

Background: Suppressor of IκB kinase ϵ (SIKE) inhibits a key innate immune effector molecule, TANK-binding kinase 1 (TBK1), through an undefined mechanism.
Results: SIKE is a TBK1 substrate.
Conclusion: SIKE controls TBK1 activity by acting as a high affinity substrate.
Significance: SIKE attenuates phosphorylation of interferon regulatory factor 3 (IRF3) by serving as an alternative, high affinity substrate for TBK1.
Keywords: Cooperativity, Enzyme Inhibitors, Enzyme Kinetics, Enzyme Mechanisms, Innate Immunity
Abstract
TANK-binding kinase 1 (TBK1) serves as a key convergence point in multiple innate immune signaling pathways. In response to receptor-mediated pathogen detection, TBK1 phosphorylation promotes production of pro-inflammatory cytokines and type I interferons. Increasingly, TBK1 dysregulation has been linked to autoimmune disorders and cancers, heightening the need to understand the regulatory controls of TBK1 activity. Here, we describe the mechanism by which suppressor of IKKϵ (SIKE) inhibits TBK1-mediated phosphorylation of interferon regulatory factor 3 (IRF3), which is essential to type I interferon production. Kinetic analyses showed that SIKE not only inhibits IRF3 phosphorylation but is also a high affinity TBK1 substrate. With respect to IRF3 phosphorylation, SIKE functioned as a mixed-type inhibitor (Ki, app = 350 nm) rather than, given its status as a TBK1 substrate, as a competitive inhibitor. TBK1 phosphorylation of IRF3 and SIKE displayed negative cooperativity. Both substrates shared a similar Km value at low substrate concentrations (∼50 nm) but deviated >8-fold at higher substrate concentrations (IRF3 = 3.5 μm; SIKE = 0.4 μm). TBK1-SIKE interactions were modulated by SIKE phosphorylation, clustered in the C-terminal portion of SIKE (Ser-133, -185, -187, -188, -190, and -198). These sites exhibited striking homology to the phosphorylation motif of IRF3. Mutagenic probing revealed that phosphorylation of Ser-185 controlled TBK1-SIKE interactions. Taken together, our studies demonstrate for the first time that SIKE functions as a TBK1 substrate and inhibits TBK1-mediated IRF3 phosphorylation by forming a high affinity TBK1-SIKE complex. These findings provide key insights into the endogenous control of a critical catalytic hub that is achieved not by direct repression of activity but by redirection of catalysis through substrate affinity.
Introduction
To defend against pathogens, multicellular organisms mount an immune response that recognizes, sequesters, and eradicates invading infectious agents. Essential to this safeguard is the innate anti-viral response mediated by type I interferons (1–3). To initiate type I interferon production, pattern recognition receptors, such as Toll-like receptor 3 (TLR3)2 (4), retinoic acid-inducible gene I (5), melanoma differentiation factor 5 (6), or DNA-dependent activator of interferon-regulatory factors (7), recognize and respond to distinct virus-associated molecular signatures, for example double-stranded RNA (dsRNA). These receptors trigger signaling cascades that converge at the kinase, TANK-binding kinase 1 (TBK1) (8–10).
TBK1 (11), also known as NF-κB-activating kinase (12) or TRAF2-associated kinase (13), is a ubiquitously expressed serine/threonine kinase of the IκB kinase family. The canonical ΙκΒ kinases, IKKα and IKKβ, phosphorylate IκBα allowing the transcription factor, NF-κB, to induce a proinflammatory response (14). TBK1, a noncanonical IκB kinase, targets the transcription factors interferon regulatory factor (IRF) 3 and 7 (9). In IRF3, seven Ser/Thr sites are phosphorylated near the C terminus, 385SSX9SXSXXXSXTS405 (15, 16). Of these seven phosphorylation sites, phosphorylated Ser-396 to Ser-405 induces IRF3 interaction with CBP/p300, whereas phosphorylation at residues Ser-385 and Ser-386 controls IRF3 dimerization (17–20). Modified IRF3 translocates to the nucleus where it binds to the interferon β enhancer and contributes to type I interferon production (15). In addition to IRF3/7, several other substrates have been identified that implicate TBK1 function in the insulin response (insulin receptor (21)), cell growth (Akt (22)) and xenophagy (optineurin (23, 24)). Not surprisingly, aberrant TBK1 activity contributes to autoimmune disorders and cancers (22, 25–27).
To become active, TBK1 requires phosphorylation at Ser-172 within the kinase domain activation loop (28). IKKβ (29) and TBK1, via a trans-phosphorylation event (30), provide this modification. Phosphorylated Ser-172 induces reorganization of the activation loop, making the active site competent to bind substrate (30). Alignment of TBK1 substrates revealed a weak consensus sequence for TBK1 phosphorylation consisting of a hydrophobic residue immediately proceeding the modified serine that is mirrored by several hydrophobic residues lining the P + 1 site (30). Kinetic analysis of TBK1 phosphorylation of Ser-36 within the IκBα peptide (residues 19–41) revealed that TBK1 functions through a rapid equilibrium, random order mechanism (31), consistent in all IκB kinases (31–33).
To regulate TBK1 activity, endogenous inhibitors that alter TBK1 ubiquitination (A20 (34)) or mask tyrosine phosphorylation sites (SHP-2 (35)) have been characterized. In addition, a yeast two-hybrid screen for interaction partners of the closely related, noncanonical IκB kinase, ΙKKϵ, identified a 207-amino acid protein dubbed suppressor of IKKϵ (SIKE) (36). When examined in the context of TLR3 signaling, SIKE associated with IKKϵ and TBK1 only prior to dsRNA stimulation or viral infection (36), but SIKE mRNA was increased after viral challenge suggesting SIKE formed a negative feedback inhibition loop (37). Moreover, SIKE was shown to disrupt IKKϵ-TBK1 interactions with the upstream signaling component, TIR domain-containing adaptor-inducing interferon-β, and the downstream substrate IRF3 via an undefined mechanism (36).
Because SIKE acts as a physiological suppressor of TBK1 at a key convergence point in the innate anti-viral response, we investigated the mechanism by which SIKE inhibits the type I interferon response and the regulatory release mechanism of SIKE from TBK1 upon viral activation of TBK1. Our studies show that SIKE acts as a mixed-type inhibitor of TBK1-mediated phosphorylation of IRF3. Surprisingly, TBK1 directly phosphorylated SIKE at six sites that partially mimic IRF3's phosphorylation motif sequence. Analysis of the initial rate data revealed potential negative cooperativity in IRF3 and SIKE reactions. Using site-directed mutagenesis and cell-based assays, we showed that SIKE phosphorylation correlates with activation of the anti-viral response and modulates the TBK1-SIKE interaction. From these findings, we propose that SIKE is not only an inhibitor of TBK1-mediated phosphorylation of IRF3 but is also a TBK1 substrate.
EXPERIMENTAL PROCEDURES
Reagents
Chemicals were purchased from Sigma unless otherwise specified. [γ-32P]ATP was purchased from PerkinElmer Life Sciences. Recombinant TBK1 was purchased from Invitrogen. Anti-phosphoserine antibody was purchased from Qiagen. Anti-FLAG-HRP was purchased from Sigma. Anti-HA antibody was purchased from Covance. Anti-full-length IRF3 and anti-actin-HRP antibodies were purchased from Santa Cruz Biotechnology. Anti-phospho-Ser-396 IRF3 antibody was purchased from Cell Signaling. Anti-rabbit-IgG-HRP and anti-mouse-IgG-HRP antibodies were purchased from Southern Biotechnology Associates, Inc.
Cell Culture
HEK293 cells were purchased from American Type Culture Collection. Cells were cultured in complete medium (RPMI 1640 supplemented with 10% low endotoxin FBS, 20 mm l-glutamine, 100 mm HEPES, 10 mm sodium pyruvate, and 1× nonessential amino acid solution) at 37 °C in 5% CO2.
Constructs
The human SIKE sequence was cloned from total RNA isolated from the 786O cell line using the One-step RT-PCR kit according to the manufacturer's protocol (Qiagen). The primers incorporated a 5′ NdeI restriction enzyme site (5′ ATTATCATATGAGCTGCACCATCGAGAAGATC 3′) and a 3′ BamHI restriction enzyme site (5′ TAATAGGATCCTTATTTGATGGCTTGGGAAGC 3′) compatible with insertion into the pET15b vector (Novagen). The SIKE72 construct (residues 72–207) was amplified using 5′ TATACATATGCTGCTGTCCCAAGAGAAC 3′ and inserted into the pET15b vector using the same strategy. Full-length SIKE was amplified from FL-SIKE pET15b using standard molecular biology techniques with primers, including a 5′ EcoRI restriction enzyme site (5′ TATAGAATTCAGAGCTGCACCATCGAGAAG 3′) and 3′ SalI restriction enzyme site (5′ TATAGTCGACTTATTTGATGGCTTGG 3′) compatible with insertion into the pCMV HA vector (Clontech). SIKE(113–207) was amplified from FL-SIKE pET15b using standard molecular biology techniques and primers with a 5′ EcoRI restriction enzyme site (5′ TATAGAATTCGGTTACAGTTAATGGTTGC 3′) or 5′ NdeI restriction enzyme site (5′ TATACATATGTTACAGTTAATGGTTGC 3′), and the 3′ primer was used for FL-SIKE pCMV HA or pET15b compatible with insertion into the pCMV HA or pET15b vector, respectively. SIKE point mutations (S133A/S133E, S185A/S185E, S187A/S187E, S188A/S188E, S190A/S190E, and S198A/S198E) and insertions (stop codons after 112 (SIKE(1–112) and 184 (SIKE(72–184)) and the N-terminal FLAG sequence into pUNO-TBK1 vector (Invivogen) were made using the QuikChange site-directed mutagenesis kit following the manufacturer's protocol (Agilent Technologies). All constructs and mutations were confirmed by DNA sequencing. GST-IRF3(173–427) pGEX4T1 vector was a kind gift from Dr. Katherine Fitzgerald.
Expression and Purification of Proteins
For pET15b SIKE72 construct expression, the vector was transformed into chemically competent BL21-CodonPlus (DE3)-RIPL following the manufacturer's protocol (Agilent Technologies). A single colony was used to inoculate an overnight culture of Luria broth plus 100 μg/ml ampicillin. The overnight culture was subcultured 1:100 into two 1-liter LB/ampicillin flasks grown at 37 °C until the cell density reached an A600 ≈0.6. Cultures were induced with 1 mm isopropyl β-d-galactopyranoside and allowed to grow for an additional 4 h at 37 °C. Cells were harvested by centrifugation at 7,000 × g, and the pellet was solubilized in guanidine hydrochloride (GdnHCl) buffer (6 m guanidine hydrochloride, 100 mm sodium phosphate, pH 8.0, 500 mm NaCl, and 1 mm 2-mercaptoethanol), 5 ml of buffer/g of cell pellet. Lysate was clarified by centrifugation at 14,000 × g. The supernatant was mixed with 5 ml of nickel-nitrilotriacetic acid-agarose resin (Qiagen), pre-equilibrated in GdnHCl buffer. The lysate/resin mixture was loaded into a column (Bio-Rad) and washed by gravity with 50 column volumes (CV) of GdnHCl buffer. Bound protein was refolded on the column using a 40-CV reverse gradient of GdnHCl buffer to 100 mm sodium phosphate, pH 8.0, 500 mm NaCl, and 1 mm 2-mercaptoethanol (Buffer 1) and eluted with 5 CV of Buffer 1 plus 500 mm imidazole. Elution fractions were separated on a HiLoad 16/60 Superdex 200 column (GE Healthcare), and peak fractions were screened for target protein by SDS-PAGE analysis. CD and thermal denaturation of WT and mutant SIKE72 proteins indicated primarily helical secondary structure consistent with a predicted coiled coil domain structure and a ≥2-state unfolding curve typical of globular, oligomeric proteins (data not shown).
For GST-IRF3(173–427) expression, pGEX4T1-IRF3(173–427) was transformed into chemically competent BL21-CodonPlus (DE3)-RIPL following the manufacturer's protocol (Agilent Technologies). Cell culture was identical to pET15b expression prior to induction. When cell density reached A600 ≈ 0.6, cultures were incubated on ice for 30 min, induced with 1 mm isopropyl β-d-1-thiogalactopyranoside, and allowed to grow at 16 °C for an additional 14 h. Cells were harvested by centrifugation at 7,000 × g, and the cell pellet was resuspended in Buffer A (2× PBS, 10 mm DTT, 1 mm EDTA), 5 ml of buffer/g of cell pellet. Cells were emulsified to lyse (Emulsiflex C3, Avestin) and clarified by centrifugation at 12,000 × g. The supernatant was mixed with glutathione-Sepharose 4b resin pre-equilibrated in Buffer A (GE Healthcare), 5 ml. The resin/supernatant solution was loaded into a column (Bio-Rad) and washed by gravity with 100 CV of Buffer A, 100 CV of 20 mm Tris, pH 7.5, 500 mm NaCl, 1 mm EDTA, 10 mm DTT, 10% glycerol (Buffer B) and eluted with 2–5-CV fractions of Buffer B plus 10 mm glutathione, pH 8.0. The elution fractions were concentrated in an Amicon Ultra-15 centrifugal filter (Millipore) to 6 mg/ml. GST-IRF(173–427) was incubated with 20 units/ml thrombin (GE Healthcare) for 24 h at 4 °C. IRF3(173–427) was separated from GST using a HiLoad 16/60 Superdex 75 column (GE Healthcare) equilibrated in 20 mm HEPES, pH 7.5, 500 mm NaCl, 1 mm 2-mercaptoethanol. Peak fractions were screened for cleaved, pure IRF3(173–427) by SDS-PAGE analysis. Fractions were pooled and stored at 4 °C. Protein concentrations were determined using the Bradford method (Bio-Rad).
In Vitro Kinase Assays
Michaelis-Menten kinetic assay reactions (50-μl volume) contained 0.1 mCi of [γ-32P]ATP, 100 μm ATP, and 0.042–20.8 μm IRF3(173–427) for IRF3-varied assays; 0.1 mCi of [γ-32P]ATP, 20 μm IRF3(173–427), and 0.48–83 μm ATP for ATP-varied assays, and 0.1 mCi of [γ-32P]ATP, 100 μm ATP, and 0.043–8.4 μm SIKE72 for SIKE-varied assays. Reactions were initiated by addition of 29.6 nm TBK1 (10 μl, 4.93 nm final concentration, pre-activated for 30 min with 100 μm ATP in assay buffer, supplemental Fig. S1A). For the ATP-varied reactions, excess cold ATP from pre-activation of TBK1 was removed by desalting column (Zeba spin, 7 molecular weight cutoff, 2 ml, Pierce) prior to initiating reactions. Reactions were incubated for 120 min at 30 °C (determined in supplemental Fig. S1B) and terminated by boiling in reducing SDS sample buffer (20 μl, Invitrogen). Duplicate samples (10 μl each) were separated by SDS-PAGE on a 4–12% Tris-BisTris polyacrylamide gel (Invitrogen). Gels were mounted on filter paper and exposed to a storage phosphor screen (Imaging Screen K, Bio-Rad). The screen was read by a Molecular Imager FX system (Bio-Rad). Band densitometry was measured and converted to 32P micromolars via a [γ-32P]ATP standard curve. Inhibition experiments were carried out using the assay methodology described for IRF3-varied and the ATP-varied assay with the addition of SIKE72 at 20.8 or 83.3 nm final concentration. All reactions were analyzed by SDS-PAGE/autoradiography as described above. A minimum of quadruplicate repeats was completed for each reaction. Data were plotted as Michaelis-Menten or double-reciprocal plots and fit to single rectangular hyperbola or linear polynomial equations, respectively, in SigmaPlot (Systat Software, Inc.). Errors were reported as standard deviation. From the intercepts and slopes derived from the double-reciprocal plot fit to a linear polynomial, inhibition constants were calculated for the SIKE + ATP or IRF3 varied data using Equations 1 and 2,
 |
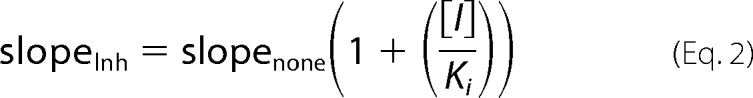 |
To examine inhibition of IRF3 phosphorylation by SIKE72, 4.17 μm IRF3(173–427), 100 μm ATP, 0.1 mCi of [γ-32P]ATP, and SIKE72 or SIKE72 mutants at indicated concentrations were prepared in assay buffer (50 μl volume). TBK1, 10 μl of 29.6 nm and 4.93 nm final concentration, pre-activated as above, was added to initiate the reaction. Reactions were incubated at 30 °C for 120 min and analyzed as described above to derive initial rates for IRF3(173–427) phosphorylation. Data were plotted as the percentage of uninhibited reaction rate versus SIKE72 concentration. Errors were reported as standard deviation. To derive the Ki, app parameter for SIKE mutants, inhibited reaction rate versus SIKE72 concentration was fit to a two-parameter rectangular hyperbola.
DNA Transfection, Immunoprecipitation, and Immunoblot Analysis
Approximately 0.5 × 106 cells were plated into 10-cm2 wells and transfected with 2.5 μg of total DNA of the different expression plasmids (1:0.9:0.6 ratio of pUNO-FLAG-TBK1/pCDNA3.1/pCMV-HA-SIKE) using Lipofectamine 2000 following the standard procedure (Invitrogen). After 24 h, cells were stimulated with 50 μg/ml polyinosinic:polycytidylic acid (poly(I:C)) for 3 h, harvested, and lysed in a lysis buffer (200 μl, 0.02 m HEPES, pH 7.4, 0.15 m NaCl, 10 mm NaF, 2 mm DTT, 2 mm EGTA, 1.5 mm MgCl2, 1 mm Na3VO4, 2.7 mg/ml β-glycerophosphate, 1 mg/ml N-ethylmaleimide, 0.5% Triton X-100, 1× Complete, EDTA-free protease inhibitor mixture (Roche Applied Science)). Lysates were cleared by centrifugation (14,000 × g for 30 min at 4 °C). Protein concentration was quantified by the Bradford method (Bio-Rad). For whole cell lysates, 50 μg of total protein per sample were boiled in sample buffer (Invitrogen), separated by SDS-PAGE (10% Tris/glycine), and transferred to nitrocellulose membrane. The membrane was blocked in 5% nonfat dry milk diluted in Tris-buffered saline (TBS) containing 0.1% Tween 20 and probed with the indicated antibodies (see supplemental Table 1). Blots were developed with chemiluminescent reagents, ECL Plus (GE Healthcare). For immunoprecipitations, cell lysates (500 μg) were incubated with 40 μl of anti-FLAG M2 antibody affinity gel (Sigma) or anti-HA resin (Sigma) for 24 h in TBS at 4 °C. Resin was washed with TBS (3× 1 ml), and bound proteins were eluted with 100 μl of 125 ng/μl FLAG peptide (Sigma) or HA peptide (Sigma). Eluted proteins were analyzed by immunoblot as described above. Each experiment was repeated in triplicate. A list of antibody dilutions for immunoblots is given in the supplemental Methods.
Phosphopeptide Mapping
Recombinantly expressed SIKE72 (500 ng) was incubated with 4.93 nm TBK1 and 100 μm ATP in assay buffer (100 μl volume) for 24 h at 30 °C. The reaction was terminated by diluting the reaction 1:1 with 6 m GdnHCl buffer. SIKE72 was isolated from the reaction by incubation with 40 μl of nickel-nitrilotriacetic acid resin, washed with GdnHCl buffer, and bound SIKE eluted with 50 μl of GdnHCl buffer plus 0.5 m imidazole. Eluent was separated by SDS-PAGE (10% Tris/glycine), and the target band was excised for analysis. The gel slice was washed and destained in 200 μl of 50% methanol overnight. The gel pieces were dehydrated in acetonitrile, rehydrated in 30 μl of 10 mm dithiothreitol in 0.1 m ammonium bicarbonate, and reduced at room temperature for 0.5 h. The DTT solution was removed, and the samples were alkylated in 30 μl of 50 mm iodoacetamide in 0.1 m ammonium bicarbonate at room temperature for 0.5 h. The reagent was removed, and the gel pieces were dehydrated in 100 μl of acetonitrile. The acetonitrile was removed, and the gel pieces were rehydrated in 100 μl of 0.1 m ammonium bicarbonate. The pieces were dehydrated in 100 μl of acetonitrile, and the acetonitrile was removed, and the pieces were completely dried by vacuum centrifugation. The gel pieces were rehydrated in 1 μg of trypsin in 50 mm ammonium bicarbonate on ice for 10 min. Any excess trypsin solution was removed, and 20 μl of 50 mm ammonium bicarbonate was added. The samples were digested overnight at 37 °C, and the peptides formed were extracted from the polyacrylamide in two 30-μl aliquots of 50% acetonitrile, 5% formic acid. Extracts, evaporated to 15 μl, were separated by C18 reversed-phase capillary column (Waters NanoAcquity) coupled with nanospray tandem mass spectrometry (LTQ-Orbitrap hybrid, Thermo Electron). The digest was analyzed using the double play capability of the instrument acquiring full scan mass spectra to determine peptide molecular weights and product ion spectra to determine amino acid sequence in sequential scans. The data were analyzed by database searching using the Sequest search algorithm against custom databases generated from the SIKE72 sequence. All potential phosphopeptides were manually examined for correct identification of the modified site.
RESULTS
SIKE Acts as a Mixed Type Inhibitor of TBK1-mediated Phosphorylation of IRF3
SIKE was originally classified as a physiological suppressor of TBK1 (36). Our studies into the mechanism of SIKE's inhibitory activity focused on the TBK1-mediated phosphorylation of IRF3. The IRF3 construct used in these studies included residues 173–427, encompassing the IRF activation domain and TBK1 phosphorylation sites (385, 386, 396, 398, 402, 404, and 405). The Michaelis-Menten plot of IRF3 as the varied substrate gave initial kinetic parameters for Km (Michaelis constant) and Vmax (maximum velocity) as 2.69 μm and 10.5 nm/min reaction, respectively (Fig. 1A and Table 1). Similarly, using ATP as the varied substrate, initial kinetic parameters for the Km and Vmax values of the reaction were 35.3 μm and 12.3 nm/min reaction, respectively (Fig. 1B and Table 1). To examine the effect of SIKE72 on IRF3 phosphorylation, SIKE72 was added at ∼20 and ∼80 nm to the IR-varied or ATP-varied (Fig. 1, C and D) assays. The primary effect of SIKE72 for IRF3-varied reactions occurred on Vmax (∼1.8-fold decrease), indicative of a noncompetitive inhibitor (Table 1). Interestingly, the Km and Vmax values calculated from double-reciprocal plots of the ATP-varied reactions indicated a 2.2–3-fold change in both Km and Vmax values consistent with a mixed-type inhibitor (Table 1). The Ki, app values were calculated from the double-reciprocal plot slope and intercept parameters using Equations 1 and 2, as described under “Experimental Procedures” (Table 2).
FIGURE 1.

SIKE is a mixed-type inhibitor of TBK1. A, Michaelis-Menten plot of TBK1-mediated phosphorylation of IRF3 with saturating ATP (100 μm), pre-activated TBK1 (4.93 nm), and IRF3 varied from 0.042 to 20.8 μm. Data were fit to a 2-parameter rectangular hyperbola (SigmaPlot). B, Michaelis-Menten plots of TBK1-mediated phosphorylation of IRF3 with increasing SIKE (0–83 nm) fit to a single rectangular 2-parameter hyperbola curve (SigmaPlot). C, Michaelis-Menten plot of TBK1-mediated phosphorylation of IRF3 with saturating IRF3 (20.8 μm), pre-activated TBK1 (4.93 nm), and ATP varied from 0 to 150 μm. Data were fit to a 2-parameter rectangular hyperbola (SigmaPlot). D, Michaelis-Menten plots of TBK1-mediated phosphorylation of IRF3 (20.8 μm) with ATP-varied (0.08–150 μm) and increasing SIKE (0–83 nm) fit to a single rectangular, 2-parameter hyperbola curve (SigmaPlot). Circle, no SIKE; triangle, 20.8 nm SIKE; square, 83 nm SIKE.
TABLE 1.
Michaelis-Menten and Lineweaver-Burk derived kinetic parameters
ND, not determined.
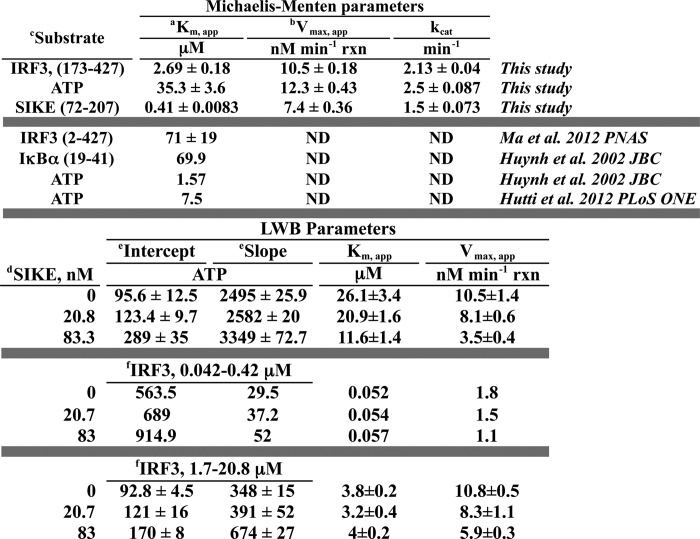
a Km,app refers to parameter describing Km for all seven IRF3 or IRF6 SIKE phosphorylation sites. Values were derived from a fitting single rectangular, 2-parameter hyperbola to the Vo versus s plots.
b Vmax,app refers to parameter describing Vmax for phosphorylation of IRF3 or SIKE at multiple sites. Values derived from fitting single rectangular, 2-parameter hyperbola to the Vo versus S plots.
c Substrates are listed with inclusive residues in parentheses.
d SIKE construct included residues 72–207.
e Intercept and slope values were derived from fitting the 1/Vo versus 1/[S] plots (supplemental Fig. 1, C and D) to a linear polynomial.
f IRF3 construct included residues 173–427 and parameters derived from data at listed concentrations.
TABLE 2.
Parameters for SIKE inhibition of TBK1-mediated IRF3 phosphorylation
| SIKE, nmb |
Ki, appa |
|||
|---|---|---|---|---|
| Intercept |
Slope |
|||
| 20.8 nm | 83.3 nm | 20.8 nm | 83.3 nm | |
| ATP | 71.7 ± 5.7 | 41.2 ± 5.0 | 589.7 ± 4.6 | 243.4 ± 5.3 |
| IRF3 (1.7–20.8 μm)c | 58.1 ± 8.1 | 100.4 ± 4.7 | ND | 88.9 ± 3.5 |
| IRF3 (0.042–0.42 μm) | 93.4 | 133.6 | 79.7 | 109.4 |
a Ki, app refers to parameter describing Ki for all IRF3 phosphorylation sites assayed. Ki derived from intercept(Inh) = intercept(none)(1 + [Inh]/Ki), where Inh = SIKE concentrations and intercept derived from linear regression of LWB plots.
b SIKE construct included residues 72–207.
c IRF3 construct included residues 173–427 and parameters derived from data at listed concentrations.
TBK1 Directly Phosphorylates SIKE in Vitro and dsRNA Stimulates SIKE Phosphorylation in Vivo
TBK1-mediated 32P incorporation was assessed by phosphor-K screen of SDS-PAGE-separated reactions with saturating ATP (100 μm), IRF3 held constant at 5 μm (approximate Km value), and increasing SIKE72 concentrations (5–5,000 nm). Analysis revealed two 32P-labeled species, IRF3 and SIKE72 (Fig. 2A). Inhibition of TBK1-mediated phosphorylation of IRF3 was apparent at 500 nm SIKE72, 10-fold less than the substrate concentration. Moreover, as IRF3 phosphorylation diminished, SIKE72 phosphorylation increased (Fig. 2A). dsRNA stimulation initiates multiple pathways leading to the activation of TBK1 (4–6). In HEK293 cells transiently transfected WT-FL SIKE, serine phosphorylation of WT-FL SIKE was observed following stimulation with poly(I:C), a synthetic dsRNA mimetic (Fig. 2B).
FIGURE 2.
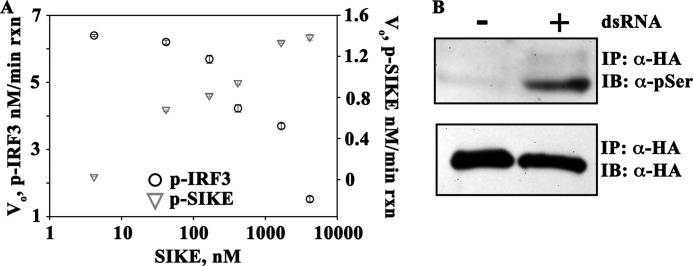
SIKE is directly phosphorylated by TBK1. A, SIKE (5–5,000 nm) was added to the TBK1 assay containing 5 μm IRF3 (∼Km concentration), 100 μm ATP, and 4.93 nm TBK1. Reactions were completed and analyzed as described under “Experimental Procedures.” ○ = p-IRF3; ▿ = p-SIKE. B, HEK293 cells were transiently transfected with HA-SIKE. Cells were stimulated for 3 h with poly(I:) (50 μg/ml). HA-SIKE was immunoprecipitated (IP) from lysates with α-HA resin. Total and phosphoserine SIKE was assessed via Western blot using α-HA or α-phospho-Ser antibody, respectively. One representative experiment is shown. IB, immunoblot.
TBK1 Phosphorylates SIKE on Six Serine Residues That Mimic the IRF3 Phosphorylation Motif
To determine the number of SIKE phosphorylation sites, we assessed TBK1-mediated phosphorylation by MALDI-TOF mass spectrometry. Over a 96-h time course, we observed a mixture of singly to triply phosphorylated SIKE72 (supplemental Fig. S2A). Under the same conditions, a single to 7 out of 7 known IRF3 phosphorylation sites were modified, whereas glyoxosomal malate dehydrogenase, not known to be phosphorylated but containing 16 preferred Ser/Thr motif sites out of 36 total Ser/Thr residues, remained unmodified (supplemental Fig. S2, B and C). To identify the positions of TBK1-mediated phosphorylation in SIKE72, we completed phosphopeptide mapping by tandem mass spectrometry. Six SIKE72 phosphoserine residues (Ser-133, Ser-185, Ser-187, Ser-188, Ser-190, and Ser-198) were identified (Fig. 3, A and B, and supplemental Fig. S3, A–F). When compared with the multiple phosphorylation sites of IRF3, SIKE72 and IRF3 share a remarkable, conserved phosphorylation motif (Fig. 3C).
FIGURE 3.

TBK1 phosphorylates SIKE on six serine residues that mimic the IRF3 phosphorylation sequence. A, mass spectrum resulting from analysis of indicated phosphopeptide sequence. Two separate ion series were recorded simultaneously, the b- and y-ion series, which represent sequencing inward from the N and C termini, respectively. Lowercase s indicates phosphorylated residue. B, summary of phosphopeptide sequences identified using a Sequest search algorithm against custom databases generated from the His6-SIKE72 sequence. ? indicates that a phosphorylation site exists in peptide but cannot be uniquely assigned to a single serine residue. C, sequence alignment of known TBK1-mediated phosphorylation sites on IRF3, required for dimerization and activation, with TBK1-mediated phosphorylation sites on SIKE found through phosphopeptide mapping. Gray letters, phosphorylated residues; *, conserved site; :, functionally conserved site.
SIKE Phosphorylation Status Alters Its Inhibitory Activity
To determine the relationship between the multiple SIKE phosphorylation sites and SIKE-mediated inhibition of IRF3 phosphorylation by TBK1, we probed these sites via truncated mutants or site-directed mutagenesis and assessed the effect of these SIKE mutations on TBK1-mediated IRF3 phosphorylation. The broad effect of SIKE phosphorylation was analyzed by the phosphomimetic mutant, Ser to Glu mutation, at the six identified sites (S6E), and phospho-knock-out mutant, Ser to Ala mutations, at the six identified sites (S6A). Subsets of phospho-knock-out mutants were created to further probe sites essential for SIKE's inhibitory activity: S4A (S185A, S187A, S188A, and S190A), S2A (S133A and S198A), and S185A. SIKE's phosphorylation sites cluster in the C-terminal portion of SIKE. To examine the role of the N-terminal sequence in SIKE inhibition, SIKE truncation mutants containing one (SIKE(72–184)) or retaining all phosphorylation sites (SIKE(113–207)) were constructed. The panel of constructs is summarized in Fig. 4A.
FIGURE 4.

Phosphorylation status modulates SIKE inhibitory activity. A, schematic diagram of SIKE constructs highlighting the six potential sites of serine phosphorylation, mutations used to probe phosphorylation sites, and truncation mutants. Serines are listed from left to right: 133, 185, 187, 188, 190, and 198. CC, coiled coil. B, WT or mutant SIKE (4–1658 nm) was added to the TBK1 assay containing 5 μm IRF3 (∼Km concentration), 100 μm ATP, and 4.93 nm TBK1. Reactions were completed and analyzed as described under “Experimental Procedures.”
Inhibition curves for the phosphomimetic and phospho-knock-out mutants showed that phosphorylation of SIKE72 reduced SIKE's ability to inhibit TBK1-mediated IRF3 phosphorylation, whereas unmodified SIKE72 had greatly enhanced inhibitory activity (supplemental Fig. S4A). Using four concentrations that defined SIKE's inhibitory effect, kinase assays were completed with the full panel of mutants (Fig. 4B). Apparent inhibition constants (Ki, app) for each mutant were derived (Table 3). The Ki, app for the phosphomimetic mutant increased ∼3-fold over WT SIKE72, suggesting that phosphorylation may reduce the affinity between TBK1-SIKE and/or enhance release of SIKE from TBK1. The Ki, app for the C-terminal 113–207 construct, retaining all phosphorylation sites, was similar to WT SIKE72. Surprisingly, the Ki, app for the N-terminal 72–184 construct was 196 nm, even though maximal inhibition was only ∼25% of the reaction. Loss of the two peripheral phosphorylation sites, Ser-133 and Ser-198, in the S2A construct also did not alter the Ki, app value, whereas phospho-knock-out of the four clustered serines, Ser-185, Ser-187, Ser-188, and Ser-190, reduced the Ki, app parameter by 2.5-fold. Within this cluster, the Ki, app for the point mutation S185A was similar to the S6A mutant (Table 3).
TABLE 3.
Parameters for mutant SIKE inhibition of TBK1-mediated IRF3 phosphorylation
| SIKE constructa | Ki, appb |
|---|---|
| nm | |
| WT | 350 ± 16 |
| 72–184 | 196 ± 7.9 |
| 113–207 | 363 ± 52 |
| S6E | 900 ± 56 |
| S6A | 77 ± 6 |
| S4A | 140 ± 7 |
| S2A | 368 ± 14 |
| S185A | 41 ± 8 |
a SIKE construct included residues 72–207 except where explicitly stated.
b Ki, app derived from fit of 2-parameter rectangular hyperbola to % inhibited Vo-pIRF3 μm/min versus SIKE concentration.
SIKE Phosphorylation Status Controls TBK1-SIKE Interaction in Vivo
To investigate the effect of SIKE phosphorylation on the TBK1-SIKE interaction, we utilized co-immunoprecipitation assays of epitope-tagged TBK1 and WT or mutant FL SIKE. Prior to stimulation with poly(I:C), all HA-tagged SIKE constructs co-immunoprecipitated with FLAG-tagged TBK1 (Fig. 5A). The S6E, S185E, and 1–112 constructs showed limited interaction with TBK1 relative to input protein. Following poly(I:) stimulation, WT-FL, 1–112, and 113–207 SIKE constructs were released from TBK1, whereas the S6A, S185A, S185E, and S6E SIKE interactions with TBK1 were unchanged from unstimulated conditions (Fig. 5A). The release of WT-FL SIKE from TBK1 correlates with increased TBK1-mediated phosphorylation of IRF3 (Fig. 5B). The reduced interaction of S6E or S185E SIKE and TBK1 has no effect on dsRNA-stimulated IRF3 phosphorylation, but the stable interaction of S6A or S185A SIKE and TBK1, irrespective of dsRNA stimulation, negates dsRNA-stimulated IRF3 phosphorylation.
FIGURE 5.

Phosphorylation status modulates TBK1/SIKE interaction and p-IRF3 in vivo. A, HEK293 cells were transiently transfected with FLAG-TBK1 and HA-tagged WT or mutant SIKE. Cells were stimulated with poly(I:) (50 μg/ml) for 3 h. FLAG-TBK1 was immunoprecipitated (IP) from lysates with α-FLAG resin, separated by SDS-PAGE, transferred to nitrocellulose, and immunoblotted (IB) with α-HA or a-FLAG antibody. Immunoblots of whole cell lysates show expression of each construct. Blots are representative of three independent experiments. B, HEK293 cells were transiently transfected with empty vector (none) or TLR3 and WT or mutant SIKE. Cells were stimulated with poly(I:) (50 μg/ml) for 3 h. Immunoblots of whole cell lysates were probed for α-phospho-Ser-396-IRF3, α-IRF3, α-HA, or α-actin antibodies. Blots are representative of two independent experiments. Densitometry was calculated using ImageJ software. Each band was corrected for background scatter, and the phospho-IRF3 and IRF3 bands were normalized to their respective actin bands. Ratio of phospho-IRF3/total IRF3 is reported under corresponding bands.
SIKE Is a TBK1 Substrate
To establish kinetic parameters for SIKE as a TBK1 substrate, TBK1 assays with saturating ATP (100 μm) and SIKE72 varied from 0.043 to 8.3 μm were completed. The Km and Vmax parameters were 0.41 μm and 7.4 nm/min reaction, respectively (Fig. 6A and Table 1). Strikingly, the double-reciprocal plot revealed a downward concave curve, similar to the double-reciprocal plot of IRF3-varied, TBK1 assays (Fig. 6, B and C, and supplemental Fig. S1C).
FIGURE 6.
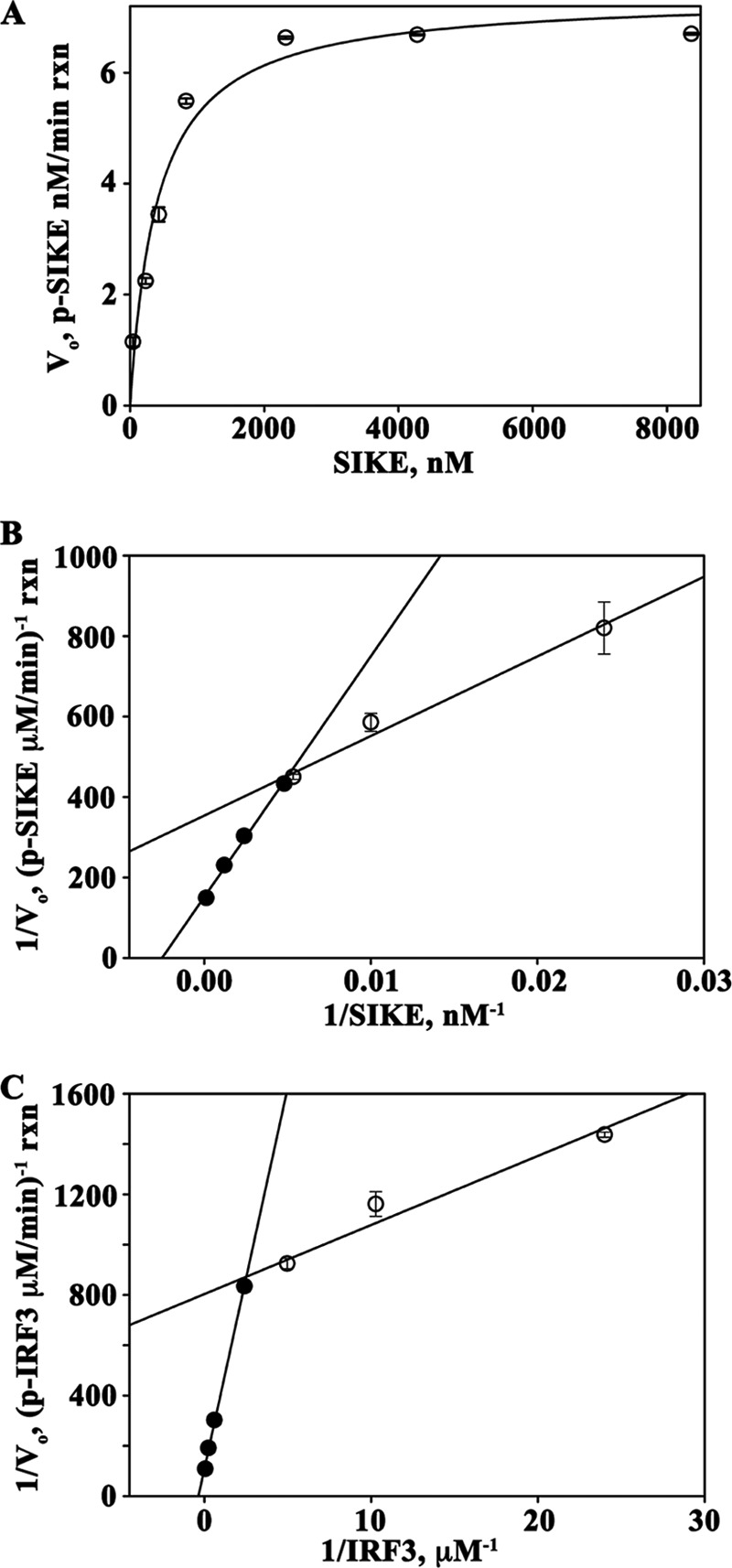
SIKE is a substrate of TBK1. A, Michaelis-Menten plot of TBK1-mediated phosphorylation of SIKE with saturating ATP (100 mm), pre-activated TBK1 (4.93 nm), and SIKE varied from 0.043 to 8.4 mm. Data were fit to a 2-parameter rectangular hyperbola (SigmaPlot). B and C, Lineweaver-Burk plots of TBK1-SIKE assays with SIKE varied from 0.043 to 8.4 mm, ATP (100 mm) (B) and TBK1-IRF3 assays with IRF3 varied from 0.042 to 20.8 mm, ATP (100 mm) (C). TBK1 was at 4.93 nm in both assays. Data for 0.042–0.42 μm (○) and 1.7–20.8 μm (●) IRF3 and 0.043–0.23 μm (○) and 0.43–8.4 μm (●) SIKE were fit to a linear polynomial equation (SigmaPlot).
DISCUSSION
Multiple innate immune receptors recognize pathogen-derived ligands and initiate an immediate inflammatory response (38, 39). Several of these signaling pathways converge to activate TANK-binding kinase 1 (TBK1) (9, 22, 24, 40, 41). Although a central player in the innate immune system's defenses that shapes the downstream innate immune response, mechanisms by which the host controls TBK1 activity are not well understood. To address this issue, we sought to define mechanistically how an endogenous inhibitor of TBK1, SIKE, blocks the TBK1-mediated anti-viral response.
The major discovery of this work is that SIKE is not only an endogenous inhibitor of TBK1-mediated phosphorylation of IRF3 but is also a TBK1 substrate. SIKE has a 6.5-fold lower Km value (0.41 μm), as compared with IRF3, with a comparable Vmax value suggesting that changes in SIKE concentration could effectively manipulate TBK1 function through altered substrate selection. Using kinetic analyses and protein interactions assays, we characterized how this novel TBK1 substrate effectively inhibits type I interferon production.
TBK1 Displays Negative Cooperativity for Substrates IRF3 and SIKE
In our studies, we examined the effect of SIKE on TBK1-mediated IRF3 phosphorylation using a macromolecular IRF3 substrate (residues 173–427). Previous studies on the TBK1 kinetic mechanism, using an inhibitor of κBα peptide as substrate, showed that the enzyme functioned via a RERO mechanism (31). Our data obtained with macromolecular substrates are consistent with this mechanism but reveal evidence suggesting the existence of negative cooperativity in substrate binding or the presence of a regulatory site for protein substrate. The equilibria for this mechanism with respect to IRF3 phosphorylation are given in Fig. 7A, and are described by Dalziel's generalized rate Equation 3 (42),
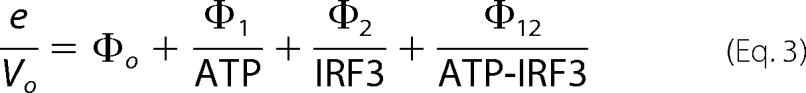 |
where e is TBK1 concentration in the reaction; Vo is the initial velocity of the reaction, and Φ are Dalziel coefficients that, for an RERO mechanism, pertain to the concentrations of specific complexes in the reaction mechanism (Φ1 = TBK1-IRF3, Φ2 = TBK1-ATP, Φ12 = TBK1, and Φ0 = TBK1-ATP-IRF3), labeled in Fig. 7A.
FIGURE 7.

A, diagram of RERO enzyme mechanism of TBK1 with binding of I (SIKE) to each enzyme complex. B, schematic of mechanisms to interpret nonlinear Lineweaver-Burk plots of TBK1-IRF3 and TBK1-SIKE data and SIKE inhibition of TBK1-mediated phosphorylation of IRF3. Ribbon diagram of TBK1 (Protein Data Bank code 4IM0 (44)) shows overall arrangement of dimeric kinase.
We first determined the base-line kinetic parameters for IRF3 and ATP as summarized in Table 1. The double-reciprocal plot of IRF3-varied substrate was not linear but a downward concave curve (Fig. 6C). This type of plot is indicative of one of three phenomena as follows: (i) substrate activation; (ii) nonidentical active sites, or (iii) negative cooperativity. In examining these three explanations, we assumed from both the related IKKβ (43) and TBK1 (30, 44, 45) crystallographic structure and our own size exclusion chromatography (data not shown) that TBK1 functions as a dimeric species (required for explanations ii and iii above). Substrate activation entails a substrate-binding site (with lower affinity than the active site) that when bound leads to activation of the reaction. For nonidentical active sites, the dimeric TBK1 would have a high and low affinity active site for substrate a priori. Finally, negative cooperativity between TBK1 active sites would occur if binding of substrate at the first active site lowered the affinity for substrate at the second active site by allosteric interactions between the subunits. Alone, the IRF3 varied assay data did not allow us to discern which mechanism is at play. Luckily, SIKE also functioned as a macromolecular TBK1 substrate. The double-reciprocal plot of the SIKE-varied assays also exhibited this characteristic downward concave curvature (Fig. 6B). As SIKE and IRF3 share no sequence homology and are distinct macromolecular substrates, the substrate activation or nonidentical active site mechanism seemed improbable as these macromolecular substrates must be similarly recognized by the kinase at a site independent of the active site or with equally reduced affinity at a nonidentical active site. Rather, negative cooperativity induced by recognition of IRF3's and SIKE's shared phosphorylation motif could explain the nonlinear double-reciprocal plots. Negative cooperativity requires communication between active sites. Interestingly, in IKKβ the two active sites appear to be independent of one another with dimerization mediated by the scaffold dimerization domain. In contrast, the TBK1 structure revealed three additional dimerization interfaces between the kinase and ubiquitin-like domains of one subunit to the scaffold dimerization domain of the contralateral subunit (Fig. 7B). These additional subunit interfaces identified in the TBK1 structures may provide a means of communicating substrate binding between active sites. Structural study of a kinase-substrate complex to complement the reported kinase-ATP analog inhibitor structures (30, 44, 45) would directly address this hypothesis.
In a RERO mechanism, the Km value of IRF3 is the Kd value for IRF3 binding to the E-ATP complex. From the two linear regions of the IRF3 double-reciprocal plot, we calculated the Km value at low (0.042–0.42) versus high (0.42–20.8) substrate concentrations as 50 nm and 3.5 μm, respectively. Similarly, the two linear regions of SIKE's double-reciprocal plot were 55.9 and 395 nm at low (42–208 nm) and high (208–8,000 nm) substrate concentrations, respectively. These data suggest that binding of substrate, either IRF3 or SIKE, to the first active site occurs with near equal affinity. The ∼10-fold difference in Km values at high substrate concentration favoring the TBK1-SIKE complex would suggest that SIKE functions primarily as a competitive inhibitor of IRF3 phosphorylation.
SIKE's Inhibitory Mechanism
For a simple RERO mechanism, an alternative substrate (as we have shown SIKE to be) should give competitive inhibition with respect to IRF3 and mixed (or noncompetitive) inhibition with respect to ATP (46). Analysis of SIKE's inhibitory effects on TBK1-mediated phosphorylation of IRF3 revealed a mixed-type inhibitor (Fig. 1 and Table 1) with respect to ATP with ∼2–3-fold decreases in Km and Vmax values (Fig. 1D and Table 1), as expected for a RERO mechanism. Surprisingly, SIKE inhibition in IRF3-varied experiments did not exhibit a competitive inhibition pattern but rather displayed primarily as a noncompetitive inhibitor, reducing Vmax by 3-fold (Fig. 1C and Table 1). To further interpret these results, we examined SIKE inhibition of the TBK1 reaction in terms of the generalized linear rate equation for a RERO mechanism developed by Dalziel (42). In the RERO mechanism, SIKE could in theory interact with any TBK1 complex and would affect the appropriate Φ parameter. The rearranged generalized linear rate Equation 4 with ATP as the varied substrate is as follows:
 |
In Equation 4, the slope effects result from SIKE interactions with E and/or E-IRF3, whereas intercept effects result from the interaction of SIKE with E-ATP and/or E-ATP-IRF3. From the Ki, app values determined from the slopes and intercepts of the double-reciprocal plots, we observed that the Ki, app derived from the intercept is ∼56 nm, whereas the Ki, app derived from the slopes is ∼420 nm. This suggests that the apparent affinity of the E-ATP or E-ATP-IRF3 complexes for SIKE is greater than the affinity of E alone or the E-IRF3 complex. Rearranging the generalized Equation 5 for IRF3 as the varied substrate gives
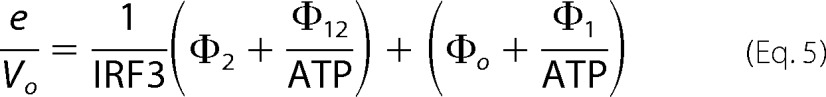 |
Here, the slope effects can be attributed to the interaction of SIKE with E and/or E-ATP, whereas intercept effects are attributable to SIKE interactions with E-IRF3 and/or E-ATP-IRF3. The Ki, app values determined from slope and intercept were 111 and 98 nm, respectively, equivalent between the two grouped complexes. Because we have shown that SIKE is directly phosphorylated by TBK1 (Figs. 2C and 6A), SIKE would be expected to bind to free enzyme or enzyme-ATP complexes but not IRF3-containing complexes. The fact that SIKE alters either or both Φ0 or Φ1 suggests that these effects must arise from either allosteric interactions between active sites or the existence of a separate regulatory site for SIKE. Both of these possibilities are consistent with the observed downward curvature of the double-reciprocal plots with either SIKE or IRF3 as the varied substrate. Because IRF3 and SIKE are quite different overall structures, these studies suggest that TBK1 substrates bind with negative cooperativity between otherwise identical sites rather than the existence of an IRF3/SIKE-binding site separate from the active sites in the dimer.
Effect of SIKE Phosphorylation on Inhibitory Function
Four of the six phosphorylated serines of SIKE align well with the phosphorylation sites of IRF3 (Fig. 3C) (15, 16). The strongest homology was evident at the SIKE sequence 184LSISSE189 composed of a cluster of three phosphorylation sites. When TBK1 substrates are compared, the phosphorylation pattern of TBK1 substrates has three forms as follows: singly as in Akt (22), insulin receptor (21), and optineurin (23), multiple sites but not clustered as in DDX3 (47), or multiple clustered sites as in IRF3 (15, 16), IRF7 (48, 49) and, as we show here, SIKE. In IRF3/7, these clustered phosphorylation sites, when modified, alter protein function, activating the transcription factor (17–20). We explored the role of these clustered SIKE phosphorylation sites in the context of inhibition of TBK1-mediated IRF3 phosphorylation. When replaced with the phosphomimetic mutations, Ser to Glu, SIKE's inhibitory activity (Ki, app ∼900 nm versus WT Ki, app = 350 nm) and interaction with TBK1 are greatly diminished (Figs. 4B and 5A). As the introduced negative charge grossly mimics SIKE's product form, these results are consistent with product release from an enzyme. Initial studies of SIKE held that SIKE maintained TBK1 in an inactive state prior to TBK1 activation at which point SIKE was released from the TBK1 complex. Here, we attribute the TBK1-SIKE interaction to SIKE's high affinity for the enzyme and SIKE's release to its post-translational modifications.
Similar to IRF3 where individual phosphorylation sites have been attributed to specific function, we probed the six SIKE phosphorylation sites to determine whether a subset of residues was essential to TBK1's preferential binding to SIKE over IRF3. With a complete phospho-knock-out mutant (Ser to Ala), SIKE's inhibitory activity increased 4.5-fold, consistent with retained TBK1 recognition. The S2A versus S4A mutants further defined the cluster of phosphorylation sites centered at the 185SXSSXS190 motif as essential to phosphorylation-dependent release of SIKE from TBK1, whereas the phosphorylation state of the S2A sites did not contribute to SIKE's release from TBK1. The single mutation, S185A, alone could enhance inhibition of TBK1-mediated IRF3 phosphorylation. In fact, the Ki, app values for the S6A (77 nm) and the single S185A (41 nm) mutants are in line with the Michaelis constant at low SIKE concentrations (55.9 nm, Fig. 6B). Additional interactions between TBK1 and SIKE or the structural context of the phosphorylation motif may also contribute to the high affinity TBK1-SIKE interaction. This is supported by the ability of SIKE(72–184) to inhibit TBK1 function and SIKE(1–112) to form an interaction with TBK1 (Figs. 4B and 5A). SIKE(72–184) retains a single phosphorylation site (Ser-133) that when mutated to Ala (S2A mutant) does not alter SIKE inhibitory activity. Therefore, inhibition by this mutant is attributed to not only Ser-133 docking to TBK1's active site, but also interactions between SIKE and TBK1 outside of the active site. Similarly, SIKE(1–112), as it contains no phosphorylation sites, must form a TBK1-SIKE interaction exclusive of the active site. These interactions independent of the active site are consistent with the noncompetitive/mixed type inhibition mediated by SIKE.
SIKE's Role in Innate Immunity
SIKE's original characterization as an endogenous inhibitor of the TBK1-mediated anti-viral response only partially explains the SIKE function. Comparison of the pseudo-Kd value for the E-ATP complex showed that SIKE is preferred as a TBK1 substrate 6.5-fold over IRF3. At equivalent concentrations, SIKE would appear to inhibit IRF3 phosphorylation. This would not be the first time a TBK1 substrate had initially been defined as an inhibitor. Optineurin, an autophagy receptor, was classified as an endogenous TBK1 inhibitor (50). Subsequent studies defined optineurin as a TBK1 substrate (23, 24). Phospho-optineurin has increased affinity for the microtubule-associated protein light chain 3, thereby promoting selective autophagy of ubiquitin-coated cytosolic bacteria and providing a mechanism by which TBK1 functions in maintaining xenophagosomes (24). If not just an endogenous regulator of TBK1-mediated phosphorylation of IRF3, what is the primary function of SIKE? Two pieces of data suggest a SIKE function. First, a related protein, fibroblast growth factor receptor 1 oncoprotein partner 2 (51), shares 56% sequence identity with SIKE but is expressed from chromosome 12 instead of chromosome 1 and lacks all six TBK1 phosphorylation sites. Fibroblast growth factor receptor 1 oncoprotein partner 2 (FGFR1OP2) associates with the actin cytoskeleton and has been linked to wound healing pathways (52). Second, we have shown by fluorescence microscopy that SIKE fused to GFP localizes to the cytosol with accumulation at actin/cytoskeleton-like structures at the cell periphery and within the lamellipodia (data not shown). We hypothesize that SIKE functions in cytoskeletal rearrangement in response to pathogen challenge through its phosphorylation by TBK1. Current studies in the laboratory are focused on testing this hypothesis.
In summary, we have revealed through kinetic analysis that SIKE is a novel, high affinity TBK1 substrate. TBK1-SIKE interactions are modulated by SIKE phosphorylation but are not limited to the typical kinase-substrate interactions within the active site. TBK1 phosphorylation of increasing IRF3 or SIKE concentrations displays negative cooperativity. The mechanism by which substrate binding is allosterically conveyed between subunits remains to be determined. With respect to IRF3 phosphorylation, SIKE effectively functioned as a mixed-type inhibitor of TBK1-mediated IRF3 phosphorylation rather than, given its status as a TBK1 substrate, as a competitive inhibitor. Together, these studies suggest that endogenous control of a critical catalytic hub is not only achieved by direct repression of activity but also by redirection of catalysis through substrate affinity. Importantly, these studies identify a novel TBK1 substrate modified in response to pathogen challenge. Further investigation into SIKE's function in the innate immune response will uncover unique pathways and provide insight into novel mechanisms of host immune defenses.
Supplementary Material
Acknowledgments
We thank the Spiegel and Kordula laboratories (Virginia Commonwealth University) for generously sharing their resources for radiolabeled assays.
This work was supported by Thomas F. and Kate Miller Jeffress Memorial Trust Grant J1016 (to J. K. B.).

This article contains supplemental Methods, Figs. 1–4, and Table 1.
- TLR3
- Toll-like receptor 3
- Akt
- protein kinase B
- DDX3
- ATP-dependent RNA helicase DEAD box protein 3
- IKK
- IκB kinase
- IRF3
- interferon regulatory factor 3
- NF-κB
- nuclear factor κB
- RERO
- rapid equilibrium, random order
- SIKE
- suppressor of IKKϵ
- TBK1
- TANK binding kinase 1
- CV
- column volume
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- GdnHCl
- guanidine hydrochloride.
REFERENCES
- 1. Kumar H., Kawai T., Akira S. (2011) Pathogen recognition by the innate immune system. Int. Rev. Immunol. 30, 16–34 [DOI] [PubMed] [Google Scholar]
- 2. Keating S. E., Baran M., Bowie A. G. (2011) Cytosolic DNA sensors regulating type I interferon induction. Trends Immunol. 32, 574–581 [DOI] [PubMed] [Google Scholar]
- 3. Wang B. X., Fish E. N. (2012) The yin and yang of viruses and interferons. Trends Immunol. 33, 190–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Matsumoto M., Kikkawa S., Kohase M., Miyake K., Seya T. (2002) Establishment of a monoclonal antibody against human Toll-like receptor 3 that blocks double-stranded RNA-mediated signaling. Biochem. Biophys. Res. Commun. 293, 1364–1369 [DOI] [PubMed] [Google Scholar]
- 5. Yoneyama M., Kikuchi M., Natsukawa T., Shinobu N., Imaizumi T., Miyagishi M., Taira K., Akira S., Fujita T. (2004) The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate anti-viral responses. Nat. Immunol. 5, 730–737 [DOI] [PubMed] [Google Scholar]
- 6. Kang D.-C., Gopalkrishnan R. V., Wu Q., Jankowsky E., Pyle A. M., Fisher P. B. (2002) mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc. Natl. Acad. Sci. U.S.A. 99, 637–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Takaoka A., Wang Z., Choi M. K., Yanai H., Negishi H., Ban T., Lu Y., Miyagishi M., Kodama T., Honda K., Ohba Y., Taniguchi T. (2007) DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448, 501–505 [DOI] [PubMed] [Google Scholar]
- 8. Chau T.-L., Gioia R., Gatot J.-S., Patrascu F., Carpentier I., Chapelle J.-P., O'Neill L., Beyaert R., Piette J., Chariot A. (2008) Are the IKKs and IKK-related kinases TBK1 and IKK-ϵ similarly activated? Trends Biochem. Sci. 33, 171–180 [DOI] [PubMed] [Google Scholar]
- 9. Fitzgerald K. A., McWhirter S. M., Faia K. L., Rowe D. C., Latz E., Golenbock D. T., Coyle A. J., Liao S.-M., Maniatis T. (2003) IKKϵ and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4, 491–496 [DOI] [PubMed] [Google Scholar]
- 10. Sharma S., tenOever B. R., Grandvaux N., Zhou G. P., Lin R., Hiscott J. (2003) Triggering the interferon anti-viral response through an IKK-related pathway. Science 300, 1148–1151 [DOI] [PubMed] [Google Scholar]
- 11. Pomerantz J. L., Baltimore D. (1999) NF-κB activation by a signaling complex containing TRAF2, TANK, and TBK1, a novel IKK-related kinase. EMBO J. 18, 6694–6704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tojima Y., Fujimoto A., Delhase M., Chen Y., Hatakeyama S., Nakayama K., Kaneko Y., Nimura Y., Motoyama N., Ikeda K., Karin M., Nakanishi M. (2000) NAK is an IκB kinase-activating kinase. Nature 404, 778–782 [DOI] [PubMed] [Google Scholar]
- 13. Bonnard M., Mirtsos C., Suzuki S., Graham K., Huang J., Ng M., Itié A., Wakeham A., Shahinian A., Henzel W. J., Elia A. J., Shillinglaw W., Mak T. W., Cao Z., Yeh W.-C. (2000) Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-κB-dependent gene transcription. EMBO J. 19, 4976–4985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu F., Xia Y., Parker A. S., Verma I. M. (2012) IKK biology. Immunol. Rev. 246, 239–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin R., Heylbroeck C., Pitha P. M., Hiscott J. (1998) Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell Biol. 18, 2986–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yoneyama M., Suhara W., Fukuhara Y., Fukuda M., Nishida E., Fujita T. (1998) Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 17, 1087–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takahasi K., Suzuki N. N., Horiuchi M., Mori M., Suhara W., Okabe Y., Fukuhara Y., Terasawa H., Akira S., Fujita T., Inagaki F. (2003) X-ray crystal structure of IRF-3 and its functional implications. Nat. Struct. Biol. 10, 922–927 [DOI] [PubMed] [Google Scholar]
- 18. Mori M., Yoneyama M., Ito T., Takahashi K., Inagaki F., Fujita T. (2004) Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. J. Biol. Chem. 279, 9698–9702 [DOI] [PubMed] [Google Scholar]
- 19. Servant M. J., Grandvaux N., tenOever B. R., Duguay D., Lin R., Hiscott J. (2003) Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA. J. Biol. Chem. 278, 9441–9447 [DOI] [PubMed] [Google Scholar]
- 20. Panne D., McWhirter S. M., Maniatis T., Harrison S. C. (2007) Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. J. Biol. Chem. 282, 22816–22822 [DOI] [PubMed] [Google Scholar]
- 21. Muñoz M. C., Giani J. F., Mayer M. A., Toblli J. E., Turyn D., Dominici F. P. (2009) TANK-binding kinase 1 mediates phosphorylation of insulin receptor at serine residue 994: a potential link between inflammation and insulin resistance. J. Endocrinol. 201, 185–197 [DOI] [PubMed] [Google Scholar]
- 22. Ou Y.-H., Torres M., Ram R., Formstecher E., Roland C., Cheng T., Brekken R., Wurz R., Tasker A., Polverino T., Tan S.-L., White M. A. (2011) TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol. Cell 41, 458–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gleason C. E., Ordureau A., Gourlay R., Arthur J. S., Cohen P. (2011) Polyubiquitin binding to optineurin is required for optimal activation of TANK-binding kinase 1 and production of interferon β. J. Biol. Chem. 286, 35663–35674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wild P., Farhan H., McEwan D. G., Wagner S., Rogov V. V., Brady N. R., Richter B., Korac J., Waidmann O., Choudhary C., Dötsch V., Bumann D., Dikic I. (2011) Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333, 228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shen R. R., Hahn W. C. (2011) Emerging roles for the noncanonical IKKs in cancer. Oncogene 30, 631–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oganesyan G., Saha S. K., Pietras E. M., Guo B., Miyahira A. K., Zarnegar B., Cheng G. (2008) IRF3-dependent type I interferon response in B cells regulates CpG-mediated antibody production. J. Biol. Chem. 283, 802–808 [DOI] [PubMed] [Google Scholar]
- 27. Hammaker D., Boyle D. L., Firestein G. S. (2012) Synoviocyte innate immune responses: TANK-binding kinase-1 as a potential therapeutic target in rheumatoid arthritis. Rheumatology 51, 610–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kishore N., Huynh Q. K., Mathialagan S., Hall T., Rouw S., Creely D., Lange G., Caroll J., Reitz B., Donnelly A., Boddupalli H., Combs R. G., Kretzmer K., Tripp C. S. (2002) IKK-i and TBK-1 are enzymatically distinct from the homologous enzyme IKK-2. Comparative analysis of recombinant human IKK-i, TBK-1, and IKK-2. J. Biol. Chem. 277, 13840–13847 [DOI] [PubMed] [Google Scholar]
- 29. Clark K., Peggie M., Plater L., Sorcek R. J., Young E. R., Madwed J. B., Hough J., McIver E. G., Cohen P. (2011) Novel cross-talk within the IKK family controls innate immunity. Biochem. J. 434, 93–104 [DOI] [PubMed] [Google Scholar]
- 30. Ma X., Helgason E., Phung Q. T., Quan C. L., Iyer R. S., Lee M. W., Bowman K. K., Starovasnik M. A., Dueber E. C. (2012) Molecular basis of Tank-binding kinase 1 activation by trans-autophosphorylation. Proc. Natl. Acad. Sci. U.S.A. 109, 9378–9383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huynh Q. K., Kishore N., Mathialagan S., Donnelly A. M., Tripp C. S. (2002) Kinetic mechanisms of IκB-related kinases (IKK) inducible IKK and TBK-1 differ from IKK-1/IKK-2 heterodimer. J. Biol. Chem. 277, 12550–12558 [DOI] [PubMed] [Google Scholar]
- 32. Burke J. R., Miller K. R., Wood M. K., Meyers C. A. (1998) The multisubunit IκB kinase complex shows random sequential kinetics and is activated by the C-terminal domain of IκBα. J. Biol. Chem. 273, 12041–12046 [DOI] [PubMed] [Google Scholar]
- 33. Peet G. W., Li J. (1999) IκB kinases α and β show a random sequential kinetic mechanism and are inhibited by staurosporine and quercetin. J. Biol. Chem. 274, 32655–32661 [DOI] [PubMed] [Google Scholar]
- 34. Parvatiyar K., Barber G. N., Harhaj E. W. (2010) TAX1BP1 and A20 inhibit anti-viral signaling by targeting TBK1-IKKi kinases. J. Biol. Chem. 285, 14999–15009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. An H., Zhao W., Hou J., Zhang Y., Xie Y., Zheng Y., Xu H., Qian C., Zhou J., Yu Y., Liu S., Feng G., Cao X. (2006) SHP-2 phosphatase negatively regulates the TRIF adaptor protein-dependent type I interferon and proinflammatory cytokine production. Immunity 25, 919–928 [DOI] [PubMed] [Google Scholar]
- 36. Huang J., Liu T., Xu L. G., Chen D., Zhai Z., Shu H. B. (2005) SIKE is an IKKϵ/TBK1-associated suppressor of TLR3- and virus-triggered IRF-3 activation pathways. EMBO J. 24, 4018–4028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Piao W., Song C., Chen H., Diaz M. A., Wahl L. M., Fitzgerald K. A., Li L., Medvedev A. E. (2009) Endotoxin tolerance dysregulates MyD88- and Toll/IL-1R domain-containing adapter inducing IFN-β-dependent pathways and increases expression of negative regulators of TLR signaling. J. Leukocyte Biol. 86, 863–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thompson M. R., Kaminski J. J., Kurt-Jones E. A., Fitzgerald K. A. (2011) Pattern recognition receptors and the innate immune response to viral infection. Viruses 3, 920–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Newton K., Dixit V. M. (2012) Signaling in innate immunity and inflammation. Cold Spring Harbor Perspect. Biol. 4, a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chien Y., Kim S., Bumeister R., Loo Y.-M., Kwon S. W., Johnson C. L., Balakireva M. G., Romeo Y., Kopelovich L., Gale M., Jr., Yeaman C., Camonis J. H., Zhao Y., White M. A. (2006) RalB GTPase-mediated activation of the IκB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell 127, 157–170 [DOI] [PubMed] [Google Scholar]
- 41. Radtke A. L., Delbridge L. M., Balachandran S., Barber G. N., O'Riordan M. X. (2007) TBK1 protects vacuolar integrity during intracellular bacterial infection. PLoS Pathog. 3, e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dalziel K. (1957) Initial steady state velocities in the evaluation of enzyme-coenzyme-substrate reaction mechanisms. Acta Chem. Scand. 11, 1706–1723 [Google Scholar]
- 43. Xu G., Lo Y.-C., Li Q., Napolitano G., Wu X., Jiang X., Dreano M., Karin M., Wu H. (2011) Crystal structure of inhibitor of κB kinase. Nature 472, 325–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tu D., Zhu Z., Zhou A. Y., Yun C.-H., Lee K.-E., Toms A. V., Li Y., Dunn G. P., Chan E., Thai T., Yang S., Ficarro S. B., Marto J. A., Jeon H., Hahn W. C., Barbie D. A., Eck M. J. (2013) Structure and ubiquitination-dependent activation of TANK-binding kinase 1. Cell Rep. 3, 747–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Larabi A., Devos J. M., Ng S.-L., Nanao M. H., Round A., Maniatis T., Panne D. (2013) Crystal structure and mechanism of activation of TANK-binding kinase 1. Cell Rep. 3, 734–746 [DOI] [PubMed] [Google Scholar]
- 46. Bell J. E., Beyer T. A., Hill R. L. (1976) The kinetic mechanism of bovine milk galactosyltransferase. The role of α-lactalbumin. J. Biol. Chem. 251, 3003–3013 [PubMed] [Google Scholar]
- 47. Soulat D., Bürckstümmer T., Westermayer S., Goncalves A., Bauch A., Stefanovic A., Hantschel O., Bennett K. L., Decker T., Superti-Furga G. (2008) The DEAD-box helicase DDX3X is a critical component of the TANK-binding kinase 1-dependent innate immune response. EMBO J. 27, 2135–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lin R., Mamane Y., Hiscott J. (2000) Multiple regulatory domains control IRF-7 activity in response to virus infection. J. Biol. Chem. 275, 34320–34327 [DOI] [PubMed] [Google Scholar]
- 49. Paz S., Sun Q., Nakhael P., Romieu-Mourez R., Goubau D., Juikunen I., Lin R., Hiscott J. (2006) Induction of IRF-3 and IRF-7 phosphorylation following activation of the RIG-I pathway. Cell. Mol. Biol. 52, 17–28 [PubMed] [Google Scholar]
- 50. Mankouri J., Fragkoudis R., Richards K. H., Wetherill L. F., Harris M., Kohl A., Elliott R. M., Macdonald A. (2010) Optineurin negatively regulates the induction of IFNβ in response to RNA virus infection. PLoS Pathog. 6, e1000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Grand E. K., Grand F. H., Chase A. J., Ross F. M., Corcoran M. M., Oscier D. G., Cross N. C. (2004) Identification of a novel gene, FGFR1OP2, fused to FGFR1 in 8p11 myeloproliferative syndrome. Genes Chromosomes Cancer 40, 78–83 [DOI] [PubMed] [Google Scholar]
- 52. Lin A., Hokugo A., Choi J., Nishimura I. (2010) Small cytoskeleton-associated molecule, fibroblast growth factor receptor 1 oncogene partner 2/would inducible transcript-3.0 (FGFR1OP2/wit3.0), facilitates fibroblast-driven would closure. Am. J. Pathol. 176, 108–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.