

Background: The Plasmodium SUB1 protease is essential for erythrocyte egress and invasion of malaria parasites and is an attractive drug target.
Results: In silico screening on three-dimensional models selected a P. vivax SUB1-competitive inhibitor, active against P. falciparum and P. berghei.
Conclusion: Combined virtual screening and biological validation identified a promising hit.
Significance: Targeting SUB1 could lead to a globally active antimalarial.
Keywords: Malaria, Parasitology, Protease, Protease Inhibitor, Structural Biology, Plasmodium vivax, Drug Target, Egress, Subtilisin, Virtual Screening
Abstract
Widespread drug resistance calls for the urgent development of new antimalarials that target novel steps in the life cycle of Plasmodium falciparum and Plasmodium vivax. The essential subtilisin-like serine protease SUB1 of Plasmodium merozoites plays a dual role in egress from and invasion into host erythrocytes. It belongs to a new generation of attractive drug targets against which specific potent inhibitors are actively searched. We characterize here the P. vivax SUB1 enzyme and show that it displays a typical auto-processing pattern and apical localization in P. vivax merozoites. To search for small PvSUB1 inhibitors, we took advantage of the similarity of SUB1 with bacterial subtilisins and generated P. vivax SUB1 three-dimensional models. The structure-based virtual screening of a large commercial chemical compounds library identified 306 virtual best hits, of which 37 were experimentally confirmed inhibitors and 5 had Ki values of <50 μm for PvSUB1. Interestingly, they belong to different chemical families. The most promising competitive inhibitor of PvSUB1 (compound 2) was equally active on PfSUB1 and displayed anti-P. falciparum and Plasmodium berghei activity in vitro and in vivo, respectively. Compound 2 inhibited the endogenous PfSUB1 as illustrated by the inhibited maturation of its natural substrate PfSERA5 and inhibited parasite egress and subsequent erythrocyte invasion. These data indicate that the strategy of in silico screening of three-dimensional models to select for virtual inhibitors combined with stringent biological validation successfully identified several inhibitors of the PvSUB1 enzyme. The most promising hit proved to be a potent cross-inhibitor of PlasmodiumSUB1, laying the groundwork for the development of a globally active small compound antimalarial.
Introduction
Effective drugs are a critical component of malaria control. Selection of Plasmodium sp. parasites resistant to multiple drugs calls for accelerated efforts to develop new anti-malarial drugs targeting novel essential parasite pathways. In the context of the agenda for malaria eradication (1), future drugs should ideally target different stages of the life cycle and be active on the various Plasmodium sp., in particular Plasmodium falciparum and Plasmodium vivax. Two main approaches have been successfully used to identify new compounds with anti-parasite activity. The first consists of phenotypic screening of chemical libraries looking for molecules that inhibit the growth of P. falciparum (2–4) or Plasmodium yoelii (53) cultures. The second approach aims at targeting a particular parasite-specific metabolic pathway (5) or an essential parasite enzyme.
Proteases are known to play an important role in numerous pathways and represent potent drug targets for several chronic infectious diseases. Anti-proteases have indeed changed the course of the epidemic of AIDS, and equivalent efficiency is expected in the treatment of hepatitis C (6). Over the last decades, Plasmodium proteases have been extensively studied and shown to play a role in hydrolysis of host erythrocyte hemoglobin in the parasite food vacuole (7–9), maturation of proteins for export into the host erythrocyte (10–13), or in the egress and subsequent invasion of merozoites (14). They are considered attractive drug target candidates.
Several parasite cysteine and serine proteases were shown to participate in the cascade of events that orchestrate the egress of merozoites from and their subsequent entry into erythrocytes (14, 15). The subtilisin-like serine protease PfSUB1 plays an essential role in both merozoite egress and invasion. Following its secretion into the parasitophorous vacuole, PfSUB1 carries out the maturation of papain-like proteases called the serine-rich repeat antigens, which, once activated, are thought to participate in the rupture of the parasitophorous vacuole and/or the infected erythrocyte membrane (16). In addition, PfSUB1 is involved in the maturation of several merozoite surface proteins, including MSP1, MSP6, and MSP7 (17), priming merozoites for invasion of erythrocytes.
The predicted active site of the various Plasmodium SUB1 orthologs displays similarity to the active sites of bacterial subtilisins but differs from that of host enzymes, a feature shared with the second merozoite subtilisin-like protease PfSUB2 (18). We and others exploited this similarity to obtain three-dimensional models of the Pf/PvSUB1 and PfSUB2 catalytic domains, built by homology with three-dimensional crystal structures of bacterial subtilisins (18–21).
In this study, we characterize the P. vivax-SUB1 (PvSUB1) and show that it displays similar cellular location, auto-processing, and enzymatic properties as its PfSUB1 ortholog. To search for potential PvSUB1 inhibitors, we used an in silico screening approach using three-dimensional models of PvSUB1 as targets. Because the predictive performance of in silico screening strongly depends on the accuracy of the protein's models, we built a suite of homology models to perform a structure-based in silico screening of a large library of small molecular mass compounds. This allowed selection of a set of compounds enriched in potentially active molecules, limiting the number of compounds to be tested experimentally, reducing time and cost to a manageable scale. Thus, we selected the 306 best predicted hits and tested their inhibitory potency on the PvSUB1 recombinant enzyme. Active compounds were then tested for their anti-parasite properties on P. falciparum blood stages in culture. The anti-parasite activity of the most promising compound was evaluated in vivo on Plasmodium berghei-infected mice, and its specific activity against the P. falciparum merozoite egress and invasion steps was demonstrated.
EXPERIMENTAL PROCEDURES
Production and Purification of the PfSUB1 and PvSUB1 Recombinant Enzymes
The sequence coding for PvSUB1, amplified from P. vivax genomic DNA (GenBankTM accession number FJ536584, Belem strain), and the codon-optimized Pfsub1 and Pbsub1 synthetic coding sequences (Genscript) were cloned into the BamHI restriction site of the pAcGP67A and pAcGP67B plasmids (BD Biosciences), respectively, downstream from and in phase with the baculovirus glycoprotein 67 signal sequence (GenBankTM accession numbers of the resulting constructs are FJ536585, JX491486, and JX848551, respectively). Spodoptera frugiperda Sf9 insect cells (Invitrogen) were grown in monolayer or suspension cultures at 27 °C, in Insect XPRESS (Lonza) supplemented with 4 mm glutamine (Invitrogen), 5% fetal bovine serum, 50 μg/ml gentamicin. We generated recombinant baculoviruses with the BaculoGold (BD Biosciences) transfection and expression system according to the manufacturer's instructions. Viral stocks were produced in Sf9 insect cells, stored at 4 °C, and amplified in 150-cm2 T flasks. The viral stock titer was obtained by end point dilution assay. For large scale protein production, Sf9 cells (1 liter at 3 × 106 cells/ml) were infected for 72 h with recombinant baculovirus at a multiplicity of infection of 10 in Insect XPRESS medium supplemented with 50 μg/ml gentamycin and 0.5 μg/ml tunicamycin (Sigma).
Culture supernatant containing the secreted PvSUB1, PfSUB1, or PbSUB1 recombinant proteins (PvSUB1r, PfSUB1r, and PbSUB1r, respectively) was harvested, centrifuged 30 min at 2150 × g to remove cells, and concentrated/diafiltrated against D-PBS 0.5 m NaCl, 5 mm imidazole (loading buffer). The proteins were purified on an AKTA purifier system (GE Healthcare). The sample was loaded onto a 3-ml TALON metal affinity resin (Clontech) equilibrated in loading buffer. After extensive washes with loading buffer, the bound protein was eluted with a linear gradient of 5–200 mm imidazole in D-PBS, 0.5 m NaCl. Fractions containing PvSUB1r, PfSUB1r, or PbSUB1r were pooled and concentrated by using Amicon Ultra 15 (molecular weight cutoff of 10,000) and size-fractionated onto a HiLoad 16/60 Superdex 75 column equilibrated with 20 mm Tris, pH 7.5, 100 mm NaCl. Fractions were monitored by absorbance (280 nm) and analyzed by Coomassie Blue staining of SDS-polyacrylamide gels and enzyme activity assay. The fractions containing the recombinant enzyme activity were pooled. Protein concentration was determined with the BCA protein assay following the manufacturer's recommendations (Bio Basic). Purified PvSUB1r, PbSUB1r, or PfSUB1r recombinant proteins were stored at −20 °C following the addition of 30% v/v of pure glycerol.
Three-dimensional modeling of PvSUB1 and PfSUB1 Catalytic Domains
The homology modeling procedure was performed with the suite of tools accessible through BisKit (22). The algorithms used for multiple alignments and for homology modeling are critical to build a reliable model. BisKit relies on 3D-Coffee (23) and Modeler (24), respectively. This choice was guided by the fact that the combination of Tcoffee (from which 3D-Coffee is derived) and Modeler produces particularly accurate models (25). The modeling of the PvSUB1 (Uniprot ID E6Y8B9) and PfSUB1 (Uniprot ID Q868D6) enzymes was restricted to their catalytic domain (residues Ile-302 to Pro-586 and Ile-360 to Pro-645, respectively) (supplemental Fig. 1).
To take advantage of the improved model accuracy when several structural templates are used simultaneously (26), we selected from the Protein Data Bank (PDB)6 a set of structures from homologs of the PvSUB1 and PfSUB1 catalytic domains. We considered only the most similar PDB entries associated with a blast e-value lower than 0.001 (27) and a resolution below 2 Å. Among them, we selected seven nonredundant structures as templates for homology modeling of PvSUB1 and PfSUB1 (PDB codes 2TEC, 1DBI, 1CGI, 1R0R, 1LW6, 1EA7, and 1IC6 with a resolution of 1.98, 1.80, 0.78, 1.10, 1.50, 0.93, and 0.98 Å respectively).
To propose a reliable multiple sequence alignment of these templates with PfSUB1 and PvSUB1 catalytic domains, we also used additional protein sequences in the alignment step. Thus, protein sequences displaying significant similarities to PfSUB1 and PvSUB1 were searched in the nonredundant sequence data bank of Swiss-Prot (ncbi.nlm.nih.gov). 73 additional homologous sequences were added to the seven template sequences for an optimal multiple sequence alignment of PfSUB1. Similarly, sequence alignment was performed for PvSUB1 with 50 additional homologous sequences. Multiple alignments were performed with 3D-Coffee, which uses structural alignments in addition to local and global sequence alignments to optimize the overall alignment of all sequences (23). For both the PfSUB1 and PvSUB1 catalytic domains, 50 models were built from their respective sequence alignments with the seven template structures with Modeler 7v7 (24).
Set Up of the Chemical Database
Virtual screening was applied on the ChemDiv chemical library, one of the largest commercial chemo-libraries available. The 508,856 molecules were filtered with the program Filter (Openeye) using standard parameters to exclude predicted aggregators and toxic compounds and to enrich the selection in “drug-like” compounds. The 149,992 selected compounds were converted into three-dimensional energy-minimized conformers with Corina (28) and used as entries in virtual screens by docking.
Virtual Screens
The ICM (29) and FlexX (30) docking programs were used to extract in silico hits from the above filtered 149,992 drug-like compounds. Indeed, prediction performances of docking programs have been shown to strongly depend on the studied biological system, and it is therefore advisable not to rely on a single program. Screens were performed with standard procedures; the 149,992 compounds were docked into a rigid binding site and ranked according to their docking scores. The docking score of a molecule is expected to reflect its affinity for the binding site, and therefore, top-ranked compounds were selected. Both programs consider ligands as flexible and binding sites as rigid. Conformational searches for ligands consider dihedral rotations and potential enantiomers and stereoisomers. ICM was applied with its standard parameters. Monte Carlo simulations were done with a thoroughness set to 1, and molecules were ranked with respect to the VLS scoring function (31). In parallel, we used FlexX and FleX-Pharm (32) to select a second pool of compounds.
PvSUB1r, PfSUB1r, and PbSUB1r Enzymatic Assays
PvSUB1r, PfSUB1r, and PbSUB1r enzymatic assays were performed as described (19) using fluorescence resonance energy transfer (FRET) substrates of PvSUB1 and PfSUB1 corresponding to their auto-maturation sequences, KLVGADDVSLA and KLVSADNIDIS, respectively. Both sequences were coupled to the fluorophore/quencher dyes Dabsyl/Edans (PvSUB1-DE or PfSUB1-DE) (excitation/emission 360/500 nm) or to the DYQ660/DY630, which fluoresce in the far-red wavelength (excitation/emission 620/680 nm) (PvSUB1-FR or PfSUB1-FR). Substrates were custom made by ThermoFisher Scientific. The enzymatic assays were performed in 20 mm Tris, pH 7.5, 25 mm CaCl2 at 37 °C. The apparent Km (Km, app) of each enzyme was obtained by following the determination of the initial velocity (Vi) of the reaction in the presence of eight different concentrations ranging from 250 to 2 μm for the Dabsyl/Edans substrates and from 80 to 0.625 μm for the DYQ660/DY630 substrates. The final mixture was distributed in duplicate into a 384-well black microtiter plate (Thermo Scientific), and the fluorescence was monitored every minute for 1 h at 37 °C with a Tecan Infinite M1000 spectrofluorimeter.
The apparent Ki (Ki, app) was determined from the analysis of the Pv/Pf/PbSUB1r activity in presence of 10 concentrations of the compounds ranging from 300 to 0.58 μm. To determine the inhibition mechanism of compound (Cpd) 2 against PvSUB1r, four different concentrations of the molecule ranging from 50 to 0.4 μm were tested in the presence of five different concentrations of the PvSUB1-FR substrate as follows: 40, 30, 20, 10, and 5 μm. All the enzymatic data were analyzed and obtained with Prism Version 5 software (GraphPad).
Parasite Culture and in Vitro and in Vivo Drug Susceptibility Assays
P. falciparum reference clones 3D7 (chloroquine-sensitive) and Dd2 (multidrug-resistant) obtained from MR4 were cultured in vitro in RPMI 1640 medium containing l-glutamine, 25 mm HEPES (Invitrogen) supplemented with 10% decomplemented human serum (AB+), 100 μm hypoxanthine (C.C.pro, Germany), 50 μg/ml gentamycin (Sigma). Parasites were cultured at 37 °C in a 5% O2, 5% CO2, and 90% N2 atmosphere. Quantitative assessment of the antimalarial activity of the selected compounds was performed as described (33) on asynchronous culture of P. falciparum clones 3D7 and Dd2 (0.5% parasitemia and 1% hematocrit), except that the parasites were in contact with the drug for 48 h and the culture medium contained 10 μm hypoxanthine. EC50 were determined by following nonlinear regression analysis with HN-NonLin Version 1.1 software.
The P. falciparum merozoite egress/invasion assay was performed as described (19, 34). Briefly, compounds to be tested were added to highly synchronized P. falciparum-3D7 segmented schizonts (0.5% parasitemia and 1% hematocrit) cultured in a 24-well plate. Aliquots of the culture at the beginning (T0) and after 12 h of culture were collected and fixed in PBS (Dulbecco), 0.04% glutaraldehyde before flow cytometry analysis. E64 (Sigma), a cysteine-protease inhibitor known to block the egress of P. falciparum merozoites in vitro (35), was used at a 10 μm final concentration, whereas Cpd2 was tested in the 60 to 1 μm range, and a mock control received 1% of sterile DMSO, the Cpd2 vehicle. Following YOYO-1 staining (36), parasitemia was assessed by flow cytometry with a FACSCalibur cytometer, and data were analyzed with FlowJo (Tree Star) software (34). The YOYO-1 fluorescence signal of dying cells was collected in the FL-1 channel after compensation of fluorescent intensity in the FL-2 channel.
In vivo anti-parasite activity was determined against the P. berghei ANKA-GFP clone (37) by using the 4-day suppressive test (38, 39). Briefly, 3-week-old Swiss mice were inoculated intraperitoneally with 2·106 parasitized red blood cells and randomly allocated to six groups of five mice. Cpd2 dissolved in 70:30 v/v Tween 80/ethanol (Sigma) and diluted 10-fold in sterile water was administered in 100, 66, or 33 mg/kg dose per mouse. Control mice received the vehicle (water containing 7% of Tween 80, 3% of ethanol). Two groups of mice received 10 and 2 mg/kg chloroquine. All treatments were administered intraperitoneally daily for 4 consecutive days. The day after the last treatment, parasitemia was determined by flow cytometry with a FACSCalibur cytometer. The signal emitted by GFP parasites was collected in the FL-1 channel, and the data were analyzed with FlowJo software.
P. vivax Parasites
P. vivax parasites were collected from Cambodian malaria patients and matured at 37 °C for 48 h in vitro in complete RPMI medium, in an atmosphere composed of 3% oxygen before purification on a Percoll gradient. Ethical clearance for collection of patients' isolates was obtained from the Cambodian National Ethics Committee for Health Research (IRB number 160 NECHR, October 28, 2010). Informed written consent was provided by all patients or their parents/guardians before inclusion.
Immunoblotting and Indirect Immunofluorescence Assays
OF1 female mice (Charles River) were immunized subcutaneously with 10 μg of purified PvSUB1 emulsified with complete Freund's adjuvant (Sigma) for the first injection and incomplete Freund's adjuvant for two subsequent injections. Serum was collected 10 days after the third injection in Capiject T-MG tubes (Terumo) and kept in 50% glycerol at −20 °C.
P. falciparum synchronous cultures at the schizont stage (prepared as above) were collected and resuspended in XT loading buffer (Bio-Rad) in the presence of 100 mm DTT. For Fig. 6, the supernatants of mechanically disrupted synchronized P. falciparum segmented schizonts following the 12 h of culture have been used for immunoblot to reveal proteins located in the parasitophorous vacuole. Proteins were separated on 4–12% Criterion XT precast gels (Bio-Rad) and transferred on a Hybond nitrocellulose membrane (Amersham Biosciences). The primary anti-PvSUB1 polyclonal serum was used at a dilution of 1:400 in TBS containing 0.2% Tween 20 and 5% milk. The 24C6 mAb specific for SERA5 and the 1C11 mAb specific for the ubiquist HSP70 were used at a dilution of 1:500 and 1:1000, respectively. Anti-mouse IgG coupled to alkaline phosphatase (Promega) was used at a dilution of 1:10,000. Following extensive washing, the membrane was revealed by using the nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate substrates (Promega).
FIGURE 6.

Evaluation of Cpd2 activity on P. falciparum merozoite egress and invasion in vitro and on the maturation of PfSUB1 natural substrate PfSERA5. A, synchronized P. falciparum segmented schizonts were cultivated for 12 h in serum-containing medium in the presence of a range of concentrations of Cpd2 as indicated or E64 (10 μm). At the end of the incubation, newly formed young trophozoites were numbered by cytometry. Results show the mean of two independent experiments and are expressed as percent inhibition of the formation of young trophozoites compared with the mock-treated culture (1% DMSO as the vehicle of Cpd2), which was similar to untreated culture (not shown). B, immunoblot of protein extracts prepared from the supernatant of mechanically disrupted, synchronized P. falciparum segmented schizonts (starting material, lane 1) cultivated for 12 h in serum-free medium (lane 2), in the presence of the vehicle (1% DMSO, lane 3), 10 μm E64 (lane 4), and a range of concentrations of Cpd2 (1, 10, 30, and 60 μm for lanes 5–8, respectively). Blots were probed with the PfSERA5-specific mAb 24C6.1F1. The PfHSP70-specific mAb 1C11 was used as a loading control. The PfSERA5 processed forms p73, p56, and p50 kDa are indicated by an arrow. Molecular mass is indicated in kDa.
Macs-purified P. falciparum 3D7 segmented schizonts and P. vivax mature stages were washed in PBS, deposited on glass slides, and air-dried. Anti-PvSUB1 polyclonal serum and the anti-AMA1 rat mAb 28G2 were used at a 1:400 and 1:1000 dilution, respectively. Secondary antibodies Alexa Fluor 488-conjugated anti-mouse IgG and Alexa Fluor 594-conjugated anti-rat IgG (Invitrogen) were used diluted to 1:1000. Antibody solutions were prepared in PBS, 1% Albumax. Hoechst 33342 (Invitrogen) diluted at 1:5000 was added to the secondary antibody solution. PBS-washed slides were sealed with Vectashield (Vector Laboratories). Images were collected on a Leica DM5000B microscope with a ×1000 magnification.
RESULTS
Characterization of P. vivax-SUB1
The PvSUB1 sequence amplified from the P. vivax Belem strain encodes 630 residues, although the reference PvSUB1-Sal1 sequence has 619 residues. This reflects the absence from PvSUB1-Sal1 of 11 residues present in all known sequences of Plasmodium SUB1 orthologs and located in the center of the sequence coding for the SUB1 active site (supplemental Fig. 1). This deletion (amino acids 417–427 of PvSUB1-Belem) includes two cysteines involved in predicted di-sulfide bridges, as indicated by the PvSUB1 three-dimensional model (see below). This suggests a sequencing error for PvSUB1-Sal1. The full-length sequence of PvSUB1-Belem displays 56.5, 55.5, and 54.7% identity to PfSUB1, PbSUB1, and PySUB1, respectively. The closer conservation of PvSUB1 to PfSUB1, compared with the rodent orthologs, was more pronounced for the enzymatic domain, with 60.3, 55, and 55.9% identity to PfSUB1, PbSUB1, and PySUB1, respectively. In the four species, the seven cysteines were conserved (excluding PvSUB1-Sal1), and there is essentially no insertion nor deletion from the first to the last residue involved in catalysis, namely from Asp-316 to Ser-526, suggesting a strong positive pressure to secure SUB1 overall structure and enzymatic activity.
Coomassie Blue staining of SDS-PAGE identified four molecular species of the purified PvSUB1 recombinant enzyme (PvSUB1r) expressed in baculovirus-infected insect cells (labeled a–d, Fig. 1A). N-terminal sequencing showed that all derived from the PvSUB1r pro-enzyme, providing key information on the processing sites. The first processing site generating PvSUB1r-a (47 kDa), thought to be the result of auto-maturation, was located at the sequence KLVGA(D/D)VSL, a sequence similar to the processing site of PfSUB1r for the P6 to P1 positions, but less conserved for the P′ positions (supplemental Fig. 1 and Fig. 1A)(20). Maturation occurs at a similar sequence in the rodent Plasmodium-SUB1, as confirmed by the N-terminal sequencing of the 46-kDa form of the PbSUB1r (Fig. 1A), with the notable exception of the presence of a glutamic acid in the P6 position of PbSUB1, rather than the lysine observed in PvSUB1 and PfSUB1. This difference suggests a potential difference of substrate specificity, because the P6 position of the substrate could contribute to subtilase specificity (40). A second processing site was identified from the N-terminal sequence of PvSUB1r-b (42 kDa), namely PPSHA(A/S)S, which is absent from PbSUB1r (Fig. 1A). Interestingly, it differs from the VEN(D/A)E processing site reported for PfSUB1r-b (supplemental Fig. 1 and Fig. 1A) (20), which is absent from PvSUB1. A third processing generated PvSUB1r-c (39 kDa, Fig. 1A) by cleavage at the site HLA(G/S)K. This has not been reported for any other Plasmodium SUB1 enzymes. Whether or not it is a biologically relevant process or a cleavage resulting from the expression of PvSUB1 in a heterologous system remains unclear. Note that the purified PfSUB1r produced in the same expression system did not contain this additional band (Fig. 1A), consistent with other reports (20). Band PvSUB1r-d (Fig. 1A) corresponded to the N-terminal pro-region, following the removal of the gp67 recombinant signal peptide. The sequence of the PvSUB1r-d 25-kDa pro-region starts with ADPGDIL, in which the first three residues derive from pAcGP67A, the natural sequence of PvSUB1 starting with GDIL (supplemental Fig. 1). Thus, just as in PfSUB1 and PbSUB1 (20, 21), the recombinant enzymatic form of PvSUB1r co-purifies with its N-terminal pro-region, suggesting that strong interactions stabilize the two domains to form an holo-enzyme.
FIGURE 1.

Expression of PbSUB1r, PfSUB1r, and PvSUB1r and characterization of endogenous PvSUB1. A, Coomassie Blue staining of SDS-polyacrylamide gels of PbSUB1r, PfSUB1r, and PvSUB1r. Proteins were produced in baculovirus-infected insect cells, HPLC-purified, and concentrated (see “Experimental Procedures”). B, immunoblot of uninfected human erythrocytes (lane 1), P. falciparum 3D7 segmented schizonts (lane 2), and in vitro maturated P. vivax segmented schizonts (lane 3) probed with an anti-PvSUB1r serum. Molecular mass markers are in kDa. C, indirect immunofluorescence assays on air-dried P. falciparum 3D7 segmented schizonts (1st row of panels), individual merozoites (2nd row of panels), and P. vivax segmented schizonts (3rd and 4th rows of panels), probed with the anti-PvSUB1 polyclonal serum (green) and the anti-AMA1 rat monoclonal antibody 28G2 (red). Merozoite nuclei are labeled with Hoescht 33342. The scale bar represents 3 μm.
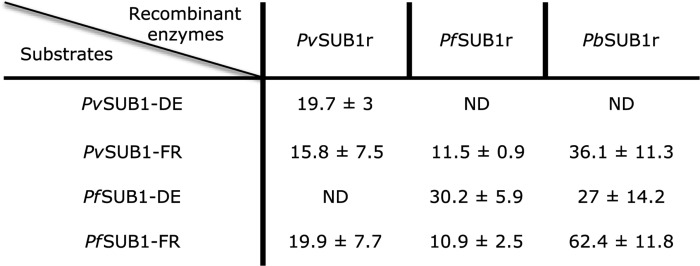
The enzymatic activity of PvSUB1r, PfSUB1r, and PbSUB1r was studied with FRET-based peptide substrates, whose sequence corresponded to the PvSUB1 and PfSUB1 primary processing site (site a), KLVGADDVSLA and KLVSADNIDIS, respectively. The apparent Km (Km, app) values of the Dabsyl/Edans (PvSUB1-DE or PfSUB1-DE) or DYQ660/DY630 (PvSUB1-FR or PfSUB1-FR)-coupled substrates were determined for PvSUB1r and PfSUB1r. Both enzymes were able to efficiently cleave both synthetic substrates with Km, app in the 10–30 μm range (Table 1). It is noteworthy that the Km, app was not significantly different for the two fluorophore/quencher pairs, reported to affect enzyme affinity for some synthetic substrates (41). Importantly, both enzymes had similar Km, app for the cognate and for the heterologous substrates. This is encouraging for future work as the PvSUB1 and PfSUB1 active sites only have 60.3% identity, and the substrates differ in their P′ sequence. It indicates that development of inhibitors equally effective on both enzymes is a realistic goal. Interestingly, the three tested Pv/PfSUB1 substrates had a lower affinity for PbSUB1r, with Km, app ranging from 27 to 62.4 μm (Table 1). This difference suggests, alongside the divergent sequence of its auto-processing site, that the rodent PbSUB1 may have slightly different substrate specificities.
TABLE 1.
Apparent Km of PvSUB1r, PfSUB1r, and PbSUB1r for synthetic FRET-based substrates
The apparent Km of PvSUB1-DE, PvSUB1-FR, PfSUB1-DE, and PfSUB1-FR for the three purified recombinant enzymes were calculated from the determination of the initial velocity (Vi) of the enzymatic reaction in presence of substrate concentrations ranging from 250 to 2 μm and 80 to 0.625 μm for the DE and FR substrates, respectively. The values are expressed in micromolars as the mean (± S.D.) of a minimum of three independent experiments. ND stands for not determined.
Mouse polyclonal serum raised against PvSUB1r cross-reacted on immunoblots of P. falciparum 3D7 segmented schizonts with the two previously reported forms of PfSUB1 at 54 and 47 kDa (Fig. 1B, lane 2) (42), and we identified two bands on protein extracts prepared from in vitro maturated P. vivax segmented schizonts (lane 3). These bands migrated with an apparent molecular mass similar to that of PvSUB1r-a and PvSUB1r-b (Fig. 1A) and with the deduced mass of the 47- and 42.8-kDa based PvSUB1 sequence, respectively, consistent with endogenous processed forms of PvSUB1. Interestingly, no endogenous PvSUB1 form equivalent to PvSUB1r-c could be detected, suggesting that either the latter is an artifact due to the expression in an heterologous system, or alternatively, this form represents a downstream step of maturation absent from the parasite preparation analyzed.
The anti-PvSUB1 polyclonal serum also cross-reacted with PfSUB1 by immunofluorescence on P. falciparum 3D7 segmented schizonts (Fig. 1C, upper P. falciparum panels) and free merozoites (lower P. falciparum panels) producing a typical punctate pattern and showing partial superposition with the signal produced by the anti-AMA1 mAb 28G2. This is in line with the subcellular localization of PfSUB1 and AMA1 in the exonemes and micronemes, respectively (16). A similar pattern was observed with air-dried P. vivax segmented schizonts (Fig. 1C). These data indicate conservation of the biochemical processing and subcellular localization of SUB1 in P. falciparum and P. vivax blood stages.
Homology Modeling of PvSUB1 and PfSUB1 Catalytic Domains
Models of the PvSUB1 and PfSUB1 catalytic domain were constructed based on seven bacterial subtilisin structures as templates (PDB entries 2TEC, 1DBI, 1CGI, 1R0R, 1LW6, 1EA7, and 1IC6, see under “Experimental Procedures” for details). These high resolution structures represented the best compromise in terms of resolution and sequence similarities with PfSUB1 and PvSUB1 catalytic domains, ranging from 22 to 35% and 25 to 36%, respectively (supplemental Table 1). Among them, PDB codes 1DBI, 1EA7, 1CGI, and 1IC6 are apo-structures (free state of the protein), whereas 1R0R, 1LW6, and 2TEC are holo-structures (bound states of the protein). When building a model with the aim of further docking studies into a receptor considered as rigid, it is recommended to select holo-structures (43). However, it has been shown that ligand binding to subtilisins does not induce significant structural rearrangements in the active site (44). The tight superposition of 21 structures of either apo or holo of the Carlsberg subtilisin (r.m.s.d. on heavy atoms <1 Å) confirmed the absence of significant conformational change upon ligand binding (data not shown) and led us to retain both holo- and apo-structures as templates to model PvSUB1 and PfSUB1 catalytic domains.
The quality of the 50 models of PvSUB1 catalytic domain obtained with Modeler was assessed with Procheck (45), and only a few residues were identified in a disabled region of the Ramachandran diagram. These residues were mainly situated in large loops corresponding to three large insertions (residues 330–353, 467–494, and 514–522) (46) in the PvSUB1 sequence (Fig. 2, A and B, and supplemental Fig. 1) but absent from the templates. This led to ab initio modeling of their structures by Modeler, although their sizes were too large to allow reliable structure prediction of these regions (47). As these loops were far from the binding site and not involved in the PvSUB1 catalytic groove, we considered that the lack of reliable information about their local topologies should not affect the subsequent docking studies. When excluding these insertion loops, the root mean square deviation (r.m.s.d.) observed between the models was equal to 2 Å on all backbone atoms, which corresponded to highly similar structures.
FIGURE 2.

Overall modeled three-dimensional structure of the PvSUB1 catalytic domain and predicted secondary structure. A, topology of the PvSUB1 catalytic domain. Numbering of the N- and C-terminal amino acids delimiting each secondary structure (purple) is indicated in black. Solid and open circles represent the putative disulfide bonds involving Cys-313–Cys-423 and Cys-402–Cys-419, respectively. Black squares indicate Ile-388, Val-383, and Asp-325 involved in the strong and conserved calcium-binding site. B, overall three-dimensional modelized structure of PvSUB1 catalytic domain (purple). The catalytic triad composed of Asp-316, His-372, and Ser-549 is shown in red. The loops formed by the three main conserved insertions in the SUB1 sequence compared with bacterial enzymes are indicated. The side chains of Ile-388, Val-383, and Asp-325 involved in the conserved strong calcium (orange sphere)-binding site are shown in gray. In green and blue are shown the residues forming the S1 and S4 sub-pockets that bind the P1 and P4 residues of the substrate. C, schematic representation of the PvSUB1 catalytic groove (purple) and its substrate (light gray). The residues forming the S1 and S4 sub-pockets are depicted in green and blue, respectively. The catalytic residues Asp-316, His-372, and Ser-549 are shown in red. D, FlexX docking pose of Cpd2 (turquoise carbon colored stick) within the PvSUB1 catalytic groove. Catalytic triad and residues participating in the S1 (green) and S4 (blue) sub-pockets are labeled in black.
The core structure of the modeled PvSUB1 catalytic domain contained a six-stranded parallel β-sheet (residues 311–316, 398–403, 430–433, 457–461, 497–503, and 526–529) corresponding to order 213456 with respect to the protein sequence (Fig. 2A). They were surrounded on both sides by two pairs of α-helices (residues 372–381 and 548–565 on one side and 413–426 and 442–454 on the other side). The model also contained one anti-parallel two-stranded β-sheet (residues 533–537 and 541–545), one α-helix (residues 571–581), and a structural feature conserved in the subtilisin family, namely two extended β-strands involving residues Ser-434–Phe-435–Ser-436 on the one side and Lys-409–Leu-410–Gly-11 on the other side (β′ and β″ in Fig. 2C), which can form an intermolecular anti-parallel β-sheet with the peptidic substrate. These strands are situated in a deep pocket that constitutes the catalytic groove of the enzyme active site, and their residues form hydrogen bonds principally with the P1–P4 residues of the substrate (40).
The PvSUB1-Belem sequence, used here as the prototypical PvSUB1, contains seven cysteine residues in positions Cys-313, Cys-402, Cys-419, Cys-423, Cys-465, Cys-478, and Cys-524 that are conserved in all known Plasmodium SUB1 sequences (supplemental Fig. 1). The four first cysteines are very close in space, which might indicate the presence of two disulfide bridges Cys-313/Cys-423 and Cys-402/Cys-419. They could stabilize the core of the protein by linking the α2 helix to the strand β3 and its parallel β1 one (Fig. 2A). Cys-465 and Cys-478 are located in the second large insertion loop (residues 467–494, supplemental Fig. 1) and are likely to form a disulfide bond. Several subtilisins contain one or more S–S bonds in various positions that stabilize the catalytic domain (40), but the three putative disulfide bounds of PvSUB1 are absent from the seven templates used to build the three-dimensional model.
The enzymatic activity of most of the subtilisins is strictly calcium-dependent, and this cation contributes to the structural stability of these enzymes. The crystal structures of the seven templates present up to four calcium ions. The strong affinity and widely conserved calcium-binding site 1, according to Siezen and Leunissen nomenclature (40), was clearly identified in the PvSUB1 models and would engage the backbone of the conserved residues Ile-388, Val-383, and the side chain of the Asp-325, as reported previously (Fig. 2A and supplemental Figs. 1 and 2) (20). The low pKa value of these groups (<4) indicates that they are most likely negatively charged, and electrostatic calculation shows a negative environment favorable for the binding of a cation. However, no clear information could be derived from the models about the positions of the three weaker sites mentioned by Siezen and Leunissen (40).
Superposition of the PvSUB1 three-dimensional models on the template structure outlines the large conserved groove that is characteristic of the substrate-binding site in subtilisins (Fig. 2B). Following the classical protease nomenclature established by Berger and Schechter (48), the P4–P2′ peptide of the protease substrate interacts with enzyme residues forming the S4, S3, S2, S1, S1′, and S2′ sites, a conserved organization illustrated by Siezen and Leunissen (40). The specificity of bacterial subtilisins is mainly due to the side chain of the substrate P4 and P1 residues (40, 49). A network of hydrogen bonds between residues P4 and P1 of the substrate and those of the enzyme S4 and S1 pockets of the catalytic groove is involved in the enzyme specificity and stabilizes the enzyme-substrate interaction. In contrast, the S3 and S2 sites are usually less distinct and form a smaller cleft, and as such they participate in the enzyme-substrate interaction to a lesser extent. In the PvSUB1 models, residues Ser-434, Gly-547, Thr-548, Ser-549, Ser-461, Ala-462, Ser-463, Asn-464, Arg-485, Tyr-486, Pro-487, and Pro-488 and residues Leu-410, Gly-411, Arg-412, Leu-413, Leu-405, Met-416, Phe-435, Ser-436, Phe-437, Asp-438, and Phe-444 were predicted to form the S1 and S4 pockets, respectively (Fig. 2, B and C). More precisely, we defined the binding site residues in PvSUB1 as those not belonging to insertion loops and equivalent to residues in 2TEC which had at least one atom less than 6 Å apart from the Eglin hexapeptide P1′–P5. As confirmed by IcmPocketFinder (50), this region corresponds to the most buried part of the active site involved in the substrate recognition and interaction of several subtilisins (40). We also included a few residues that define superficial S1′ and S2′ sub-pockets formed in PvSUB1 (data not shown). Altogether, the binding pocket used in docking studies included residues: Asp-316 to Ser-317, Leu-405 to Asp-406, His-408, Leu-410 to Leu-413, Met-416, Ser-434 to Asp-438, Phe-444, Ser-461 to Cys-465, and Leu-545 to Asn-546.
Selection of PvSUB1 Three-dimensional models and Virtual Docking Screens
The r.m.s.d. between the 50 PvSUB1 models calculated on all atoms of the binding pocket was lower than 1 Å, as expected from the very small deviation observed in the corresponding region of the templates. In the binding site region, PvSUB1 models differed mainly in the orientation of the long side chains of three residues (Lys-409, Arg-412, and Phe-435), which are not conserved in the templates, resulting in diverging orientations between the models. Two models were selected in which the side chains of Lys-409 and Arg-412 were not pointing into the active site. Although they could participate in PvSUB1 ligand specificity, we excluded models where these long and basic side chains pointed into the catalytic groove, because such orientations corresponded to strong assumptions that could drastically affect the docking results. In addition, a third model was used in which the Phe-435 residue, deeply buried in the binding pocket, was mutated to an alanine to eliminate the ambiguity of the position of its large side chain. Overall, this led to the choice of three PvSUB1 models that were used in the virtual screens.
We used the FlexX, FlexX-Pharm, and ICM software packages to perform docking screens on the 149,992 pre-selected drug-like compounds from the ChemDiv library (San Diego). Compared with ICM, which required 30 s to 1 min to dock a molecule, FlexX is faster, allowing processing of different docking conditions in a reasonable amount of time. Therefore, we performed three separate virtual screens with FlexX on the three selected PvSUB1 three-dimensional models, but only one with ICM. In addition, a pharmacophore was also deduced from the known structures of subtilisins crystallized with an inhibitor. Different structures of holo-subtilisins complexed with ligands (1LW6, 1R0R, and 2TEC) revealed that five conserved hydrogen bonds were involved in the interaction between the catalytic grooves and the bound ligands, defining a putative pharmacophore by the presence of some of these hydrogen bonds between the protein and a ligand. However, a preliminary test using four of these hydrogen bonds as pharmacophore restraints was too drastic to find docking solutions. A virtual screening on the 149,992 filtered compounds was thus performed with FleX-Pharm under a pharmacophore restraint defined by the presence of two of the five conserved hydrogen bonds (32). We selected 306 compounds that had the best docking scores for either of the docking protocols (see “Experimental Procedures” and Fig. 3). These compounds embraced a wide chemical space represented by 220 different chemical scaffolds.
FIGURE 3.
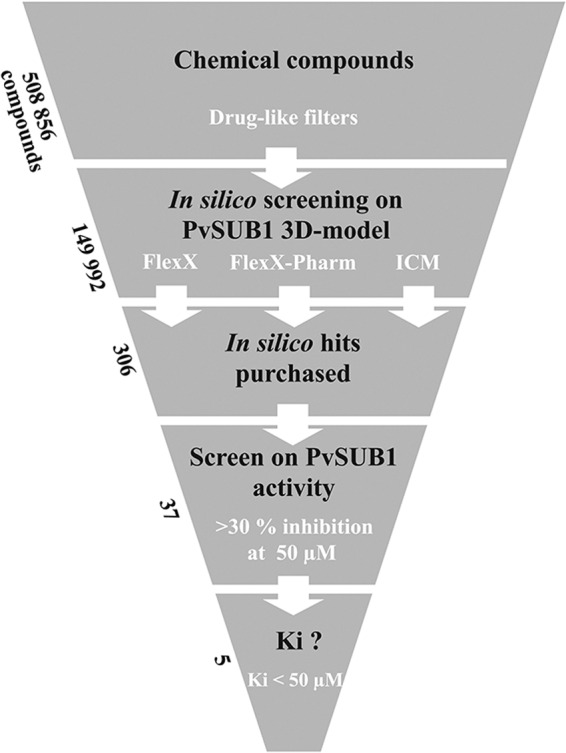
Flow chart for the in silico screen and experimental assays. The ChemDiv chemical library was filtered in silico to exclude compounds predicted to form aggregates and to select for compounds predicted to be drug-like. The resulting 149,992 compounds were used for docking on three three-dimensional models of PvSUB1 active site with FlexX, FlexX-Pharm, or ICM. The set of 306 best-scored compounds was tested for inhibition of PvSUB1r enzymatic activity at a concentration of 50 μm. The 37 compounds displaying >30% inhibition entered a dose-response assay, resulting in five compounds with an apparent Ki value of <50 μm that were tested on P. falciparum in vitro culture. Their structures and properties are presented in Table 2.
Experimental Evaluation of Virtual PvSUB1 Inhibitors on PvSUB1r, PfSUB1r, and PbSUB1r and P. falciparum Cultures
The 306 selected compounds were first tested for autofluorescence at the Dabsyl/Edans compatible wavelength at the final concentration of 50 μm. The 219 compounds that displayed no significant autofluorescence were screened at a 50 μm final concentration on the PvSUB1r enzymatic assay by using the PvSUB1-DE substrate, whereas the PvSUB1-FR substrate was used for the 87 remaining compounds. The 37 compounds that inhibited PvSUB1r activity by >30% entered into a secondary screen consisting of a dose-dependent assay at five concentrations, ranging from 250 to 0.4 μm. Five compounds (Cpd1–5) reproducibly displayed an apparent Ki of <50 μm and were further analyzed (Fig. 3). Cpd1 and Cpd2 were identified with FlexX; Cpd4, and Cpd5 were identified with ICM; Cpd3 was selected with FlexX, and the pharmacophore constraint (see under “Experimental Procedures”).
The apparent Ki of the five selected compounds for PvSUB1r ranged from 30.1 to 6 μm (Table 2). Although Cpd4 and Cpd5 displayed a Ki, app value of 30.1 and 14.7 μm for PvSUB1r, respectively, they did not significantly inhibit the intra-erythrocytic P. falciparum clone 3D7 cycle in vitro, with an EC50 value of >50 μm. In contrast, Cpd1 and -3 displayed a Ki, app for PvSUB1r and an EC50 against P. falciparum-3D7 growth in vitro within the same range, namely 7.6 and 1.1 μm and 30.1 and 22.3 μm, respectively. Interestingly, Cpd2 inhibited PvSUB1r (Ki, app of 6 μm) and displayed a potent anti-P. falciparum activity, with an EC50 of 370 and 450 nm on the chloroquine-sensitive 3D7 clone and chloroquine-resistant Dd2 clone, respectively (Table 2 and Fig. 4A).
TABLE 2.
Structure and characteristics of the five inhibitory compounds
The chemical structure, ChemDiv accession number, molecular mass, calculated logP of the compounds tested on the PvSUB1r activity, and the in vitro culture of P. falciparum 3D7 and Dd2 clones, are presented. The method allowing their selection and the score obtained, together with the PvSUB1 three-dimensional models on which they have been selected are indicated. The apparent Ki for PvSUB1r and IC50 on the parasite growth are expressed as the mean (± S.D.) of a minimum of three independent experiments.
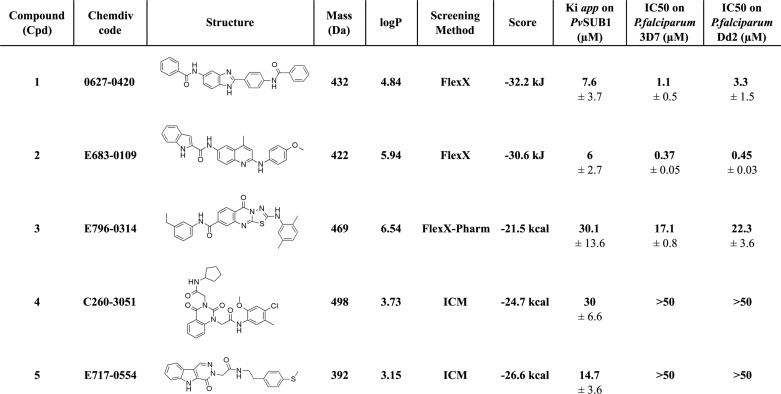
FIGURE 4.

Inhibition of PvSUB1r, PfSUB1, and PbSUB1 enzymatic activity by Cpd2. Dose-dependent inhibition of PvSUB1r (A), PfSUB1r (B), and PbSUB1 (D) by Cpd2. Results presented are the mean (± S.D.) of at least three independent experiments. Lineweaver-Burk representation of PvSUB1r inhibition by Cpd2 (C) suggests inhibition of PvSUB1 via a substrate competitive mechanism. The enzymatic assay was performed on four different concentrations of Cpd2 (50 to 0.4 μm, as indicated) and on five different concentrations of the PvSUB1-FR substrate ranging from 40 to 5 μm.
To further characterize Cpd2, we tested its inhibition potency against PfSUB1r and PvSUB1r. The apparent Ki measured for PfSUB1r (5.7 μm, Fig. 4B) was similar to the one observed for PvSUB1r, although for PbSUB1r it was significantly higher (42 μm, Fig. 4D). Moreover, among the five selected compounds, Cpd2 was the only one displaying a clear, reproducible competitive inhibition of PvSUB1r (Fig. 4C). This is consistent with the fact that the catalytic groove of the enzyme, which accommodates the substrate, was defined as the target of the chemical compounds during the in silico screening.
In the best docking pose of Cpd2 generated by FlexX, Cpd2 occupied almost completely the PvSUB1 catalytic groove. Comparison with the binding mode of bacterial subtilisin substrates indicated that the central quinoline group was positioned between the β′ and β″ strands of the PvSUB1 catalytic groove (Fig. 2, C and D). The indole and “paramethoxyaniline” moieties of Cpd2 interacted with the PvSUB1 S4 (blue) and S1 (green) sub-pockets, respectively (Fig. 2D). More precisely, the backbone of Ser-434 and Ser-436 and the side chain of Asn-464 (involved in the classical oxyanion hole) formed three hydrogen bonds maintaining Cpd2 in the S1 pocket, whereas Gly-411 backbone stabilized Cpd2 in the S4 pocket through two additional hydrogen bonds.
In Vivo Activity against P. berghei Blood Stages
To further characterize the anti-parasite activity of Cpd2, we investigated its activity in vivo on P. berghei blood stages by using the Peters' 4-day suppressive assay. Following infection with 2·106 P. berghei-GFP blood stage parasites, parasitemia reached ∼5% at D4 in the group of mice that received the vehicle. In contrast, there was a dose-dependent reduction of parasitemia in mice treated with Cpd2 (Fig. 5), and no obvious sign of toxicity could be observed (data not shown). The ED50 value of Cpd2 against P. berghei could thus be estimated as ∼40 mg/kg, consistent with the EC50 value of Cpd2 measured on P. falciparum growth in vitro. For comparison, parasitemia was cured in the group of mice treated with 10 mg/kg chloroquine, although it was reduced by ∼50% in mice treated with 2 mg/kg chloroquine, a figure consistent with the ED50 value for chloroquine against P berghei (51).
FIGURE 5.

Evaluation of the anti-P. berghei activity of Cpd2 in vivo using the 4-day Peters' assay. At day 0, mice were infected intraperitoneally with 2·106 P. berghei-GFP blood stage parasites and received the first treatment 2 h after and then at the same hour at days 1–3. The Cpd2 dose per injection is indicated for each group of mice. Parasitemia was determined by FACS analysis. Chloroquine (CQ) and vehicle were used as positive and negative controls, respectively. Each group was composed of a minimum of five mice; the results show the mean parasitemia at day 4 (± S.E.) for each group.
Inhibition of the Egress/Invasion Steps of P. falciparum in Vitro
We then investigated whether Cpd2 indeed inhibited P. falciparum merozoite egress and invasion of human erythrocytes in vitro, the process in which PfSUB1 has been shown to play a dual and essential role (16). We used the recently developed inhibition of ring formation assay (19, 34) and treated highly synchronous schizont cultures with Cpd2. There was a dose-dependent inhibition of egress/invasion with >80 and 10% inhibition for 60 and 1 μm, respectively (Fig. 6A), that correlated with a dose-dependent accumulation of segmented, unruptured schizont (supplemental Fig. 3). As expected (35), the cysteine protease inhibitor E64 at a dose of 10 μm inhibited by >80% the egress-invasion steps of P. falciparum merozoites in vitro.
These results are consistent with Cpd2 inhibiting the egress invasion steps of P. falciparum merozoites in vitro. However, the level of inhibition observed in presence of 1 μm of Cpd2 was not fully consistent with the EC50 of 370–450 nm obtained by use of the classical Desjardins assay, which measures the inhibition of P. falciparum erythrocytic cycle, regardless of the affected stage (33).
To demonstrate that Cpd2 inhibited the endogenous PfSUB1, we monitored the processing of one of its natural substrates, SERA5 (16). Immunoblot analysis of culture supernatants, also containing proteins from the parasitophorous vacuole following mechanical rupture of segmented schizonts, showed a dose-dependent reduction of the p73 and p50 SERA5 processing fragments in Cpd2-treated cultures (Fig. 6B, lanes 5–8), whereas the expected processing pattern was observed in the untreated culture (lane 2) and in parasites treated with the DMSO vehicle (lane 3). Treatment with 10 μm E64 did not significantly inhibit SERA5 processing, although the p56 form corresponding to the last processing step accumulated as reported by others (16). This indicated that Cpd2 did inhibit the endogenous PfSUB1 activity. This obviously does not rule out inhibition of additional parasite enzymes by Cpd2, the combination of which may account for the efficient anti-parasite activity on asynchronous cultures.
DISCUSSION
The agenda of malaria control demands the identification of new anti-parasite candidates acting efficiently against P. falciparum and P. vivax. Most anti-malarial candidates under development target intracellular stages of the parasite life cycle. The SUB1 subtilisin-like protease has emerged as a new potential drug target, implicated at a critical, still untargeted step of the life cycle. In line with the high degree of sequence homology of the catalytic region, PvSUB1 and PfSUB1 had similar enzymatic properties, with similar affinity for the various substrates derived from primary processing sites (Table 1). Consistent with this, homology models of the catalytic domain of PvSUB1 and PfSUB1 presented a similar overall structure, provided the long inserts were excluded. All features described for the PvSUB1 model hold for the PfSUB1 model and are in agreement with the SUB1 models recently proposed by Withers-Martinez et al. (21). As an example, the residues identified as forming the S1, S4, and S1′ sub-pockets of the PvSUB1 catalytic groove are equivalent to those recently published (21). However, as mentioned above, close analysis of the active site showed that Lys-409, Arg-412, and Phe-435, three residues with long side chains, were not conserved in the templates. Different orientations of these side chains were therefore observed among the 50 PvSUB1 models. To take into account this ambiguity and to try to reduce its potential impact on the prediction performances, virtual screening was performed on three different models of PvSUB1, including one in which the influence of the Phe-435 side chain was abrogated by mutating this residue for an alanine. In addition, to better characterize the quality of these models, we modeled the structures of the seven templates used for modeling PvSUB1 in a sort of “cross-validation” study; each of the seven templates was modeled based on the six others. We superposed the modeled templates to their crystallographic structures, and we observed that the r.m.s.d. values in the active site were below 1 Å in all cases, i.e. low (data not shown). Therefore, we expect the r.m.s.d. between the active site of our SUB1 models and their “true” structures to be in the same range. In addition, the binding pockets of PvSUB1 and PfSUB1 showed a high primary sequence identity and a strong three-dimensional structural similarity, as illustrated by the r.m.s.d. in the range of 0.2 Å for the catalytic groove residues. We thus performed docking studies only on one of the proteins, namely PvSUB1, under the assumption that a ligand of the PvSUB1 active site would also be a ligand for PfSUB1 active site, with similar binding modes.
Despite the modest overall sequence similarity between PvSUB1 and bacterial subtilisins, we identified 37 new inhibitors of the recombinant PvSUB1 enzymatic activity from the in silico screen based on PvSUB1 three-dimensional models. Among them, five compounds displayed an apparent Ki of <50 μm for PvSUB1r and were shown to be active against P. falciparum in vitro. They belong to different chemical families than the first reported PfSUB1 nonpeptidic inhibitor known as MRT12113 (16) and display similar or better affinity for SUB1 than the recently identified quinolhydrazone derivatives, whose anti-parasite activity has not been reported (52). Taken together, these compounds define a new chemical diversity to develop a SUB1-specific anti-malarial compound. The most promising hit, Cpd2, displayed a similar affinity to both P. falciparum and P. vivax recombinant SUB1 enzymes and behaved as a competitive inhibitor of PvSUB1 (Fig. 4) and PfSUB1 (data not shown) enzymatic activities. We conclude that Cpd2 likely targets the SUB1 active site, thus competing with the enzyme substrate, although a formal confirmation would only be provided by a co-crystallization of Cpd2 with either PvSUB1 or PfSUB1.
The affinity of Cpd2 for PbSUB1r was significantly lower than those obtained for P. vivax and P. falciparum SUB1. Similar results have recently been obtained with a peptidyl α-ketoamide inhibitor based on one of the cleavage sites of PfSERA4, a natural PfSUB1 substrate. This ketoamide inhibited PfSUB1 and PvSUB1 recombinant enzymes with equivalent IC50 values of 7.3 and 12 μm, respectively, although the IC50 value for recombinant PbSUB1 was >100 μm (21). Together with the lower sequence similarity of its catalytic domain with respect to those of PfSUB1 and PvSUB1, and its different affinity to similar substrates, this is the third evidence that PbSUB1 catalytic groove is expected to display substantial structural differences to those of PfSUB1 and PvSUB1.
Cpd2 was active against P. falciparum parasites in vitro, irrespective of their susceptibility to chloroquine, as well as against P. berghei in vivo. Cpd2 was shown to target the endogenous P. falciparum SUB1, as indicated by the observed inhibition of parasite egress, as well as SERA5 maturation in vitro. Altogether, these data qualify Cpd2 as the so far most promising anti-SUB1 hit, although its affinity and specificity have to be significantly optimized before qualifying it as a new anti-malarial lead compound.
This study illustrates the benefit of in silico screening and provides clues on ways to improve its efficiency. The relatively low yield of potent inhibitors selected may reflect the hurdles brought by the absence of an experimental three-dimensional structure of SUB1. Rigidity of the binding site during docking remains an important limitation, because it prevents considering induced rearrangement upon binding. Indeed, a minor variation of the predicted binding site structure may drastically affect the docking. This illustrates the general challenge of finding specific inhibitors based on homology models. Data presented by Withers-Martinez et al. (21) suggest that a less buried region of the SUB1 catalytic groove also participates in the specificity and involves interactions with the P1′–P2′ part of the substrate. These residues are solvent-exposed, and the fact that the structural templates are too divergent and show too little sequence similarity prevented us from including them in the definition of the binding pocket for virtual screening. It will obviously be interesting to include them in future studies, in case better structural templates are available, to gain specificity.
It is encouraging that, even in the absence of SUB1 x-ray structure, the present in silico and experimental studies led to a dramatic cut on the number of molecules that underwent biochemical evaluation. Among the >500,000 initially considered molecules, 306 molecules were indeed experimentally tested, and among the 37 active compounds, five proved to inhibit PvSUB1 recombinant activity and P. falciparum growth in vitro. Among them, Cpd2 displays a consistent activity against PfSUB1 and PvSUB1 and constitutes a novel promising hit against a yet untargeted biological stage of both P. vivax and P. falciparum human-infecting parasites.
Supplementary Material
Acknowledgments
We are grateful to J. D'Alayer and P. Lenormand for the N-terminal sequencing of recombinant proteins.
This work was supported in part by the “Fond Dédié: Combattre les Maladies Parasitaires” from Sanofi-Aventis and the French Ministry of Research and a grant from the Institut Carnot-Pasteur Maladies Infectieuses.

This article contains supplemental Figs. 1–3 and Table 1.
- PDB
- Protein Data Bank
- r.m.s.d.
- root mean square deviation
- FR
- far red
- Cpd
- compound
- ICM
- internal coordinate mechanics.
REFERENCES
- 1. malERA Consultative Group on Drugs (2011) A research agenda for malaria eradication: drugs. PLoS Med. 8, e1000402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rottmann M., McNamara C., Yeung B. K., Lee M. C., Zou B., Russell B., Seitz P., Plouffe D. M., Dharia N. V., Tan J., Cohen S. B., Spencer K. R., González-Páez G. E., Lakshminarayana S. B., Goh A., Suwanarusk R., Jegla T., Schmitt E. K., Beck H. P., Brun R., Nosten F., Renia L., Dartois V., Keller T. H., Fidock D. A., Winzeler E. A., Diagana T. T. (2010) Spiroindolones, a potent compound class for the treatment of malaria. Science 329, 1175–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gamo F. J., Sanz L. M., Vidal J., de Cozar C., Alvarez E., Lavandera J. L., Vanderwall D. E., Green D. V., Kumar V., Hasan S., Brown J. R., Peishoff C. E., Cardon L. R., Garcia-Bustos J. F. (2010) Thousands of chemical starting points for antimalarial lead identification. Nature 465, 305–310 [DOI] [PubMed] [Google Scholar]
- 4. Guiguemde W. A., Shelat A. A., Garcia-Bustos J. F., Diagana T. T., Gamo F. J., Guy R. K. (2012) Global phenotypic screening for antimalarials. Chem. Biol. 19, 116–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Déchamps S., Shastri S., Wengelnik K., Vial H. J. (2010) Glycerophospholipid acquisition in Plasmodium–a puzzling assembly of biosynthetic pathways. Int. J. Parasitol. 40, 1347–1365 [DOI] [PubMed] [Google Scholar]
- 6. Klibanov O. M., Williams S. H., Smith L. S., Olin J. L., Vickery S. B. (2011) Telaprevir: a novel NS3/4 protease inhibitor for the treatment of hepatitis C. Pharmacotherapy 31, 951–974 [DOI] [PubMed] [Google Scholar]
- 7. Rosenthal P. J. (2011) Falcipains and other cysteine proteases of malaria parasites. Adv. Exp. Med. Biol. 712, 30–48 [DOI] [PubMed] [Google Scholar]
- 8. Ang K. K., Ratnam J., Gut J., Legac J., Hansell E., Mackey Z. B., Skrzypczynska K. M., Debnath A., Engel J. C., Rosenthal P. J., McKerrow J. H., Arkin M. R., Renslo A. R. (2011) Mining a cathepsin inhibitor library for new antiparasitic drug leads. PLoS Negl. Trop. Dis. 5, e1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Saify Z. S., Azim M. K., Ahmad W., Nisa M., Goldberg D. E., Hussain S. A., Akhtar S., Akram A., Arayne A., Oksman A., Khan I. A. (2012) New benzimidazole derivatives as antiplasmodial agents and plasmepsin inhibitors: synthesis and analysis of structure-activity relationships. Bioorg. Med. Chem. Lett. 22, 1282–1286 [DOI] [PubMed] [Google Scholar]
- 10. Russo I., Babbitt S., Muralidharan V., Butler T., Oksman A., Goldberg D. E. (2010) Plasmepsin V licenses Plasmodium proteins for export into the host erythrocyte. Nature 463, 632–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boddey J. A., Hodder A. N., Günther S., Gilson P. R., Patsiouras H., Kapp E. A., Pearce J. A., de Koning-Ward T. F., Simpson R. J., Crabb B. S., Cowman A. F. (2010) An aspartyl protease directs malaria effector proteins to the host cell. Nature 463, 627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meyers M. J., Goldberg D. E. (2012) Recent advances in plasmepsin medicinal chemistry and implications for future antimalarial drug discovery efforts. Curr. Top. Med. Chem. 12, 445–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goldberg D. E., Cowman A. F. (2010) Moving in and renovating: exporting proteins from Plasmodium into host erythrocytes. Nat. Rev. Microbiol. 8, 617–621 [DOI] [PubMed] [Google Scholar]
- 14. Dowse T. J., Koussis K., Blackman M. J., Soldati-Favre D. (2008) Roles of proteases during invasion and egress by Plasmodium and Toxoplasma. Subcell. Biochem. 47, 121–139 [DOI] [PubMed] [Google Scholar]
- 15. Blackman M. J. (2008) Malarial proteases and host cell egress: an ‘emerging’ cascade. Cell. Microbiol. 10, 1925–1934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yeoh S., O'Donnell R. A., Koussis K., Dluzewski A. R., Ansell K. H., Osborne S. A., Hackett F., Withers-Martinez C., Mitchell G. H., Bannister L. H., Bryans J. S., Kettleborough C. A., Blackman M. J. (2007) Subcellular discharge of a serine protease mediates release of invasive malaria parasites from host erythrocytes. Cell 131, 1072–1083 [DOI] [PubMed] [Google Scholar]
- 17. Koussis K., Withers-Martinez C., Yeoh S., Child M., Hackett F., Knuepfer E., Juliano L., Woehlbier U., Bujard H., Blackman M. J. (2009) A multifunctional serine protease primes the malaria parasite for red blood cell invasion. EMBO J. 28, 725–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barale J. C., Blisnick T., Fujioka H., Alzari P. M., Aikawa M., Braun-Breton C., Langsley G. (1999) Plasmodium falciparum subtilisin-like protease 2, a merozoite candidate for the merozoite surface protein 1–42 maturase. Proc. Natl. Acad. Sci. U.S.A. 96, 6445–6450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bastianelli G., Bouillon A., Nguyen C., Crublet E., Pêtres S., Gorgette O., Le-Nguyen D., Barale J. C., Nilges M. (2011) Computational reverse-engineering of a spider-venom-derived peptide active against Plasmodium falciparum SUB1. PLoS One 6, e21812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Withers-Martinez C., Saldanha J. W., Ely B., Hackett F., O'Connor T., Blackman M. J. (2002) Expression of recombinant Plasmodium falciparum subtilisin-like protease-1 in insect cells. Characterization, comparison with the parasite protease, and homology modeling. J. Biol. Chem. 277, 29698–29709 [DOI] [PubMed] [Google Scholar]
- 21. Withers-Martinez C., Suarez C., Fulle S., Kher S., Penzo M., Ebejer J. P., Koussis K., Hackett F., Jirgensons A., Finn P., Blackman M. J. (2012) Plasmodium subtilisin-like protease 1 (SUB1): Insights into the active-site structure, specificity and function of a pan-malaria drug target. Int. J. Parasitol. 42, 597–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grünberg R., Nilges M., Leckner J. (2007) BisKit–a software platform for structural bioinformatics. Bioinformatics 23, 769–770 [DOI] [PubMed] [Google Scholar]
- 23. O'Sullivan O., Suhre K., Abergel C., Higgins D. G., Notredame C. (2004) 3DCoffee: combining protein sequences and structures within multiple sequence alignments. J. Mol. Biol. 340, 385–395 [DOI] [PubMed] [Google Scholar]
- 24. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 25. Dalton J. A., Jackson R. M. (2007) An evaluation of automated homology modelling methods at low target template sequence similarity. Bioinformatics 23, 1901–1908 [DOI] [PubMed] [Google Scholar]
- 26. Larsson P., Wallner B., Lindahl E., Elofsson A. (2008) Using multiple templates to improve quality of homology models in automated homology modeling. Protein Sci. 17, 990–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 28. Sadowski J. (1997) A hybrid approach for addressing ring flexibility in 3D database searching. J. Comput. Aided Mol. Des. 11, 53–60 [DOI] [PubMed] [Google Scholar]
- 29. Totrov M., Abagyan R. (1997) Flexible protein-ligand docking by global energy optimization in internal coordinates. Proteins Suppl. 1, 215–220 [DOI] [PubMed] [Google Scholar]
- 30. Rarey M., Kramer B., Lengauer T., Klebe G. (1996) A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 261, 470–489 [DOI] [PubMed] [Google Scholar]
- 31. Schapira M., Totrov M., Abagyan R. (1999) Prediction of the binding energy for small molecules, peptides and proteins. J. Mol. Recognit. 12, 177–190 [DOI] [PubMed] [Google Scholar]
- 32. Hindle S. A., Rarey M., Buning C., Lengaue T. (2002) Flexible docking under pharmacophore type constraints. J. Comput. Aided Mol. Des. 16, 129–149 [DOI] [PubMed] [Google Scholar]
- 33. Desjardins R. E., Canfield C. J., Haynes J. D., Chulay J. D. (1979) Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 16, 710–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bouillon A., Gorgette O., Mercereau-Puijalon O., Barale J.-C. (2013) in Methods in Molecular Biology: Malaria Methods and Protocols (Ménard R., ed) pp. 523–534, Springer Science+Business Media, LLC; [DOI] [PubMed] [Google Scholar]
- 35. Salmon B. L., Oksman A., Goldberg D. E. (2001) Malaria parasite exit from the host erythrocyte: a two-step process requiring extraerythrocytic proteolysis. Proc. Natl. Acad. Sci. U.S.A. 98, 271–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li Q., Gerena L., Xie L., Zhang J., Kyle D., Milhous W. (2007) Development and validation of flow cytometric measurement for parasitemia in cultures of P. falciparum vitally stained with YOYO-1. Cytometry A 71, 297–307 [DOI] [PubMed] [Google Scholar]
- 37. Ishino T., Orito Y., Chinzei Y., Yuda M. (2006) A calcium-dependent protein kinase regulates Plasmodium ookinete access to the midgut epithelial cell. Mol. Microbiol. 59, 1175–1184 [DOI] [PubMed] [Google Scholar]
- 38. Peters W., Robinson B. L. (1999) in Handbook of Animal Models of Infection (Zak O., Sande M. E., eds) pp. 757–773, Academic Press, New York [Google Scholar]
- 39. Fidock D. A., Rosenthal P. J., Croft S. L., Brun R., Nwaka S. (2004) Antimalarial drug discovery: efficacy models for compound screening. Nat. Rev. Drug Discov. 3, 509–520 [DOI] [PubMed] [Google Scholar]
- 40. Siezen R. J., Leunissen J. A. (1997) Subtilases: The superfamily of subtilisin-like serine protease. Protein Sci. 6, 501–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grum-Tokars V., Ratia K., Begaye A., Baker S. C., Mesecar A. D. (2008) Evaluating the 3C-like protease activity of SARS-Coronavirus: recommendations for standardized assays for drug discovery. Virus Res. 133, 63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Blackman M. J., Fujioka H., Stafford W. H., Sajid M., Clough B., Fleck S. L., Aikawa M., Grainger M., Hackett F. (1998) A subtilisin-like protein in secretory organelles of Plasmodium falciparum merozoites. J. Biol. Chem. 273, 23398–23409 [DOI] [PubMed] [Google Scholar]
- 43. McGovern S. L., Shoichet B. K. (2003) Information decay in molecular docking screens against holo, apo, and modeled conformations of enzymes. J. Med. Chem. 46, 2895–2907 [DOI] [PubMed] [Google Scholar]
- 44. Gros P., Teplyakov A. V., Hol W. G. (1992) Effects of eglin-c binding to thermitase: three-dimensional structure comparison of native thermitase and thermitase eglin-c complexes. Proteins 12, 63–74 [DOI] [PubMed] [Google Scholar]
- 45. Laskowski I. R., MacArthur M. W., Moss D. S., Thornton J. M. (1993) PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 46. Jean L., Withers-Martinez C., Hackett F., Blackman M. J. (2005) Unique insertions within Plasmodium falciparum subtilisin-like protease-1 are crucial for enzyme maturation and activity. Mol. Biochem. Parasitol. 144, 187–197 [DOI] [PubMed] [Google Scholar]
- 47. Fiser A., Sali A. (2003) ModLoop: automated modeling of loops in protein structures. Bioinformatics 19, 2500–2501 [DOI] [PubMed] [Google Scholar]
- 48. Berger A., Schechter I. (1970) Mapping the active site of papain with the aid of peptide substrates and inhibitors. Philos. Trans. R. Soc. Lond B Biol. Sci. 257, 249–264 [DOI] [PubMed] [Google Scholar]
- 49. Grøn H., Meldal M., Breddam K. (1992) Extensive comparison of the substrate preferences of two subtilisins as determined with peptide substrates which are based on the principle of intramolecular quenching. Biochemistry 31, 6011–6018 [DOI] [PubMed] [Google Scholar]
- 50. Nicola G., Smith C. A., Abagyan R. (2008) New method for the assessment of all drug-like pockets across a structural genome. J. Comput. Biol. 15, 231–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Safeukui I., Mangou F., Malvy D., Vincendeau P., Mossalayi D., Haumont G., Vatan R., Olliaro P., Millet P. (2004) Plasmodium berghei: dehydroepiandrosterone sulfate reverses chloroquino-resistance in experimental malaria infection; correlation with glucose-6-phosphate dehydrogenase and glutathione synthesis pathway. Biochem. Pharmacol. 68, 1903–1910 [DOI] [PubMed] [Google Scholar]
- 52. Gemma S., Giovani S., Brindisi M., Tripaldi P., Brogi S., Savini L., Fiorini I., Novellino E., Butini S., Campiani G., Penzo M., Blackman M. J. (2012) Quinolylhydrazones as novel inhibitors of Plasmodium falciparum serine protease PfSUB1. Bioorg. Med. Chem. Lett. 22, 5317–5321 [DOI] [PubMed] [Google Scholar]
- 53. Meister S., Plouffe D. M., Kuhen K. L., Bonamy G. M., Wu T., Barnes S. W., Bopp S. E., Borboa R., Bright A. T., Che J., Cohen S., Dharia N. V., Gagaring K., Gettayacamin M., Gordon P., Groessl T., Kato N., Lee M. C., McNamara C. W., Fidock D. A., Nagle A., Nam T. G., Richmond W., Roland J., Rottmann M., Zhou B., Froissard P., Glynne R. J., Mazier D., Sattabongkot J., Schultz P. G., Tuntland T., Walker J. R., Zhou Y., Chatterjee A., Diagana T. T., Winzeler E. A. (2011) Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science 334, 1372–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.