Pks13/FadD32 cross-talk for the biosynthesis of mycolic acids in Mycobacterium tuberculosis. J. Biol. Chem. 284, 19255–19264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goyal A., Verma P., Anandhakrishnan M., Gokhale R. S., Sankaranarayanan R. (2012) Molecular basis of the functional divergence of fatty acyl-AMP ligase biosynthetic enzymes of Mycobacterium tuberculosis. J. Mol. Biol. 416, 221–238 [DOI] [PubMed] [Google Scholar]
- 14. Wang F., Langley R., Gulten G., Wang L., Sacchettini J. (2007) Identification of a type III thioesterase reveals the function of an operon crucial for Mtb virulence. Chem. Biol. 14, 543–551 [DOI] [PubMed] [Google Scholar]
- 15. Hotter G. S., Wards B. J., Mouat P., Besra G. S., Gomes J., Singh M., Bassett S., Kawakami P., Wheeler P. R., de Lisle G. W., Collins D. M. (2005) Transposon mutagenesis of Mb0100 at the ppe1-nrp locus in Mycobacterium bovis disrupts phthiocerol dimycocerosate (PDIM) and glycosylphenol-PDIM biosynthesis, producing an avirulent strain with vaccine properties at least equal to those of M. bovis BCG. J. Bacteriol. 187, 2267–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chhabra A., Haque A. S., Pal R. K., Goyal A., Rai R., Joshi S., Panjikar S., Pasha S., Sankaranarayanan R., Gokhale R. S. (2012) Nonprocessive [2 + 2]e- off-loading reductase domains from mycobacterial nonribosomal peptide synthetases. Proc. Natl. Acad. Sci. U.S.A. 109, 5681–5686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Billman-Jacobe H., McConville M. J., Haites R. E., Kovacevic S., Coppel R. L. (1999) Identification of a peptide synthetase involved in the biosynthesis of glycopeptidolipids of Mycobacterium smegmatis. Mol. Microbiol. 33, 1244–1253 [DOI] [PubMed] [Google Scholar]
- 18. Rengarajan J., Bloom B. R., Rubin E. J. (2005) Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc. Natl. Acad. Sci. U.S.A. 102, 8327–8332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sassetti C. M., Rubin E. J. (2003) Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. U.S.A. 100, 12989–12994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Raman S., Puyang X., Cheng T. Y., Young D. C., Moody D. B., Husson R. N. (2006) Mycobacterium tuberculosis SigM positively regulates Esx secreted protein and nonribosomal peptide synthetase genes and down regulates virulence-associated surface lipid synthesis. J. Bacteriol. 188, 8460–8468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Röttig M., Medema M. H., Blin K., Weber T., Rausch C., Kohlbacher O. (2011) NRPSpredictor2–a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 39, W362–W367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gulick A. M. (2009) Conformational dynamics in the acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem. Biol. 4, 811–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 24. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 26. Vagin A., Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 27. Gibrat J. F., Madej T., Bryant S. H. (1996) Surprising similarities in structure comparison. Curr. Opin. Struct. Biol. 6, 377–385 [DOI] [PubMed] [Google Scholar]
- 28. Conti E., Franks N. P., Brick P. (1996) Crystal structure of firefly luciferase throws light on a superfamily of adenylate-forming enzymes. Structure 4, 287–298 [DOI] [PubMed] [Google Scholar]
- 29. Andersson C. S., Lundgren C. A., Magnúsdóttir A., Ge C., Wieslander A., Martinez Molina D., Högbom M. (2012) The Mycobacterium tuberculosis very-long-chain fatty acyl-CoA synthetase: structural basis for housing lipid substrates longer than the enzyme. Structure 20, 1062–1070 [DOI] [PubMed] [Google Scholar]
- 30. May J. J., Kessler N., Marahiel M. A., Stubbs M. T. (2002) Crystal structure of DhbE, an archetype for aryl acid activating domains of modular nonribosomal peptide synthetases. Proc. Natl. Acad. Sci. U.S.A. 99, 12120–12125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gulick A. M., Lu X., Dunaway-Mariano D. (2004) Crystal structure of 4-chlorobenzoate:CoA ligase/synthetase in the unliganded and aryl substrate-bound states. Biochemistry 43, 8670–8689 [DOI] [PubMed] [Google Scholar]
- 32. Hisanaga Y., Ago H., Nakagawa N., Hamada K., Ida K., Yamamoto M., Hori T., Arii Y., Sugahara M., Kuramitsu S., Yokoyama S., Miyano M. (2004) Structural basis of the substrate-specific two-step catalysis of long chain fatty acyl-CoA synthetase dimer. J. Biol. Chem. 279, 31717–31726 [DOI] [PubMed] [Google Scholar]
- 33. Kochan G., Pilka E. S., von Delft F., Oppermann U., Yue W. W. (2009) Structural snapshots for the conformation-dependent catalysis by human medium-chain acyl-coenzyme A synthetase ACSM2A. J. Mol. Biol. 388, 997–1008 [DOI] [PubMed] [Google Scholar]
- 34. Shah M. B., Ingram-Smith C., Cooper L. L., Qu J., Meng Y., Smith K. S., Gulick A. M. (2009) The 2.1 Å crystal structure of an acyl-CoA synthetase from Methanosarcina acetivorans reveals an alternate acyl-binding pocket for small branched acyl substrates. Proteins 77, 685–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yonus H., Neumann P., Zimmermann S., May J. J., Marahiel M. A., Stubbs M. T. (2008) Crystal structure of DltA. Implications for the reaction mechanism of non-ribosomal peptide synthetase adenylation domains. J. Biol. Chem. 283, 32484–32491 [DOI] [PubMed] [Google Scholar]
- 36. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 37. Marahiel M. A., Stachelhaus T., Mootz H. D. (1997) Modular peptide synthetases involved in nonribosomal peptide synthesis. Chem. Rev. 97, 2651–2674 [DOI] [PubMed] [Google Scholar]
- 38. Fraga H., Esteves da Silva J. C., Fontes R. (2004) Identification of luciferyl adenylate and luciferyl coenzyme a synthesized by firefly luciferase. ChemBioChem 5, 110–115 [DOI] [PubMed] [Google Scholar]
- 39. Linne U., Schäfer A., Stubbs M. T., Marahiel M. A. (2007) Aminoacyl-coenzyme A synthesis catalyzed by adenylation domains. FEBS Lett. 581, 905–910 [DOI] [PubMed] [Google Scholar]
- 40. Gulick A. M., Starai V. J., Horswill A. R., Homick K. M., Escalante-Semerena J. C. (2003) The 1.75 A crystal structure of acetyl-CoA synthetase bound to adenosine-5′-propylphosphate and coenzyme A. Biochemistry 42, 2866–2873 [DOI] [PubMed] [Google Scholar]
- 41. Mohanty D., Sankaranarayanan R., Gokhale R. S. (2011) Fatty acyl-AMP ligases and polyketide synthases are unique enzymes of lipid biosynthetic machinery in Mycobacterium tuberculosis. Tuberculosis 91, 448–455 [DOI] [PubMed] [Google Scholar]
- 42. Mitchell C. A., Shi C., Aldrich C. C., Gulick A. M. (2012) Structure of PA1221, a nonribosomal peptide synthetase containing adenylation and peptidyl carrier protein domains. Biochemistry 51, 3252–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lambert C., Léonard N., De Bolle X., Depiereux E. (2002) ESyPred3D: Prediction of protein 3D structures. Bioinformatics 18, 1250–1256 [DOI] [PubMed] [Google Scholar]
- 44. Abe T., Hashimoto Y., Hosaka H., Tomita-Yokotani K., Kobayashi M. (2008) Discovery of amide (peptide) bond synthetic activity in acyl-CoA synthetase. J. Biol. Chem. 283, 11312–11321 [DOI] [PubMed] [Google Scholar]
- 45. Luft T., Li S. M., Scheible H., Kammerer B., Heide L. (2005) Overexpression, purification, and characterization of SimL, an amide synthetase involved in simocyclinone biosynthesis. Arch. Microbiol. 183, 277–285 [DOI] [PubMed] [Google Scholar]
- 46. Steffensky M., Li S. M., Heide L. (2000) Cloning, overexpression, and purification of novobiocic acid synthetase from Streptomyces spheroides NCIMB 11891. J. Biol. Chem. 275, 21754–21760 [DOI] [PubMed] [Google Scholar]
- 47. Pojer F., Li S. M., Heide L. (2002) Molecular cloning and sequence analusis of the clorobiocin biosynthetic gene cluster: new insights into the biosynthesis of aminocoumarin antibiotics. Microbiology 148, 3901–3911 [DOI] [PubMed] [Google Scholar]
- 48. Schmutz E., Steffensky M., Schmidt J., Porzel A., Li S. M., Heide L. (2003) An unusual amide synthetase (CouL) from the coumermycin A1 biosynthetic gene cluster from Streptomyces rishiriensis DSM 40489. Eur. J. Biochem. 270, 4413–4429 [DOI] [PubMed] [Google Scholar]
- 49. Miao V., Brost R., Chapple J., She K., Gal M. F., Baltz R. H. (2006) The lipopeptide antibiotic A54145 biosynthetic gene cluster from Streptomyces fradiae. J. Ind. Microbiol. Biotechnol. 33, 129–140 [DOI] [PubMed] [Google Scholar]
- 50. Hansen D. B., Bumpus S. B., Aron Z. D., Kelleher N. L., Walsh C. T. (2007) The loading module of mycosubtilin: an adenylation domain with fatty acid selectivity. J. Am. Chem. Soc. 129, 6366–6367 [DOI] [PubMed] [Google Scholar]
- 51. Miao V., Coëffet-Legal M. F., Brian P., Brost R., Penn J., Whiting A., Martin S., Ford R., Parr I., Bouchard M., Silva C. J., Wrigley S. K., Baltz R. H. (2005) Daptomycin biosynthesis in Streptomyces roseosporus: cloning and analysis of the gene cluster and revision of peptide stereochemistry. Microbiology 151, 1507–1523 [DOI] [PubMed] [Google Scholar]
- 52. Duitman E. H., Hamoen L. W., Rembold M., Venema G., Seitz H., Saenger W., Bernhard F., Reinhardt R., Schmidt M., Ullrich C., Stein T., Leenders F., Vater J. (1999) The mycosubtilin synthetase of Bacillus subtilis ATCC6633: a multifunctional hybrid between a peptide synthetase, an amino transferase, and a fatty acid synthase. Proc. Natl. Acad. Sci. U.S.A. 96, 13294–13299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Heinzelmann E., Berger S., Müller C., Härtner T., Poralla K., Wohlleben W., Schwartz D. (2005) An acyl-CoA dehydrogenase is involved in the formation of the Δcis3 double bond in the acyl residue of the lipopeptide antibiotic friulimicin in Actinoplanes friuliensis. Microbiology 151, 1963–1974 [DOI] [PubMed] [Google Scholar]
- 54. Baltz R. H., Brian P., Miao V., Wrigley S. K. (2006) Combinatorial biosynthesis of lipopeptide antibiotics in Streptomyces roseosporus. J. Ind. Microbiol. Biotechnol. 33, 66–74 [DOI] [PubMed] [Google Scholar]