

Abstract
Aims
Prasugrel is a novel thienopyridine P2Y12 adenosine diphosphate (ADP) receptor antagonist that inhibits ADP-mediated platelet activation and aggregation. Accordingly, it may be useful in reducing platelet-related ischaemia in sickle cell disease (SCD). Exposure to prasugrel's active metabolite (Pras-AM) and its antiplatelet activity in SCD have not been investigated.
Methods
Thirteen adult patients with SCD and an equal number of matched healthy control subjects were studied before and after 12 days of 5.0 or 7.5 mg day−1 prasugrel treatment. Platelet reactivity was assessed by light transmission aggregometry (LTA), impedance aggregometry (MEA), VerifyNow® P2Y12, vasodilator-stimulated phosphoprotein (VASP) phosphorylation and Plateletworks. Exposure to Pras-AM was also assessed.
Results
At baseline, patients with SCD showed increased platelet reactivity vs. healthy control subjects with VerifyNow (408 vs. 323 P2Y12 reaction units (PRU), respectively, P = 0.003) and MEA (106 vs. 77 area under the aggregation curve (AU.min), P = 0.002); lower platelet reactivity index with VASP flow cytometry (59 vs. 79% platelet reactivity index (PRI), P = 0.018); and no significant differences with LTA, VASP enzyme-linked immunosorbent assay or Plateletworks. Relative to baseline, prasugrel significantly reduced platelet reactivity by all assays in both populations (all P < 0.05). Prasugrel was well tolerated, with no bleeding-related events in patients with SCD. The mean concentration–time profiles of Pras-AM were comparable between healthy subjects and patients with SCD following a single 10 mg prasugrel dose and following the 12th dose of 7.5 or 5 mg prasugrel.
Conclusions
Results demonstrate that in response to prasugrel, patients with SCD and healthy subjects have similar degrees of platelet inhibition and exposure to Pras-AM, and provide a basis for further study of prasugrel in patients with SCD.
Keywords: adenosine diphosphate, P2Y12 receptor antagonist, platelet, prasugrel, sickle cell
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Sickled red blood cells are prone to haemolysis, resulting in release of adenosine diphosphate (ADP), and ADP plays a central role in platelet activation and aggregation, which may contribute to vaso-occlusion in vaso-occlusive crisis.
Previous studies of platelet inhibitory agents have shown mixed results in their ability to control platelet reactivity and decrease the frequency of vaso-occlusive crisis in patients with sickle cell disease (SCD).
Substantial data exist on the pharmacodynamic and pharmacokinetic profiles of the ADP receptor antagonist prasugrel both in healthy subjects and in patients with coronary artery disease, but prasugrel has not been studied in patients with SCD.
WHAT THIS STUDY ADDS
In patients with SCD, prasugrel produced dose-dependent exposure to its active metabolite, which in turn reduced platelet reactivity to ADP.
This reduction in platelet reactivity was detected using a variety of assays, including point-of-care tests.
Introduction
Sickle cell disease (SCD) is a common haemoglobinopathy that affects millions of people worldwide 1. Coagulation, platelet and adhesion markers are increased in patients with SCD, suggesting that these patients are in a ‘hypercoagulable state’ 2–4. Much of the morbidity and mortality in SCD arises from complications of vaso-occlusive crisis (VOC), which results not only in pain in obstructed areas, but also in organ damage from associated ischaemia 5, 6. These crises are associated with increased activity of platelets 2, 7, leucocytes 8 and the coagulation system 2, 7. Moreover, sickled red blood cells are prone to haemolysis, resulting in release of adenosine diphosphate (ADP) 5. Adenosine diphosphate plays a central role in platelet activation and aggregation 9, and platelet aggregates, together with erythrocytes and leucocytes, may further contribute to vessel occlusion 10. Moreover, platelets in patients with SCD have been reported to exhibit an exaggerated response to ADP 2, and several studies have found evidence of platelet activation in both children and adults with SCD 10–13.
Previous studies of platelet inhibitory agents have shown mixed results in their ability to control platelet reactivity and decrease the frequency of VOC in SCD. Decreases in platelet aggregation, inflammatory markers 14 and haemolysis 15 and moderate therapeutic benefit 16, 17 have been reported in some studies, while minimal effects on platelet activation markers 14 and/or no impact on the frequency of VOC 18–20 have been reported in others.
Prasugrel is an ADP-receptor antagonist, which has been shown to inhibit platelet activation and aggregation effectively via irreversible antagonism of the P2Y12 ADP receptor 9. Given the potential role of ADP in the manifestation of VOC in patients with SCD, prasugrel may reduce the frequency and severity of VOC and persistently lessen platelet activation to help reduce chronic low-level ischaemia in these patients. This, in turn, might help prevent the progressive, irreversible organ dysfunction, morbidity and mortality associated with this disease.
While substantial data exist on the pharmacodynamic and pharmacokinetic profiles of prasugrel in healthy subjects and in patients with coronary artery disease, prasugrel has not been studied in patients with SCD. The pharmacodynamic objectives of this phase 1 study were to examine the platelet-inhibitory effects of prasugrel in adults with SCD compared with its activity in healthy adult subjects. In addition, given that certain platelet function assays offer different advantages in the clinical setting, we sought to assess the suitability of five independent assays, including point-of-care tests, for detection of prasugrel-mediated platelet inhibition in patients with SCD. The pharmacokinetic objective was to characterize the exposure to the active and inactive metabolites of prasugrel in patients with SCD compared with healthy subjects. A detailed description of the metabolism and activation of prasugrel has previously been published 21.
Methods
Study design
This was an open-label, single-centre trial conducted by Eli Lilly and Company from July 2010 to February 2011 (H7T-MC-TAEJ, NCT01178099). Participants were given a single 10 mg oral dose of prasugrel to examine the pharmacokinetic profile of this dose. This was followed by 12 ± 2 days of treatment with oral prasugrel at 5 mg day−1 for patients <60 kg and at 7.5 mg day−1 for patients ≥60 kg.
Participants
Patients with SCD genotypes HbSS, HbSC, HbSb0- or HbSβ+-thalassaemia with no occurrence of a VOC (defined as an event requiring medical intervention in an emergency department, infusion centre or as an inpatient service) within 1 month of screening were eligible for enrolment. Patients with SCD also had to be between 18 and 60 years of age and weigh between 50 and 100 kg. Females of child-bearing age had to be using a reliable method of birth control. Healthy control subjects were matched for gender, age within 10 years and weight category (<60 kg and ≥60 kg, within 10 kg). Exclusion criteria for both patients with SCD and healthy subjects included the following: severe hepatic or renal dysfunction; history of stroke, transient ischaemic attack or intracranial haemorrhage; bleeding disorders; haematocrit <18%; previous allergic reaction to thienopyridines; and aspirin, warfarin or thienopyridine use within 10 days of enrolment or anticipated use during the course of the trial.
This study was conducted in accordance with applicable laws and regulations, Good Clinical Practices (GCPs), and the ethical principles that have their origin in the Declaration of Helsinki. The study protocol was approved by appropriate ethical review boards, informed consent was obtained from all patients or their legal guardians prior to study entry, and the study was registered with ClinicalTrials.gov (NCT01178099).
Platelet reactivity measures
Details of the precision, accuracy, sensitivity and specificity of each of the analytical methods described below can be found in the associated cited publications. To assess potential differences between groups before and after treatment and to assess response to prasugrel within each population, venous blood samples were collected at baseline (1 h before the 10 mg dose of prasugrel) and on day 12, 24 h after the previous 5 or 7.5 mg dose of prasugrel. Maximal platelet aggregation (MPA) was assessed by light transmission aggregometry (LTA) using platelet-rich and platelet-poor plasma (PRP and PPP) prepared by differential centrifugation of whole blood collected in 3.2% sodium citrate, as previously described 22. Aggregation was assessed with an AggRAM aggregometer (Helena Laboratories, Beaumont, TX, USA) and recorded over 6 min following ADP addition and completed within 3 h of blood collection. Individual subjects' PRP and PPP were used to calibrate the aggregometer to 0% light transmittance and 100% light transmittance, respectively. Platelet aggregation was induced with 5 and 20 μm ADP; platelet counts in platelet-rich plasma were not adjusted.
The point-of-care device VerifyNow P2Y12 (VN-P2Y12; Accumetrics, San Diego, CA, USA) was used according to the manufacturer's instructions. Blood samples were collected in 3.2% citrate vaccuette tubes (Greiner Bio-One, Monroe, NC) and analysed within a window of ≥15 min to ≤4 h postcollection. Three measurements were reported by VN-P2Y12: P2Y12 reaction units (PRU), BASE and ‘device-reported’ percentage inhibition, calculated by the device based on PRU and BASE values. In addition, PRU values at baseline (i.e. day 1 predose) and day 12 were used to calculate the percentage inhibition, reported here as ‘calculated’ percentage inhibition. Further details on percentage inhibition parameters can be found elsewhere 23, 24.
The platelet reactivity index (PRI) was calculated using the vasodilator-associated stimulated phosphoprotein (VASP) phosphorylation assay. Following the manufacturer's (Stago/BioCytex, Marseille, France) instructions, 80 μl of 3.2% citrated blood samples were incubated with prostaglandin E1 (PGE1) alone or PGE1 plus ADP. For flow cytometry, VASP phosphorylation was measured using a BD FACSCalibur flow cytometer with Cell Quest Pro software (BD Biosciences, Franklin Lakes, NJ, USA) within 24 h of blood collection. For the enzyme-linked immunosorbent assay (ELISA), ELISA VASP/P2Y12 (BioCytex), samples were lysed following incubation with PGE1 alone or PGE1 plus ADP, stabilized, and frozen for up to 9 weeks prior to analysis. Further details on the flow cytometric and ELISA VASP assays have recently been reported 25.
Whole blood impedance aggregometry was performed using the Multiplate device (Dynabyte Medical, Munich, Germany) multiple electrode aggregometry (MEA) system, as recently described 26. Three millilitres of hirudinized whole blood was diluted 1:1 with saline and activated with 6.5 or 20 μm ADP. Aggregation was allowed to proceed for 6 min, and data were recorded as the area under the aggregation curve (AU.min) as measured by the device. Platelet aggregation was also assessed in whole blood using Plateletworks (Helena Laboratories, Beaumont, TX, USA). Aggregation was measured as the decrease in single platelet count in blood samples collected into EDTA or citrate and stimulated with ADP as per the manufacturer's instructions.
Pharmacokinetic analysis
To assess potential population differences in exposure to prasugrel's active metabolite (Pras-AM) and inactive metabolites (R-95913, R-106583 and R-119251), two 4 ml venous blood samples were collected into EDTA tubes at the following time points: 0.25, 0.5, 0.75, 1.0, 1.5, 2.0, 4.0, 6.0 and 8.0 h following the 10 mg prasugrel dose on day 1. On day 12, after samples were collected for pharmacodynamic assays and after the final 5 or 7.5 mg dose of prasugrel, blood samples were collected at the same time points as those listed above for day 1. Samples for the active metabolite were treated with 25 μl of 500 μm 3′-methoxyphenacyl bromide in acetonitrile within 30 s of collection to derivatize and stabilize the active metabolite 27. Plasma samples prepared from these blood samples were stored at approximately −70°C in polypropylene tubes until they were shipped to a central laboratory for analysis.
Plasma concentrations of the metabolites of prasugrel were determined using validated liquid chromatography methods and tandem mass spectrometric detection, as previously described 27. Pharmacokinetic parameter estimates were calculated by noncompartmental methods of analysis using WinNonlin Version 5.2. (Pharsight, Cary, NC) The primary pharmacokinetic parameter of interest was the area under the Pras-AM plasma concentration–time curve from dosing until the last measurable concentration [AUC(0–tlast)], as calculated by noncompartmental methods. Other pharmacokinetic parameters assessed were the maximal observed concentration (Cmax) and observed time of Cmax (tmax).
Tolerability and safety
All subjects who were enrolled in the study and received at least one dose of prasugrel were included in the safety analyses. Treatment-emergent adverse events, defined as those events that first emerged or worsened after initiation of prasugrel, were recorded regardless of the relation to prasugrel or study procedures. Serious adverse events were also recorded and were defined as death, initial or prolonged hospitalization, a life-threatening experience, persistent or significant disability or incapacity, congenital anomaly or birth defects, or considered significant by the investigator. Routine laboratory measurements (e.g. haematology and clinical chemistry) and vital signs were measured at screening, baseline and follow-up (∼2 weeks after last prasugrel dose). Discontinuations due to adverse events were also recorded.
Statistical analysis
Values are presented as means and SD for continuous variables and as counts (percentages) for categorical variables, unless noted otherwise. The haematology values at baseline and platelet aggregation results at baseline and day 12 were compared between two populations (patients with SCD vs. healthy subjects) using a Student's unpaired t-test. The platelet reactivity changes from baseline to day 12 within each population were analysed using a Student's paired t-test. Population differences in changes from baseline were compared using an analysis of covariance model, with dose, population and dose-by-population interaction as fixed effects and baseline measurement as the covariate; the results are presented as the least-squares mean difference between patients with SCD and healthy subjects with 95% confidence interval (95% CI). For all statistical comparisons, a P value <0.05 was considered statistically significant.
Results
Demographics
Twenty-six subjects were enrolled: 13 patients with SCD and 13 healthy subjects. The mean age was 29.1 years for patients with SCD (range, 19–50 years) and 26.7 years for healthy subjects (range, 19–42 years). Patients with SCD had a mean weight of 63 kg, compared with 65 kg for healthy subjects. All patients with SCD were of African descent, while the majority of healthy subjects were Caucasian (Table 1). All patients with SCD were of the HbSS genotype. As expected, there were notable, significant differences in several baseline haematology values in patients with SCD compared with healthy control subjects, including lower haematocrit, haemoglobin and erythrocyte count, and higher lymphocyte, monocyte and platelet counts (all P < 0.001; Table 2). Four patients each in the SCD and healthy subject groups weighed <60 kg and received prasugrel 5 mg day−1, and nine patients in each group weighed ≥60 kg and received 7.5 mg day−1. Over the course of the study, SCD subjects received 0.107 ± 0.015 mg kg−1 of prasugrel, which was similar to that received by healthy subjects, 0.104 ± 0.012 mg kg−1 (P = 0.66).
Table 1.
Baseline demographic characteristics of healthy subjects and patients with sickle cell disease
| Characteristic/measurement | Healthy subjects (n = 13) | Sickle cell disease (n = 13) |
|---|---|---|
| Age (years) | ||
| Mean (SD) | 26.7 (5.7) | 29.1 (9.3) |
| Range (minimum–maximum) | 19–42 | 19–50 |
| Males [n (%)] | 7 (53.8) | 8 (61.5) |
| Weight (kg) | ||
| Mean (SD) | 64.5 (9.6) | 63.3 (9.5) |
| Range (minimum–maximum) | 50.5–82.1 | 52.2–87.2 |
| Ethnicity [n (%)] | ||
| African descent | 3 (23.1) | 13 (100.0) |
| Asian | 1 (7.7) | 0 (0.0) |
| Caucasian | 9 (69.2) | 0 (0.0) |
Table 2.
Baseline haematology measurements for healthy subjects and patients with sickle cell disease
| Measure | Healthy subjects [n = 13; mean (SD)] | Sickle cell disease [n = 13; mean (SD)] | P value* |
|---|---|---|---|
| Haematocrit (%) | 41 (5) | 24 (3) | <0.001 |
| Haemoglobin (mmol l−1) | 8.7 (1.0) | 5.2 (0/5) | <0.001 |
| Neutrophils (×103 μl−1) | 3.5 (1.5) | 4.5 (1.4) | 0.081 |
| Mean cell volume (fl) | 88.2 (3.4) | 87.2 (15.0) | 0.817 |
| Erythrocyte count (×106 μl−1) | 4.7 (0.5) | 2.8 (0.7) | <0.001 |
| Leucocytes (×103 μl−1) | 6.0 (2.0) | 9.6 (2.7) | <0.001 |
| Lymphocytes (×103 μl−1) | 1.8 (0.5) | 3.6 (1.3) | <0.001 |
| Monocytes (×103 μl−1) | 0.5 (0.1) | 1.2 (0.5) | <0.001 |
| Eosinophils (×103 μl−1) | 0.2 (0.1) | 0.3 (0.2) | 0.095 |
| Basophils (×103 μl−1) | 0.02 (0.04) | 0.04 (0.05) | 0.199 |
| Platelets (×103 μl−1) | 234.6 (50.0) | 375.8 (112.6) | <0.001 |
| Mean platelet volume (fl) | 10.4 (0.7) | 10.1 (0.8) | 0.198 |
| MCHC (mmol l−1-Fe) | 21.1 (0.6) | 21.8 (0.6) | 0.003 |
| Reticulocyte count (×103 μl−1) | 43.6 (28.7) | 252.8 (120.1) | <0.001 |
| Prothrombin time (s) | 11.1 (0.6) | 11.5 (0.7) | 0.099 |
| APTT (s) | 28.1 (1.7) | 25.2 (2.0) | <0.001 |
| Platelet distribution index | 12.2 (1.5) | 11.0 (1.4) | 0.034 |
Abbreviations are as follows: APTT, activated partial thromboplastin time; and MCHC, mean cell haemoglobin concentration.
P values are from Student's unpaired t-test.
One patient with SCD was withdrawn from the study by the investigating physician immediately after receiving the 10 mg dose owing to poor vein quality. Blood from this patient was collected at baseline prior to treatment with prasugrel; these data are included in the pharmacodynamic analyses. No further samples were collected from this patient for either the pharmacokinetic or pharmacodynamic analyses. All other participants completed the study. The two dose groups (5 and 7.5 mg day−1) were combined for all pharmacodynamic analyses because there were only four subjects in each group receiving 5 mg day−1, and the doses were similar when considered on a milligram per kilogram basis.
Light transmission aggregometry
In response to 5 μm ADP, there were no significant differences between the populations in baseline MPA values: healthy subjects, 80 ± 23% MPA and patients with SCD, 73 ± 14% MPA (P = 0.376; Table 3 and Figure 1A). After 12 days of treatment with prasugrel, mean MPA in response to 5 μm ADP decreased to 29 ± 13% MPA in healthy subjects (P < 0.001) and to 40 ± 21% MPA in patients with SCD (P = 0.026; Table 3 and Figure 1A). There was no significant difference in MPA between the populations at day 12 (29 vs. 40% MPA, P = 0.143; Table 3 and Figure 1A). The difference in the magnitude of change from baseline was not significant [16 (95% CI −1, 34) percentage points, P = 0.070; Table 4]. Similar results were obtained with 20 μm ADP as the agonist, with no significant baseline differences between the groups, no significant differences between the groups at day 12, and a similar decrease in mean MPA following prasugrel administration (Tables 3 and 4 and Figure 1B).
Table 3.
Baseline and day 12 mean (±SD) platelet reactivity values in healthy subjects and patients with sickle cell disease
| Measure | Time | Healthy subjects (n = 13) | Sickle cell disease (n = 13) | P value* |
|---|---|---|---|---|
| LTA/MPA, 5 μm ADP (%) | Baseline | 80.4 ± 23.2 | 73.1 ± 14.4 | 0.376 |
| Day 12 | 29.4 ± 12.5 | 40.4 ± 20.5 | 0.143 | |
| P value† | <0.001 | 0.026 | ||
| LTA/MPA, 20 μm ADP (%) | Baseline | 85.0 ± 15.9 | 73.4 ± 12.9 | 0.064 |
| Day 12 | 40.8 ± 15.3 | 50.7 ± 25.6 | 0.269 | |
| P value† | <0.001 | 0.003 | ||
| VerifyNow (PRU) | Baseline | 322.5 ± 74.2 | 407.9 ± 39.6 | 0.003 |
| Day 12 | 111.5 ± 81.8 | 213.9 ± 72.2 | 0.003 | |
| P value† | <0.001 | <0.001 | ||
| VerifyNow, device-reported inhibition (%) | Baseline | 12.0 ± 11.0 | 1.0 ± 2.2 | 0.004 |
| Day 12 | 71.3 ± 19.7 | 47.8 ± 15.9 | 0.003 | |
| P value† | <0.001 | <0.001 | ||
| VerifyNow, calculated inhibition (%) | Day 12 | 65.8 ± 23.4 | 47.8 ± 18.6 | 0.067 |
| P value† | <0.001 | <0.001 | ||
| MEA, 6.5 μm ADP (AU. min) | Baseline | 76.5 ± 13.2 | 106.4 ± 27.9 | 0.002 |
| Day 12 | 27.0 ± 10.7 | 49.1 ± 24.9 | 0.008 | |
| P value† | <0.001 | <0.001 | ||
| MEA, 20 μm ADP (AU.min) | Baseline | 85.0 ± 19.0 | 108.1 ± 27.7 | 0.022 |
| Day 12 | 30.9 ± 15.7 | 48.0 ± 23.6 | 0.046 | |
| P value† | <0.001 | <0.001 | ||
| Plateletworks, 20 μm ADP (%) | Baseline | 91.3 ± 9.8 | 74.0 ± 30.0 | 0.061 |
| Day 12 | 37.9 ± 16.2 | 23.3 ± 21.2 | 0.081 | |
| P value† | <0.001 | 0.009 | ||
| VASP, flow cytometry (PRI%) | Baseline | 78.91 ± 10.03 | 59.36 ± 23.31 | 0.018 |
| Day 12 | 29.03 ± 18.18 | 16.07 ± 13.58 | 0.078 | |
| P value† | <0.001 | 0.002 | ||
| VASP, ELISA (PRI%) | Baseline | 94.70 ± 2.39 | 92.03 ± 5.65 | 0.159 |
| Day 12 | 24.26 ± 17.32 | 24.08 ± 12.98 | 0.978 | |
| P value† | <0.001 | <0.001 |
Abbreviations are as follows: ADP, adenosine diphosphate; AU.min, area under the aggregation curve; ELISA, enzyme-linked immunosorbent assay; LTA, light transmission aggregometry; MEA, multielectrode aggregometry; MPA, maximal platelet aggregation; PRI, platelet reactivity index; PRU, P2Y12 reaction units; SCD, sickle cell disease; and VASP, vasodilator-stimulated phosphoprotein.
P values for between-group comparisons are from Student's unpaired t-test.
P values within groups from baseline to day 12 are from Student's paired t-test.
Figure 1.
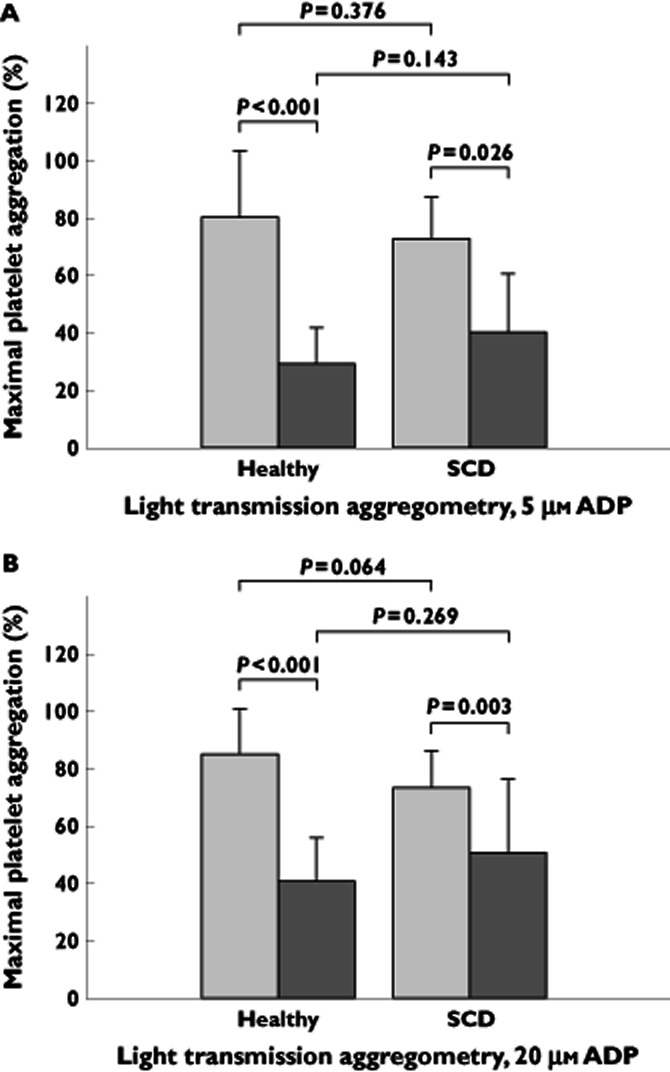
Light transmission platelet aggregometry in healthy subjects and patients with sickle cell disease (SCD) before and after prasugrel treatment. Maximal platelet aggregation, measured by light transmission aggregometry, in response to 5 μm (A) and 20 μm (B) ADP at baseline before prasugrel treatment and following 12 days of prasugrel treatment in healthy subjects (n = 13) and in patients with sickle cell disease (n = 13).  , baseline;
, baseline;  , day 12
, day 12
Table 4.
Least squares (LS) mean change from baseline to day 12 in platelet reactivity assays in healthy subjects and patients with sickle cell disease
| Change from baseline [LS mean (SEM)] | Sickle cell disease – Healthy [LS mean (95% CI)] | P value* | ||
|---|---|---|---|---|
| Healthy subjects (n = 13) | Sickle cell disease (n = 13) | |||
| LTA/MPA, 5 μm ADP (%) | −48.55 (5.11) | −32.27 (6.33) | 16.28 (−1.49, 34.05) | 0.070 |
| LTA/MPA, 20 μm ADP (%) | −41.89 (5.28) | −31.82 (5.55) | 10.07 (−6.62, 26.77) | 0.219 |
| VerifyNow (PRU) | −231.00 (25.28) | −148.17 (28.31) | 82.83 (−6.23, 171.88) | 0.066 |
| VerifyNow, device-reported inhibition (%) | 59.97 (5.78) | 40.89 (6.29) | −19.08 (−39.07, 0.92) | 0.060 |
| VerifyNow, calculated inhibition (%) | 62.52 (6.93) | 43.85 (7.76) | −18.67 (−43.07, 5.72) | 0.124 |
| MEA, 6.5 mm ADP (AU.min) | −58.60 (6.49) | −48.79 (7.31) | 9.81 (−13.83, 33.46) | 0.396 |
| MEA, 20 mm ADP (AU.min) | −63.88 (6.61) | −52.01 (7.39) | 11.87 (−10.85, 34.59) | 0.288 |
| Plateletworks, 20 μm ADP (%) | −42.35 (6.84) | −61.21 (7.04) | −18.86 (−40.50, 2.78) | 0.083 |
| VASP, flow cytometry (PRI%) | −40.69 (5.76) | −58.52 (7.16) | −17.83 (−38.53, 2.88) | 0.086 |
| VASP, ELISA (PRI%) | −67.34 (4.77) | −64.44 (4.54) | 2.90 (−11.57, 17.38) | 0.679 |
Abbreviations are as follows: ADP, adenosine diphosphate; AU.min, area under the aggregation curve; ELISA, enzyme-linked immunosorbent assay; LTA, light transmission aggregometry; MEA, multielectrode aggregometry; MPA, maximal platelet aggregation; PRI, platelet reactivity index; PRU, P2Y12 reaction units; and VASP, vasodilator-stimulated phosphoprotein.
P values are from an analysis of covariance model, with dose, baseline measurement, subject group, and dose-by-subject group interaction as fixed effects.
VerifyNow P2Y12
At baseline, patients with SCD had higher PRU values compared with healthy subjects (408 ± 40 vs. 323 ± 74, respectively, P = 0.003; Table 3 and Figure 2A). After treatment with prasugrel, mean PRU was decreased to 214 ± 72 in patients with SCD (P < 0.001) and to 112 ± 82 in healthy subjects (P < 0.001; Table 3 and Figure 2A). Following treatment with prasugrel, PRU was significantly greater in patients with SCD compared with that seen in healthy subjects (214 vs. 112, respectively, P = 0.003; Table 3 and Figure 2A), but the difference in the magnitude of change from baseline was not significant [83 (95% CI −6, 172) PRU, P = 0.066; Table 4]. Mean device-reported inhibition at baseline was lower in patients with SCD compared compared with that in healthy subjects (1 ± 2 vs. 12 ± 11%, P = 0.004, Table 3), with significant differences also noted between populations on day 12 (48 ± 16% in patients with SCD vs. 71 ± 20% in healthy subjects, P = 0.003; Table 3 and Figure 2B). The difference in the magnitude of change from baseline was not significant [−19 (95% CI −39, 1) percentage points, P = 0.060; Table 4]. There was no significant difference between the mean calculated percentage inhibition on day 12 for patients with SCD (48 ± 19%) and for healthy subjects (66 ± 23%, P = 0.067; Table 3 and Figure 2B) or in the difference in magnitude of change from baseline [−19 (95% CI −43, 6) percentage points, P = 0.124; Table 4].
Figure 2.
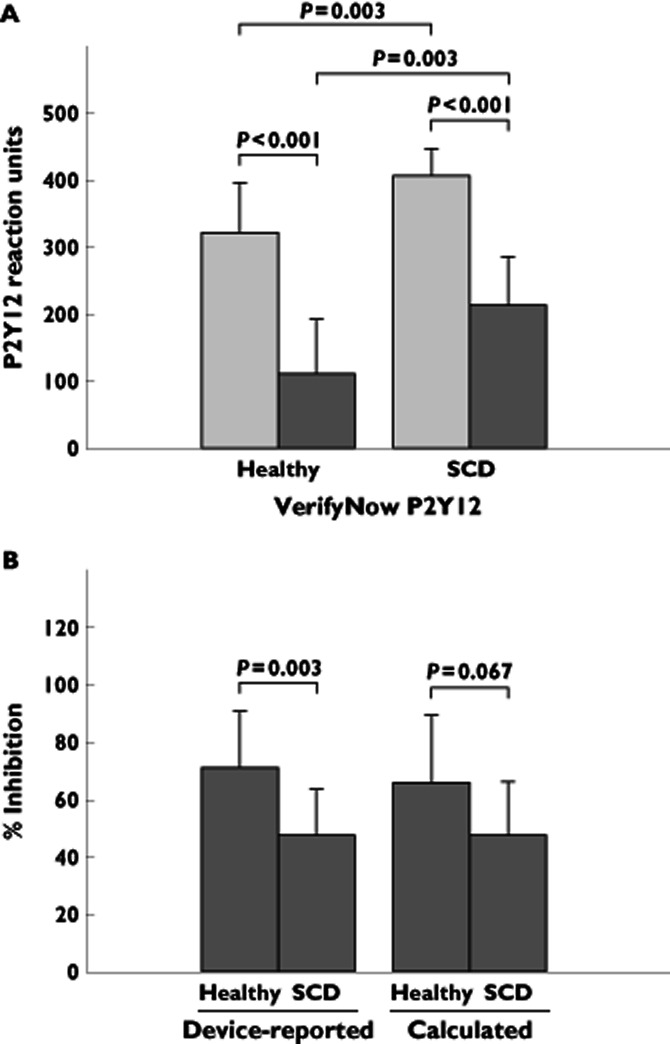
Whole blood platelet aggregation by VerifyNow P2Y12 in healthy subjects and patients with SCD before and after prasugrel treatment. P2Y12 reactivity units (A) before initiation of prasugrel treatment and following 12 days of prasugrel treatment and device-reported and calculated percentage inhibition (B) following 12 days of prasugrel treatment in healthy subjects (n = 13) and in patients SCD (n = 13).  , baseline;
, baseline;  , day 12
, day 12
Impedance aggregometry
Aggregation in response to 6.5 μm ADP was greater at baseline in patients with SCD compared with healthy subjects (106 ± 28 vs. 77 ± 13 AU.min, P = 0.002; Table 3 and Figure 3A). Mean aggregation was significantly greater in healthy subjects compared with patients with SCD on day 12 (49 vs. 27 AU.min, P = 0.008; Table 3 and Figure 3A), but there was no significant difference in the magnitude of change from baseline [10 (95% CI −14, 33) AU.min, P = 0.396; Table 4]. Comparable results were obtained with samples stimulated with 20 μm ADP (Tables 3 and 4).
Figure 3.
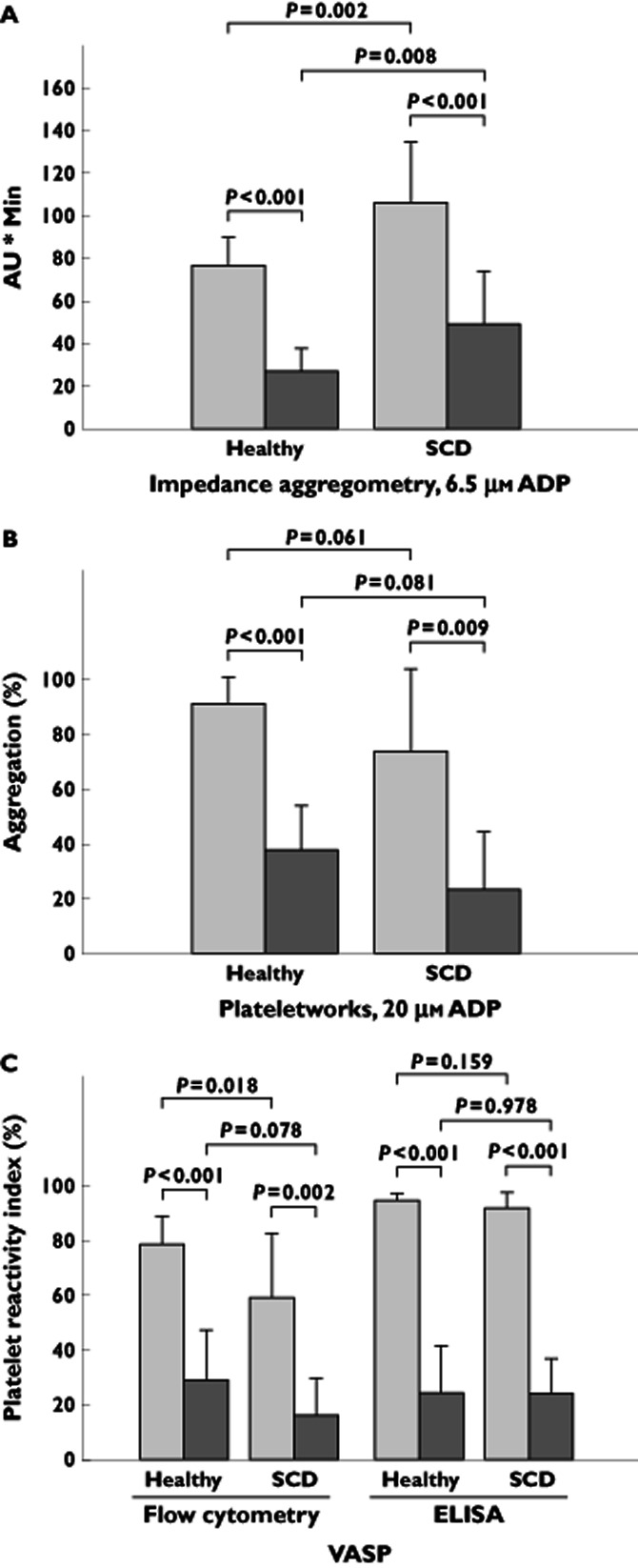
Whole blood impedance aggregometry, single platelet aggregometry and vasodilator-stimulated phosphoprotein (VASP) platelet reactivity index, in healthy subjects and in patients with SCD before and after prasugrel treatment. Area under the aggregation curve (AU.min) as measured by impedance aggregometry in response to 6.5 μm ADP (A), platelet aggregation as measured by Plateletworks (B) and platelet reactivity index as measured by VASP phosphorylation assessed with flow cytometry and an enzyme-linked immunosorbent assay (ELISA) (C), at baseline before initiation of prasugrel treatment and following 12 days of prasugrel treatment in healthy subjects (n = 13) and in patients with SCD (n = 13).  , baseline;
, baseline;  , day 12
, day 12
Plateletworks
At baseline, there was no statistically significant difference between healthy subjects and patients with SCD in platelet aggregation (91 ± 10 vs. 74 ± 30%, P = 0.061; Table 3 and Figure 3B). Mean aggregation was decreased following treatment with prasugrel to 38 ± 16% in healthy subjects (P < 0.001) and to 23 ± 21% in patients with SCD (P = 0.009; Table 3 and Figure 3B). The populations did not significantly differ in aggregation on day 12 (38 vs. 23%, P = 0.081; Table 3 and Figure 3B) or in the difference in magnitude of change from baseline [−19 (95% CI −41, 3) percentage points, P = 0.083; Table 4].
VASP assay
At baseline, healthy subjects had greater PRI compared with patients with SCD when assessed with flow cytometry (79 ± 10 vs. 59 ± 23%, P = 0.018; Table 3 and Figure 3C), but there were no significant differences between the populations using ELISA (95 ± 2 vs. 92 ± 6%, P = 0.159). Following treatment with prasugrel, mean PRI by flow cytometry decreased to 29 ± 18% in healthy subjects (P < 0.001) and to 16 ± 14% in patients with SCD (P < 0.001; Table 3 and Figure 3C). Mean PRI by ELISA decreased to 24 ± 17% in healthy subjects (P < 0.001) and to 24 ± 13% in patients with SCD (P = 0.002). There was no significant difference between the populations in PRI on day 12 (flow cytometry, 29 vs. 16%, P = 0.078; and ELISA, 24 vs. 24%, P = 0.978; Table 3 and Figure 3C), or in the difference in magnitude of change from baseline [flow cytometry, −18 (95% CI −39, 3) percentage points, P = 0.086; and ELISA, 3 (95% CI −12, 17) percentage points, P = 0.679; Table 4].
Pharmacokinetic analysis
In both healthy subjects and patients with SCD, exposure to Pras-AM increased with increasing prasugrel dose, except for patients with SCD who had lower Cmax estimates for the 7.5 mg dose (38.2 ng ml−1) than for the 5 mg dose (42.0 ng ml−1; Table 5). Following treatment with the single 10 mg prasugrel dose, and following the 12th dose of 7.5 or 5 mg prasugrel, the mean concentration–time profiles of Pras-AM were comparable between healthy subjects and patients with SCD, with no notable differences between groups. For the 5 mg dose, the AUC(0–tlast) was 40 ng h ml−1 for healthy subjects, compared with 36 ng h ml−1 for patients with SCD; the Cmax was 41 ng ml−1 for healthy subjects, compared with 42 ng ml−1 for patients with SCD; and the tmax was 0.6 h for healthy subjects, compared with 0.5 h for patients with SCD. At lower prasugrel doses, clearance was numerically greater in patients with SCD compared with healthy subjects (138.8 vs. 125.6 l h−1 for the 5 mg dose and 166.1 vs. 148.7 l h−1 for the 7.5 mg dose), while at the 10 mg prasugrel dose the clearance of Pras-AM was similar between groups (149.7 vs. 148.1 l h−1 for patients with SCD and healthy subjects, respectively). Concentrations of the inactive metabolites were consistent with the Pras-AM profile; however, exposure to the inactive precursor metabolite, R-95913, was approximately 50% greater for patients with SCD than for healthy subjects (data not shown); despite this, there were no differences in the exposure to Pras-AM.
Table 5.
Pharmacokinetic estimates of prasugrel's active metabolite (Pras-AM) in healthy subjects and in patients with sickle cell disease across the three doses of prasugrel
| Parameter | Healthy subjects | Patients with sickle cell disease | ||||
|---|---|---|---|---|---|---|
| 10 mg (n = 13) | 7.5 mg (n = 9) | 5 mg (n = 4) | 10 mg (n = 12) | 7.5 mg (n = 8) | 5 mg (n = 4) | |
| Pras-AM AUC(0–tlast) (ng h ml−1) | 67.5 (28) | 50.4 (23) | 39.8 (21) | 66.8 (36) | 45.2 (32) | 36.0 (7) |
| Pras-AM Cmax (ng ml−1) | 71.8 (49) | 62.5 (21) | 40.9 (20) | 63.0 (48) | 38.2 (59) | 42.0 (19) |
| Pras-AM CL/F (l h−1) | 148.1 (28) | 148.7 (23) | 125.6 (21) | 149.7 (36) | 166.1 (32) | 138.8 (7) |
| Median (minimum–maximum) | ||||||
|---|---|---|---|---|---|---|
| Pras-AM tmax (h) | 0.75 (0.50–1.00) | 0.75 (0.50–0.75) | 0.63 (0.50–1.00) | 0.65 (0.25–2.08) | 0.50 (0.25–1.50) | 0.51 (0.50–1.00) |
Data expressed as geometric mean (%CV).
Abbreviations are as follows: AUC(0–tlast), area under the plasma concentration–time curve from time of dosing to the sampling time of the last quantifiable concentration; Cmax, maximal observed concentration; CL, clearance; CV, coefficient of variance; F, bioavailability; Pras-AM, prasugrel active metabolite; and tmax, time of Cmax.
Safety and tolerability
No serious adverse events were reported during the study, and no subjects discontinued the study due to an adverse event. Headache was the most commonly reported adverse event, occurring in 69% (n = 9) of patients with SCD and in 54% (n = 7) of healthy subjects. Dizziness occurred in one subject from each group (7%) and, of the following, one event each occurred in patients with SCD: chest pain, musculoskeletal stiffness, oropharyngeal pain and vomiting (7% for each). Two bleeding-related adverse events, contusion (excessive bruising) and vaginal haemorrhage, were reported in two healthy subjects, and no bleeding-related adverse events were reported in patients with SCD.
Discussion
In this open-label, single-centre trial, 13 adult patients with SCD and 13 healthy subjects demonstrated a similar degree of platelet inhibition in response to prasugrel treatment. This result was observed using multiple platelet assays, suggesting that any one of them could be used to monitor platelet inhibition in SCD, with the optimal choice depending on circumstances. The pharmacokinetic characteristics of and exposure to active and inactive metabolites were similar in SCD compared with healthy subjects. To date, the study of prasugrel has been limited to healthy volunteers and to patients with stable or unstable coronary artery disease. The results reported here represent the first experience of prasugrel in patients with SCD, and serve as a precursor to future studies of the safety and efficacy of prasugrel in the treatment of patients with SCD.
A hallmark of SCD is the sickling and lysis of erythrocytes, resulting in release of intracellular contents. Erythrocytes contain substantial amounts of adenine nucleotides, including ADP, which is released following cell lysis 28. ADP is an effective driver of platelet activation and aggregation 29, and may consequently contribute to platelet involvement in the occlusive ischaemic aspects of SCD, including VOC 10. One might therefore expect that there would be a role for antiplatelet therapy, such as with aspirin or thienopyridyl P2Y12 ADP receptor antagonists, in the treatment of patients with SCD. However, limited benefit has been observed in small trials in which patients with SCD were treated with aspirin 15, 16, 18, 20, a cyclo-oxygenase-1 inhibitor, or ticlopidine 17, 19, an early generation P2Y12 antagonist of low potency with a variable antiplatelet effect 30. Antiplatelet agents acting via more appropriate targets or those of greater potency may provide more substantial benefit 14. Accordingly, prasugrel, a more effective P2Y12 receptor antagonist, presents an attractive candidate to control ADP-induced platelet activation and aggregation better in patients with SCD.
The present study adds to the somewhat sparse literature regarding antiplatelet therapy in patients with SCD, which in part suggests pharmacodynamic and clinical benefit from these agents. Treatment with ticlopidine has been reported to decrease platelet activation 19 and the incidence of VOC 17, though these studies had limited sample sizes. Likewise, treatment with aspirin has been shown to alter haematology measurements 15 and to possibly decrease the incidence of VOC when combined with dipyridamole 16. Eptifibatide has been shown to inhibit platelet aggregation, but not in vivo markers of platelet activation, in patients with SCD 14. In the present study, we found that treatment with prasugrel inhibited both ADP-induced platelet activation and aggregation ex vivo. The difference between our results and those of Lee et al. 14 may reflect the distinct sites of action of prasugrel and eptifibatide. By inhibiting proximal ADP signalling pathways, prasugrel attenuates several consequences of ADP-induced activation, including aggregation, while eptifibatide acts on the more distal GPIIb–IIIa fibrinogen receptor.
In this study, five independent assays were employed and demonstrated that overall platelet reactivity to ADP was reduced by a similar amount in patients with SCD to that in healthy subjects following prasugrel administration. No major differences were observed between groups in the change in platelet reactivity from baseline to day 12 of prasugrel administration, though the power of between-group comparisons was limited by the small sample size. For example, the 19 percentage-point difference in VerifyNow P2Y12 device-reported inhibition between patients with SCD and healthy subjects appeared large but was not statistically significant. Few differences between patients with SCD and healthy subjects were found when comparing absolute values for measures of platelet reactivity on day 12 (e.g. MEA, VerifyNow). Several assays showed a trend towards greater ‘residual’ platelet reactivity at day 12 in the SCD population, but this was not a consistent finding and may reflect the greater reactivity seen in this population at baseline. For example, MEA (6.5 μm ADP) values were significantly higher on day 12 in patients with SCD than in healthy subjects (49 vs. 27, P = 0.008; Table 3); however, at baseline (prior to prasugrel administration), MEA values were also substantially higher in the SCD population (106 vs. 76.5, P = 0.002; Table 3). Thus, the treatment effect (i.e. the change from baseline) was not significantly different (−49 vs. −59, P = 0.396; Table 4). Similar trends were observed for the VerifyNow P2Y12 assay. In contrast to the aforementioned whole blood assays, LTA results did not demonstrate any significant difference in baseline reactivity between groups, though a similar reduction in reactivity was observed following 12 days of prasugrel administration. Between-group differences in red blood cell-derived ADP are a possible explanation for the difference in platelet reactivity detected at baseline using the two assay types. However, patients with SCD and healthy subjects differed in several baseline haematological parameters, including haematocrit and counts of RBCs, platelets, leucocytes and reticulocytes. Of note, differences in platelet reactivity between groups at baseline were not seen with all whole blood assays (e.g. Plateletworks); therefore, the relative merit of whole blood assays vs. LTA remains unclear.
Anaemia in patients with SCD is another factor to consider regarding the subtle differences between groups seen in response to prasugrel; as expected, baseline haematocrit and haemoglobin values were substantially lower in patients with SCD than in healthy subjects (Table 2). Several reports indicate that patients with coronary artery disease who have anaemia and are on dual antiplatelet therapy (aspirin plus clopidogrel) have higher levels of ADP reactivity than those who are not anaemic 31–33.
All of the thienopyridines, including prasugrel, are prodrugs that require in vivo metabolic transformation to active metabolites, which bind to and antagonize the platelet P2Y12 ADP receptor. While there was no a priori reason to believe prasugrel metabolism would differ in patients with SCD compared with healthy subjects, given the complex pathophysiological changes associated with SCD, a pharmacokinetic assessment was considered an essential aspect of the present study. The results demonstrated dose-dependent exposure to Pras-AM, and no major differences in the pharmacokinetic exposure parameters for Pras-AM in patients with SCD compared with healthy subjects. These data are consistent with the platelet function assay results, in which no major differences between groups were found.
As discussed earlier, regardless of the assay employed, prasugrel consistently inhibited platelet reactivity to ADP. The general agreement between assays is important, as many platelet activity assays are useful in research settings but may be difficult to implement in multicentre clinical trials. For example, LTA is considered an historic standard for evaluating platelet aggregation, but its need for specialized equipment and trained staff and its lack of standardized procedures make its use in the clinic difficult 23. However, previous studies have shown reasonable correlation between results obtained with LTA, VerifyNow P2Y12 23 and VASP 34, and both VerifyNow P2Y12 and VASP have advantages in the clinical setting. VerifyNow is a simple, automated, cartridge-based whole blood point-of-care device that can be used at the bedside, if necessary. The VASP assay requires flow cytometric facilities, but has the advantage of blood sample stability for up to 72 h following collection, allowing time for shipment to a central laboratory for standardized analysis. More recently, an ELISA-based VASP assay has been developed, for which samples can be frozen for several weeks prior to final analysis. Since future multicentre prasugrel studies in patients with SCD are planned, the value and practicality of these assays is of interest.
We cannot fully explain why platelet reactivity was higher in healthy subjects as detected by VASP flow cytometry but not the VASP ELISA. It is possible that haemolysed blood samples are more common in SCD subjects, which may interfere with flow cytometric method but not with the VASP ELISA. However, no conclusions should be drawn until these results are replicated in larger study cohorts.
Prasugrel was safe, with no serious adverse events or discontinuation due to an adverse event in either population and no bleeding-related adverse events in patients with SCD. Thus, prasugrel appeared to be well tolerated in patients with SCD, a finding that encourages further clinical investigation; however, we acknowledge that this study was of short duration and involved a small number of patients.
The small sample size in this study limited the power of between-group comparisons, and while differences were generally quite small, they may have become statistically significant with a larger sample size. Also, while the clinical impact of prasugrel was not examined in this study, previous small clinical trials of ticlopidine, a related thienopyridine, have reported both attenuated platelet reactivity as assessed by LTA 19 and reductions in the rate and severity of VOC 17. These findings, in combination with the data reported here, encourage further study of prasugrel for this disease. Indeed, a phase 2 study of prasugrel in patients with SCD demonstrated similar decreases in platelet inhibition and was suggestive of clinical benefit during treatment 35.
In conclusion, we found that in patients with SCD, prasugrel produced dose-dependent exposure to its active metabolite, Pras-AM, which in turn reduced platelet reactivity to ADP, and that this effect could be monitored by a variety of assays, including point-of-care tests. The results of this study provide the basis for future investigation of the efficacy and safety of prasugrel in patients with SCD.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: J.A.J., C.Z., D.S.S., K.J.W., D.R.L., C.D.P. had employment with and held stock in Eli Lilly & Company in the previous 3 years and had no other relationships or activities that could appear to have influenced the submitted work; A.L.F. received consultancy fees and grants from Eli Lilly & Company and Daiichi Sankyo Co., Ltd in the previous 3 years and had no other relationships or activities that could appear to have influenced the submitted work; T.G.M. had employment by and held stock in Quintiles, Inc., who were contracted by Eli Lilly for assistance on this investigation; S.J. received funding from Eli Lilly for consumables and work hours of laboratory staff; and J.H. declared no conflicts of interest.
Appreciation is expressed to Janice Carlson PhD, and Tamara Ball MD, for writing and editorial contributions. Both are scientific writers employed full time by PharmaNet/i3, a division of InVentiv Health. Eli Lilly and Company contracted the technical writing of this manuscript with PharmaNet/i3. Also acknowledged are Julie Sherman AAS of Eli Lilly for contributions to figure creation; to Timothy Costigan PhD and Lori Heath MSc, both of Eli Lilly, for critical review of this manuscript; and to Laura Altobelli MS of PharmaNet/i3 for editorial review.
References
- 1.World Health Organization [Internet] Genes and human disease. 2012. Available at http://www.who.int/genomics/public/geneticdiseases/en/index2.html (last accessed 6 December 2012)
- 2.Francis RB, Jr, Johnson CS. Vascular occlusion in sickle cell disease: current concepts and unanswered questions. Blood. 1991;77:1405–1414. [PubMed] [Google Scholar]
- 3.Ataga KI, Orringer EP. Hypercoagulability in sickle cell disease: a curious paradox. Am J Med. 2003;115:721–728. doi: 10.1016/j.amjmed.2003.07.011. [DOI] [PubMed] [Google Scholar]
- 4.Ataga KI, Cappellini MD, Rachmilewitz EA. Beta-thalassaemia and sickle cell anaemia as paradigms of hypercoagulability. Br J Haematol. 2007;139:3–13. doi: 10.1111/j.1365-2141.2007.06740.x. [DOI] [PubMed] [Google Scholar]
- 5.Wun T, Paglieroni T, Tablin F, Welborn J, Nelson K, Cheung A. Platelet activation and platelet-erythrocyte aggregates in patients with sickle cell anemia. J Lab Clin Med. 1997;129:507–516. doi: 10.1016/s0022-2143(97)90005-6. [DOI] [PubMed] [Google Scholar]
- 6.Charneski L, Congdon HB. Effects of antiplatelet and anticoagulant medications on the vasoocclusive and thrombotic complications of sickle cell disease: a review of the literature. Am J Health Syst Pharm. 2010;67:895–900. doi: 10.2146/ajhp090229. [DOI] [PubMed] [Google Scholar]
- 7.Stuart MJ, Setty BN. haemostatic alterations in sickle cell disease: relationships to disease pathophysiology. Pediatr Pathol Mol Med. 2001;20:27–46. [PubMed] [Google Scholar]
- 8.Lum AF, Wun T, Staunton D, Simon SI. Inflammatory potential of neutrophils detected in sickle cell disease. Am J Hematol. 2004;76:126–133. doi: 10.1002/ajh.20059. [DOI] [PubMed] [Google Scholar]
- 9.Jakubowski JA, Winters KJ, Naganuma H, Walletin L. Prasugrel: a novel thienopyridine antiplatelet agent. A review of preclinical and clinical studies and the mechanistic basis for its distinct antiplatelet profile. Cardiovasc Drug Rev. 2007;25:357–374. doi: 10.1111/j.1527-3466.2007.00027.x. [DOI] [PubMed] [Google Scholar]
- 10.Beurling-Harbury C, Schade SG. Platelet activation during pain crisis in sickle cell anemia patients. Am J Hematol. 1989;31:237–241. doi: 10.1002/ajh.2830310404. [DOI] [PubMed] [Google Scholar]
- 11.Wun T, Paglieroni T, Rangaswami A, Franklin PH, Welborn J, Cheung A, Tablin F. Platelet activation in patients with sickle cell disease. Br J Haematol. 1998;100:741–749. doi: 10.1046/j.1365-2141.1998.00627.x. [DOI] [PubMed] [Google Scholar]
- 12.Tomer A. Platelet activation as a marker for in vivo prothrombotic activity: detection by flow cytometry. J Biol Regul Homeost Agents. 2004;18:172–177. [PubMed] [Google Scholar]
- 13.Lee SP, Ataga KI, Orringer EP, Phillips DR, Parise LV. Biologically active CD40 ligand is elevated in sickle cell anemia: potential role for platelet-mediated inflammation. Arterioscler Thromb Vasc Biol. 2006;26:1626–1631. doi: 10.1161/01.ATV.0000220374.00602.a2. [DOI] [PubMed] [Google Scholar]
- 14.Lee SP, Ataga KI, Zayed M, Manganello JM, Orringer EP, Phillips DR, Parise LV. Phase I study of eptifibatide in patients with sickle cell anaemia. Br J Haematol. 2007;139:612–620. doi: 10.1111/j.1365-2141.2007.06787.x. [DOI] [PubMed] [Google Scholar]
- 15.Osamo NO, Photiades DP, Famodu AA. Therapeutic effect of aspirin in sickle cell anaemia. Acta Haematol. 1981;66:102–107. doi: 10.1159/000207105. [DOI] [PubMed] [Google Scholar]
- 16.Chaplin H, Jr, Alkjaersig N, Fletcher AP, Michael JM, Joist JH. Aspirin-dipyridamole prophylaxis of sickle cell disease pain crises. Thromb Haemost. 1980;43:218–221. [PubMed] [Google Scholar]
- 17.Cabannes R, Lonsdorfer J, Castaigne JP, Ondo A, Plassard A, Zohoun I. Clinical and biological double-blind-study of ticlopidine in preventive treatment of sickle-cell disease crises. Agents Actions Suppl. 1984;15:199–212. [PubMed] [Google Scholar]
- 18.Greenberg J, Ohene-Frempong K, Halus J, Way C, Schwartz E. Trial of low doses of aspirin as prophylaxis in sickle cell disease. J Pediatr. 1983;102:781–784. doi: 10.1016/s0022-3476(83)80258-3. [DOI] [PubMed] [Google Scholar]
- 19.Semple MJ, Al-Hasani SF, Kioy P, Savidge GF. A double-blind trial of ticlopidine in sickle cell disease. Thromb Haemost. 1984;51:303–306. [PubMed] [Google Scholar]
- 20.Zago MA, Costa FF, Ismael SJ, Tone LG, Bottura C. Treatment of sickle cell diseases with aspirin. Acta Haematol. 1984;72:61–64. doi: 10.1159/000206360. [DOI] [PubMed] [Google Scholar]
- 21.Farid NA, Kurihara A, Wrighton SA. Metabolism and disposition of the thienopyridine antiplatelet drugs ticlopidine, clopidogrel, and prasugrel in humans. J Clin Pharmacol. 2010;50:126–142. doi: 10.1177/0091270009343005. [DOI] [PubMed] [Google Scholar]
- 22.Jakubowski JA, Stampfer MJ, Vaillancourt R, Deykin D. Cumulative antiplatelet effect of low-dose enteric coated aspirin. Br J Haematol. 1985;60:635–642. doi: 10.1111/j.1365-2141.1985.tb07467.x. [DOI] [PubMed] [Google Scholar]
- 23.Jakubowski JA, Payne CD, Li YG, Brandt JT, Small DS, Farid NA, Salazar DE, Winters KJ. The use of the VerifyNow P2Y12 point-of-care device to monitor platelet function across a range of P2Y12 inhibition levels following prasugrel and clopidogrel administration. Thromb Haemost. 2008;99:409–415. doi: 10.1160/TH07-09-0575. [DOI] [PubMed] [Google Scholar]
- 24.Jakubowski JA, Zhou C, Egan B, Wells M, Kotob-Yahfoufi M, Sugidachi A, Dahlen JR. Modification of the VerifyNow® P2Y12 test BASE channel to accommodate high levels of P2Y12 antagonism. Platelets. 2011;22:619–625. doi: 10.3109/09537104.2011.579203. [DOI] [PubMed] [Google Scholar]
- 25.Jakubowski JA, Bourguet N, Boulay-Moine D, Sugidachi A, Yamaguchi S, Barragan P, Zhou C, Moulard M. Comparison of a new ELISA assay with the flow cytometric assay for platelet vasodilator-associated stimulated phosphoprotein (VASP) phosphorylation in whole blood to assess P2Y12 inhibition. Thromb Haemost. 2012;107:388–395. doi: 10.1160/TH11-04-0282. [DOI] [PubMed] [Google Scholar]
- 26.Sibbing D, Braun S, Morath T, Mehilli J, Vogt W, Schömig A, Kastrati A, von Beckerath N. Platelet reactivity after clopidogrel treatment assessed with point-of-care analysis and early drug-eluting stent thrombosis. J Am Coll Cardiol. 2009;53:849–856. doi: 10.1016/j.jacc.2008.11.030. [DOI] [PubMed] [Google Scholar]
- 27.Farid NA, McIntosh M, Garofolo F, Wong E, Shwajch A, Kennedy M, Young M, Sarkar P, Kawabata K, Takahashi M, Pang H. Determination of the active and inactive metabolites of prasugrel in human plasma by liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 2007;21:169–179. doi: 10.1002/rcm.2813. [DOI] [PubMed] [Google Scholar]
- 28.Gaarder A, Jonsen J, Laland S, Hellem A, Owren PA. Adenosine diphosphate in red cells as a factor in the adhesiveness of human blood platelets. Nature. 1961;192:531–532. doi: 10.1038/192531a0. [DOI] [PubMed] [Google Scholar]
- 29.Born GV. Adenosine diphosphate is a mediator of platelet aggregation in vivo: an editorial view. Circulation. 1985;72:741–746. doi: 10.1161/01.cir.72.4.741. [DOI] [PubMed] [Google Scholar]
- 30.Cattaneo M. Platelet P2 receptors: old and new targets for antithrombotic drugs. Expert Rev Cardiovasc Ther. 2007;5:45–55. doi: 10.1586/14779072.5.1.45. [DOI] [PubMed] [Google Scholar]
- 31.Toma C, Zahr F, Moguilanski D, Grate S, Semaan RW, Lemieux N, Lee JS, Cortese-Hassett A, Mulukutla S, Rao SV, Marroquin OC. Impact of anemia on platelet response to clopidogrel in patients undergoing percutaneous coronary stenting. Am J Cardiol. 2012;109:1148–1153. doi: 10.1016/j.amjcard.2011.11.049. [DOI] [PubMed] [Google Scholar]
- 32.Elsenberg EH, van Werkum JW, van de Wal RM, Zomer AC, Bouman HJ, Verheugt FW, Berg JM, Hackeng CM. The influence of clinical characteristics, laboratory and inflammatory markers on ‘high on treatment platelet reactivity’ as measured with different platelet function tests. Thromb Haemost. 2009;102:719–727. doi: 10.1160/TH09-05-0285. [DOI] [PubMed] [Google Scholar]
- 33.Voisin S, Bongard V, Tidjane MA, Lhermusier T, Carrié D, Sié P. Are P2Y12 reaction unit (PRU) and % inhibition index equivalent for the expression of P2Y12 inhibition by the VerifyNow assay? Role of haematocrit and haemoglobin levels. Thromb Haemost. 2011;106:227–229. doi: 10.1160/TH11-01-0046. [DOI] [PubMed] [Google Scholar]
- 34.Jakubowski JA, Payne CD, Li YG, Farid NA, Brandt JT, Small DS, Salazar DE, Winters KJ. A comparison of the antiplatelet effects of prasugrel and high-dose clopidogrel as assessed by VASP-phosphorylation and light transmission aggregometry. Thromb Haemost. 2008;99:215–222. doi: 10.1160/TH07-09-0555. [DOI] [PubMed] [Google Scholar]
- 35.Wun T, Soulieres D, Krishnamurti L, Kutlar A, Zhou C, Heath LE, Nwachuku CE, Jakubowski JA, Winters KJ, Ataga K, Riesmeyer JS. A randomized, double-blind, adaptive phase 2 multi-center study of prasugrel compared to placebo in adults with sickle cell disease [abstract]. 53rd American Society of Hematology Annual Meeting and Exposition, 2011. Abstract 847.