

Abstract
Aims
To report the first three studies with SCH 900435, a selective glycine-1 re-uptake inhibitor in development for treating schizophrenia, using systematic evaluations of pharmacodynamics to understand the observed effects.
Methods
Three double-blind, placebo-controlled studies (single, visual effect and multiple dose) were performed. In the single and multiple dose study SCH 900435 (0.5–30 mg) was given to healthy males and frequent pharmacokinetic and pharmacodynamic measurements were performed. The visual effects study incorporated visual electrophysiological measures of macular, retinal and intracranial visual pathway function.
Results
In the single dose study (highest difference, 95% CI, P) increases in smooth pursuit eye movements (8, 12 mg (−6.09, 10.14, −2.04, 0.013), 30 mg), pupil : iris ratio (20 and 30 mg (−0.065, 0.09, −0.04, <0.0001)), VAS colour perception (30 mg (−9.48, 13.05, −5.91, <0.0001)) and changes in spontaneous reports of visual disturbance were found, while FSH (8 mg (0.42, 0.18, 0.66, 0.0015), 12, 20 mg), LH (8–30 mg (1.35, 0.65, 2.05, 0.0003)) and EEG alpha2 activity decreased (12, 20, 30 mg (0.27, 0.14, 0.41, 0.0002)). A subsequent dedicated visual effects study demonstrated that visual effects were transient without underlying electrophysiological changes. This provided enough safety information for starting a multiple ascending dose study, showing less visual symptoms after twice daily dosing and titration, possibly due to tolerance.
Conclusions
Several central nervous system (CNS) effects and gonadotropic changes resulted from administration of 8 mg and higher, providing evidence for CNS penetration and pharmacological activity of SCH 900435. Antipsychotic activity in patients, specificity of the reported effects for this drug class and possible tolerance to visual symptoms remain to be established.
Keywords: antipsychotic, central nervous system, glycine-1 re-\uptake inhibitor, healthy volunteers, schizophrenia
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Augmentation of glutamatergic systems might be an effective way of treating schizophrenia. Due to major concerns over potentially serious adverse events of indiscriminate glutamatergic stimulation, indirect strategies to stimulate N-methyl-D-aspartate (NMDA) glutamate receptor function are being investigated. One example of these strategies is glycine-1 re-uptake inhibition.
WHAT THIS STUDY ADDS
This article reports the first three studies with a highly selective glycine-1 re-uptake inhibitor in development for the treatment of schizophrenia. A novel glycine re-uptake inhibitor showed a steep dose–response curve for adverse events in animals and unanticipated, initially disconcerting visual symptoms in humans. However, an adaptive research strategy consisting of frequent interim analyses of pharmacokinetic characteristics and of data-intensive pharmacodynamic effects, and a return to dedicated animal studies, enabled the drug to be safely reintroduced in healthy volunteers.
Introduction
Traditional models of schizophrenia emphasize the importance of dopaminergic (DA) dysregulation, particularly with regard to the positive symptoms 1, 2. An alternative model is based on the effects of non-competitive antagonists of N-methyl-D-aspartate (NMDA) glutamate receptors, which induce psychotic symptoms in healthy subjects and exacerbate symptoms in schizophrenic patients. These effects seem to resemble schizophrenia more closely than those induced by dopamine activation 3–6. Moreover, dysfunction of glutamatergic neuronal systems would be consistent with the dopamine hypothesis of schizophrenia 7–11. Due to major concerns over potentially serious adverse events of indiscriminate glutamatergic stimulation, which could affect key functions such as learning, memory and neuronal excitation and cell death, research has focused on alternative strategies to augment NMDA receptor function 12. One alternative pathway is through the glycine receptor site, an obligatory co-agonist at the NMDA receptor 3. Direct glycine agonists appear to show some effect in treatment-resistant schizophrenia 3, but this requires gram level doses. Inhibition of presynaptic GlyT1 re-uptake transporters can also increase local endogenous glycine concentrations 13, 14. As reviewed by Javitt 3, glycine re-uptake inhibitors (e.g. d-cycloserine, glycine and d-serine) have been effective in a variety of rodent schizophrenia models 15–19 and in schizophrenic patients 20–22, but these agents are neither potent nor selective. SCH 900435 (formally called Org 25935) is a selective and highly potent GlyT1 re-uptake inhibitor (Figure 1), which effectively increases extracellular glycine concentrations in rat brain regions and spinal cord, but does not affect strychnine-sensitive GlyT2 transporters. Based on its general facilitation of NMDA receptor activity throughout the nervous system, its effects could be expected to influence a wide range of central nervous system (CNS) effects. D'Souza et al. showed that SCH 900435 (Org 25935) reduced the ketamine-induced increases in measures of psychosis (Positive and Negative Syndrome Scale (PANSS)) and perceptual alterations (Clinician Administered Dissociative Symptoms Scale (CADSS)) 23. Lastly, preclinical studies suggested that SCH 900435 could have a steep dose–effect relationship for adverse events. A rather abrupt transition from a no-adverse effect level (NOAEL) to severe CNS effects was observed in rats (NOAEL of 6.8 mg kg−1 and severe effects like tremors, bradypnoea and laboured respiration at doubling this dose) and dogs (NOAEL of 0.5 mg kg−1 and severe clinical signs at 1.5 mg kg−1 like increased arterial blood pressure). The onset of these symptoms was fast within 1–2 h after dosing and long lasting (≥5 h) (data on file). Therefore, a strategy was chosen for the early development of SCH 900435 that allowed for careful stepwise assessment of the pharmacological and clinical characteristics of the drug. During the first administration of SCH 900435 in humans, each dose escalation step was based on a detailed evaluation of the interim analyses of pharmacokinetics (PK), pharmacodynamics (PD) and clinical effects of the preceding doses. A multimodal test battery was used to measure frequently a wide range of effects, covering most drug sensitive CNS domains and vital functions. This article describes the three studies performed in human volunteers to determine a dosing regimen for SCH 900435 that was expected to be safe and therapeutically relevant for subsequent patient dose finding studies. Each study was approved by an independent Ethics Review Board and full written informed consent was obtained from all subjects.
Figure 1.
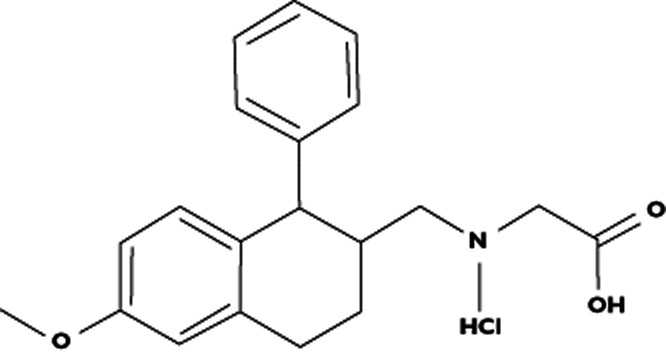
Structural formula of SCH 900435
Single ascending dose (SAD) study
Subjects and methods
Sixteen healthy male volunteers between 18 and 45 years were recruited for the first administration of single ascending doses of SCH 900435 at the Centre for Human Drug Research in Leiden, the Netherlands. Exclusion criteria and study restrictions included the use of agents known to affect CNS performance (including nicotine, alcohol or drugs) and evidence of relevant clinical or psychiatric abnormalities. Subjects remained in-house and were followed-up until 72 h after the last study drug administration. The study was double-blind, placebo controlled and randomized. Each subject was assigned to one of four dosing groups in a four way crossover study (three active ascending doses and one placebo dose, which was inserted randomly), with a minimum wash-out period of 1 week (Table 1A). Based on preclinical evaluations, the original dosing regimen was planned to cover a range from 0.5 mg (the human starting dose based on animal safety data) to 135 mg (predicted to lead to plasma concentrations in the anticipated therapeutic range). According to the protocol, doses could be adapted based on investigator-blinded interim assessments.
Table 1.
Dosing groups with corresponding SCH 900435 (mg) and placebo (P) treatment in single ascending dose study
| A) Dosing as originally planned in protocol | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Group I | Group II | Group III | Group IV | Group V | |||||||||||||||
| 0.5 | 2.0 | 8.0 | P | 8.0 | 12 | 18 | P | 18 | 27 | 40 | P | 40 | 60 | 90 | P | 90 | 135 | P | |
| 0.5 | 2.0 | P | 8.0 | 8.0 | 12 | P | 18 | 18 | 27 | P | 40 | 40 | 60 | P | 90 | 90 | P | 135 | |
| 0.5 | P | 2.0 | 8.0 | 8.0 | P | 12 | 18 | 18 | P | 27 | 40 | 40 | P | 60 | 90 | P | 90 | 135 | |
| P | 0.5 | 2.0 | 8.0 | P | 8.0 | 12 | 18 | P | 18 | 27 | 40 | P | 40 | 60 | 90 | ||||
| B) Actual dosing during study | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Group I | Group II | Group III | Group IV | ||||||||||||
| 0.5 | 1.0 | 2.0 | P | 2.0 | 3.0 | 5.0 | P | 5.0 | 8.0 | 12 | P | 12 | 20 | 30 | P |
| 0.5 | 1.0 | P | 2.0 | 2.0 | 3.0 | P | 5.0 | 5.0 | 8.0 | P | 12 | 12 | 20 | P | 30 |
| 0.5 | P | 1.0 | 2.0 | 2.0 | P | 3.0 | 5.0 | 5.0 | P | 8.0 | 12 | 12 | P | 20 | 30 |
| P | 0.5 | 1.0 | 2.0 | P | 2.0 | 3.0 | 5.0 | P | 5.0 | 8.0 | 12 | P | 12 | 20 | 30 |
Adverse events, laboratory safety parameters, ECG, oral body temperature, blood pressure and heart rate measurements were performed regularly throughout the study. Blood samples were obtained pre-dose and frequently up to 72 h after SCH 900435 administration. Frequent measurements were obtained from a multimodal CNS test battery containing a wide range of drug-responsive CNS domains up to 8 h after dosing: Visual analogue scales (VAS measuring subjective alertness, mood, calmness, psychedelic effects, sleep quality), pharmaco-electroencephalography (pharmaco-EEG), saccadic pursuit eye movements (measure of alertness), smooth pursuit eye movements (measure of motor coordination), adaptive tracking (visuo-motor coordination), body sway (postural stability), pupillometry (pupil : iris ratio) and neuroendocrine effects (serum prolactin, LH, FSH and testosterone concentrations).
Both a general treatment effect and a simple linear effect were tested for the PD parameters. AUEs (area under effect–time curves) were calculated per subject and divided by the corresponding time span, resulting in a weighted average response used for ancova analysis. If the general treatment effect was significant, contrasts of all treatments vs. placebo were calculated. Differences between treatments were defined as statistically significant at a P value of 0.05 or lower.
If the treatment effect was significant and the linear effect suggested a dose–effect relationship, an additional regression analysis of the relevant parameter on log dose was performed. The slope of the regression was considered significantly different from zero at a P value of 0.05 or lower. Conclusions about PK with respect to dose proportionality/dose independence were primarily based on descriptive statistics of PK parameters and on summary plots. An additional exploratory analysis of variance (anova) was performed to test for dose proportionality and dose independence. Detailed methods (pharmacodynamic measurements and statistics) are described in appendix I (online).
Results and discussion
Sixteen healthy males were included in the single dose study. Two subjects were withdrawn due to visual symptoms (described in more detail below) and 14 subjects completed the study. The mean (range) age of the subjects was 24 (19–32) years.
After interim assessment of the first dose, it was observed that exposure in terms of plasma AUC was 10-fold higher in humans than that which was allometrically predicted based upon a human AUC (12 ng ml−1 h) at the NOAEL of the dog (0.5 mg kg−1), i.e. the most sensitive species, with a safety factor of 30. No pharmacodynamic or adverse effects were noted after the starting dose, but the original anticipated dose range was reduced from 0.5–135 mg to 0.5–30 mg (Table 1B).
No statistically significant changes in vital signs, respiratory function, physical examination or laboratory parameters were observed during the entire study. There were no serious adverse events (SAEs). The most frequently reported adverse events (AEs) during the in-house study period were dizziness, somnolence, headache, fatigue and abnormal vision (Table 2). All were mild to moderate and self-limiting. At 3 mg, one subject developed anxiety and other psychological effects (emotional lability and hypersensitivity to noises) in addition to visual changes. The effects started 18 min after drug intake and lasted almost 4 h and led to his withdrawal from the second treatment group. In the subsequent two dosing groups, four of 12 subjects reported visual changes (one each at 3 and 8 and two at 12 mg), often described as spots of enhanced contrast or intensity accompanied by blurred vision and dizziness. These symptoms occurred around 30 min after drug administration, and disappeared within minutes to a few hours after the start of the symptoms. They were considered to be drug-related, but were of limited duration and intensity. All four subjects in the fourth dosing group reported spots in the visual fields (starting at 20 or 30 mg) similar to those observed in previous groups. It was decided to withdraw one of these subjects as he reported recurrent symptoms, which, although moderate, increased in the third visit (30 mg) compared with the second visit (20 mg). Ophthalmologic examinations did not reveal any subjective or objective visual system abnormalities, either during the study or at follow-up. The number of visual AEs was higher in subjects who used higher doses.
Table 2.
Most frequent adverse events in single ascending dose study (percentage of subjects reporting adverse events)
| Reported AE* | SCH 900435 dose | Placebo (%) | 0.5 mg (%) | 30 mg (%) | 0.5–30 mg (%) | 0.5–30 mg + placebo (%) |
|---|---|---|---|---|---|---|
| n of subjects | 16 | 4 | 4 | 16 | 32 | |
| n of AEs | 10 | 5 | 15 | 85 | 95 | |
| Dizziness | 1 (6.3) | 0 (0.0) | 3 (75.0) | 14 (87.5) | 15 (46.9) | |
| Somnolence | 2 (12.50) | 1 (25.0) | 3 (75.0) | 13 (81.3) | 15 (46.9) | |
| Headache | 1 (6.3) | 0 (0.0) | 0 (0.0) | 12 (75.0) | 13 (40.6) | |
| Fatigue | 2 (12.5) | 1 (25.0) | 1 (25.0) | 8 (50.0) | 10 (31.3) | |
| Abnormal vision | 0 (0.0) | 0 (0.0) | 3 (75.0) | 6 (37.5) | 6 (18.8) |
Not all AEs from each dose are shown for reasons of clarity; there were no relevant differences in these omitted groups.
Peak concentrations of SCH 900435 (26.7 ng ml−1 for 0.5 mg to 1170 ng ml−1 for 30 mg) were reached between 30 and 50 min after dosing. Pharmacokinetics (PK) were dose-linear over the tested dose range. The mean concentration vs. time profiles (Figure 2) showed that the last parts of the curves for all doses ran essentially parallel and that the terminal elimination phase started approximately 16 h after dosing. The terminal half-lives varied from 6.6 to 7.8 h, and clearance from 4.5 to 6.1 l h−1.
Figure 2.
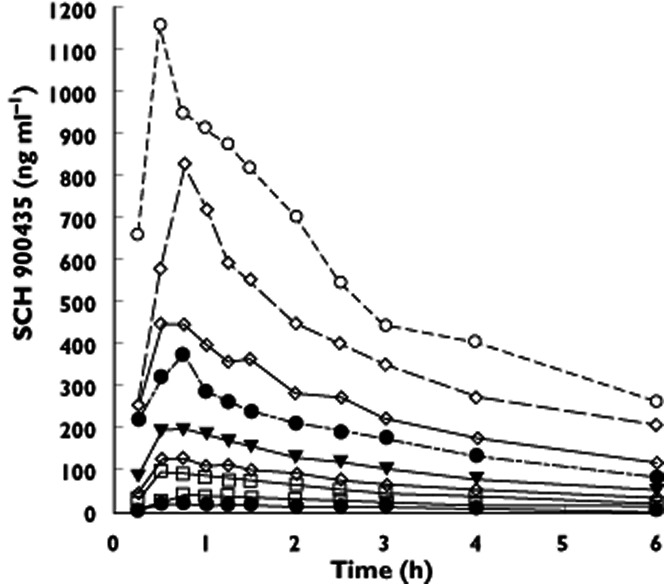
Mean concentration vs. time plot depicted as linear concentration scale in single ascending dose study.  , 0.5 mg;
, 0.5 mg;  , 1 mg;
, 1 mg;  , 2 mg;
, 2 mg;  , 3 mg;
, 3 mg;  , 5 mg;
, 5 mg;  , 8 mg;
, 8 mg;  , 12 mg;
, 12 mg;  , 20 mg;
, 20 mg;  , 30 mg
, 30 mg
In agreement with the adverse event reports, significant increases were observed in the ‘colours’ item of the Bowdle visual analogue scale (Table 3). The increases did not exhibit a linear dose–response relationship across all doses (Table 4) but were consistent at 30 mg (Figure 3). After a maximum at approximately 40 min, all effects had disappeared by 3 h following dosing (Figure 4). Pupillometry demonstrated an increase of pupil : iris ratio at 20 mg SCH 900435 (Table 3), with the largest effect at 30 mg (Table 3, Figure 5).
Table 3.
Contrasts with placebo on dose groups ancova analysis single ascending dose study (reported for doses SCH 900435 with statistically significant contrasts and higher)
| Parameter | Units | Dose (mg) | LSM Estimate placebo | LSM estimate SCH 900435 | Estimate of difference | P value * | 95% CI |
|---|---|---|---|---|---|---|---|
| Pupil : iris ratio right eye (0–90 min) † | NA | 20 | 0.41 | 0.44 | −0.030 | 0.0145 | −0.05, −0.01 |
| 30 | 0.48 | −0.065 | <0.0001 | −0.09, −0.04 | |||
| VAS colours (0–4 h) | mm | 30 | 1.45 | 10.94 | −9.48 | <0.0001 | −13.05, −5.91 |
| VAS dizzy (0–4 h) | mm | 12 | 43.72 | 49.57 | −5.85 | 0.0170 | −10.59, −1.10 |
| 20 | 50.34 | −6.62 | 0.0388 | −12.88, −0.36 | |||
| 30 | 52.14 | −8.42 | 0.0118 | −14.89, −1.95 | |||
| LH (0–final hours) | U l−1 | 8 | 3.04 | 2.21 | 0.82 | 0.0025 | 0.31, 1.34 |
| 12 | 2.69 | 0.35 | 0.0864 | −0.05, 0.74 | |||
| 20 | 2.10 | 0.93 | 0.0009 | 0.41, 1.46 | |||
| 30 | 1.90 | 1.14 | 0.0002 | 0.58, 1.70 | |||
| LH (0–4 h) | U l−1 | 8 | 2.93 | 2.17 | 0.76 | 0.0335 | 0.06, 1.45 |
| 12 | 2.15 | 0.78 | 0.0062 | 0.24, 1.32 | |||
| 20 | 1.77 | 1.16 | 0.0017 | 0.46, 1.86 | |||
| 30 | 1.58 | 1.35 | 0.0003 | 0.65, 2.05 | |||
| FSH (0–final hours) | U l−1 | 8 | 2.91 | 2.50 | 0.42 | 0.0015 | 0.18, 0.66 |
| 12 | 2.67 | 0.25 | 0.0131 | 0.06, 0.45 | |||
| 20 | 2.63 | 0.28 | 0.0301 | 0.03, 0.54 | |||
| 30 | 2.68 | 0.24 | 0.1068 | −0.05, 0.52 | |||
| Testosterone (0–final hours) | nmol l−1 | 8 | 5.88 | 4.93 | 0.95 | 0.0110 | 0.24, 1.66 |
| 12 | 5.45 | 0.43 | 0.1269 | −0.13, 0.99 | |||
| 20 | 4.66 | 1.22 | 0.0024 | 0.47, 1.98 | |||
| 30 | 5.26 | 0.62 | 0.1440 | −0.22, 1.46 | |||
| EEG alpha2 (0–4 h) | μV | 12 | 3.45 | 3.35 | 0.11 | 0.0449 | 0.00, 0.21 |
| 20 | 3.28 | 0.17 | 0.0099 | 0.044, 0.30 | |||
| 30 | 3.18 | 0.27 | 0.0002 | 0.14, 0.41 | |||
| Smooth pursuit (0–final hours) | % | 8 | 50.94 | 58.00 | −7.06 | 0.0172 | −12.80, −1.32 |
| 12 | 54.43 | −3.49 | 0.1195 | −7.93, 0.95 | |||
| 20 | 51.16 | −0.22 | 0.9397 | −6.01, 5.57 | |||
| 30 | 57.26 | −6.32 | 0.0433 | −12.45, −0.20 | |||
| Smooth pursuit (0–4 h) | % | 8 | 50.281 | 56.17 | −5.89 | 0.0289 | −11.14, −0.64 |
| 12 | 56.37 | −6.09 | 0.0042 | −10.14, −2.04 | |||
| 20 | 53.00 | −2.71 | 0.3086 | −8.01, 2.59 | |||
| 30 | 57.50 | −7.22 | 0.0130 | −12.85, −1.60 |
Significant P values are indicated in bold (lower than 0.05);
Only results for the right eye were shown as these were similar to the left eye.
Table 4.
Regression analysis table of dose-linearity single ascending dose study
| Parameter | Units | P value slope* | Estimate of the slope | 95% CI |
|---|---|---|---|---|
| Log pupil : iris ratio right eye (0–90 min) | NA | 0.573 | 0.004 | 0.017, −0.010 |
| Log VAS colours (0–4 h) | NA | 0.157 | 0.081 | 0.198, −0.035 |
| VAS dizzy (0–4 h) | mm | 0.062 | 2.302 | 4.722, −0.119 |
| LH (0–final hours) | U l−1 | 0.032 | −0.356 | −0.031, −0.682 |
| LH (0–4 h) | U l−1 | 0.004 | −0.501 | −0.164, −0.838 |
| FSH (0–final hours) | U l−1 | 0.020 | −0.193 | −0.042, −0.344 |
| Testosterone (0–final hours) | nmol l1 | 0.012 | −0.424 | −0.101, −0.747 |
| EEG alpha2 (0–4 h) | μV | 0.142 | −0.053 | 0.020, −0.126 |
| Smooth pursuit (0–4 h) | % | 0.001 | 3.305 | 5.125, 1.485 |
Statistically significant P values (lower than 0.05) are indicated in bold. NA, not applicable.
Figure 3.
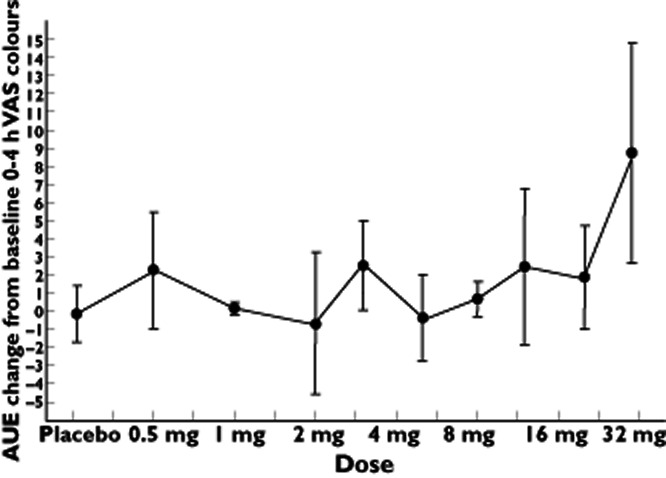
Average graph of AUE change from baseline 0–4 h VAS colours by log dose (SD as error bars) in single ascending dose study
Figure 4.
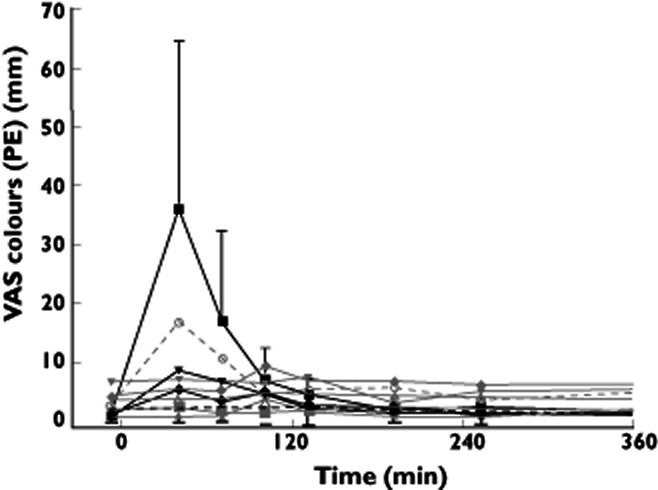
Adjusted (for baseline) mean time profile of different doses of VAS colours with 95% CI for highest dose and lower 95% CI for lowest dose in single ascending dose study.  , placebo;
, placebo;  , 0.5 mg;
, 0.5 mg;  , 1.0 mg;
, 1.0 mg;  , 2.0 mg;
, 2.0 mg;  , 3.0 mg;
, 3.0 mg;  , 5.0 mg;
, 5.0 mg;  , 8.0 mg;
, 8.0 mg;  , 12 mg;
, 12 mg;  , 20 mg;
, 20 mg;  , 30 mg
, 30 mg
Figure 5.
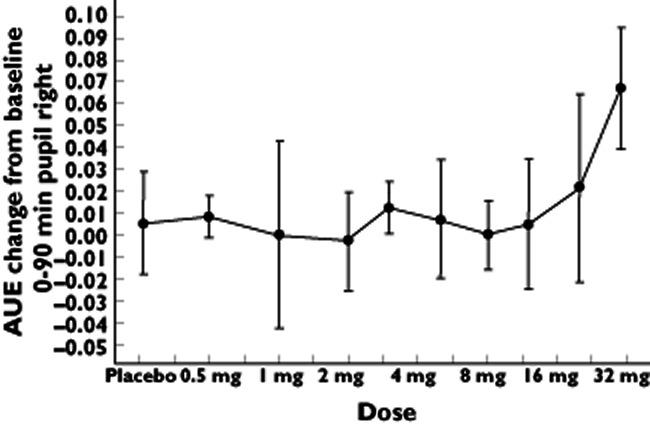
Average graph of AUE change from baseline 0–90 min pupil : iris ratio right eye by log dose (SD as error bars) in single ascending dose study
Smooth pursuit (0–4 h) showed a statistically significant increase at 8, 12, and 30 mg (largest difference) (Table 3). The regression analysis showed a statistically significant dose–response relationship for smooth pursuit eye movements (Table 4). It should be noted that the smaller number of subjects in the higher dose groups (the 20 mg dose group) may have prevented the detection of a treatment effect from reaching statistical significance.
Both LH and FSH decreased significantly compared with placebo (Table 3, Figure 6). LH (0–final hours) started to diminish after 8, 20 and 30 mg and FSH (0–final hours) after 8 mg and higher. The effect was more pronounced with higher doses. The absence of a significant effect in the 30 mg group for FSH could have been caused by the small group size (as was also the case for the smooth pursuit eye movements). Testosterone showed a decrease at 8 and 20 mg (Table 3). The LH concentration decrease followed SCH 900435 administration within 1 h and was virtually normalized after 10 h. FSH concentrations diminished after 5 to 6 h, and remained reduced during the full observation period. Regression analysis showed indications for linear dose response relationships for all three hormones (Table 4).
Figure 6.
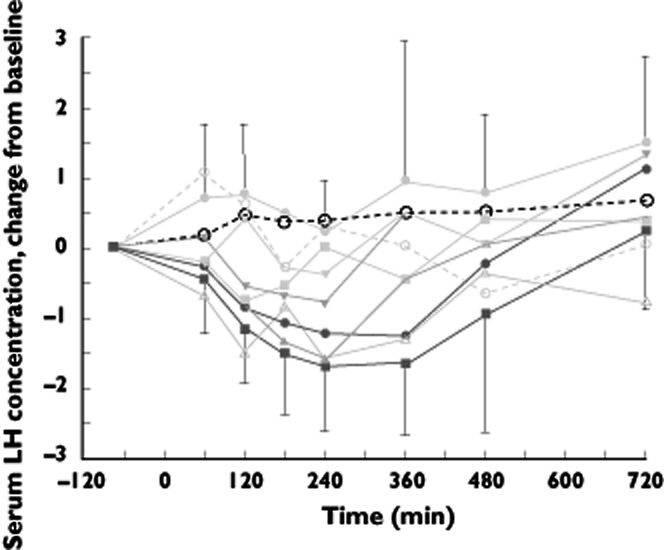
Adjusted (for baseline) mean time profile of different doses of LH with 95% CI for highest dose and lower 95% CI for lowest dose in single ascending dose study.  , placebo;
, placebo;  , 0.5 mg;
, 0.5 mg;  , 1.0 mg;
, 1.0 mg;  , 2.0 mg;
, 2.0 mg;  , 3.0 mg;
, 3.0 mg;  , 5.0 mg;
, 5.0 mg;  , 8.0 mg;
, 8.0 mg;  , 12 mg;
, 12 mg;  , 20 mg;
, 20 mg;  , 30 mg
, 30 mg
EEG alpha 2 power (0–4 h) decreased significantly after doses of 12 mg and higher.
Although the results should be treated with caution because of the well documented consequences of multiple hypothesis testing (only unadjusted 95% confidence intervals (CI) were calculated) signs of pharmacological activity in this single ascending dose study were evident from doses starting around 8 mg. Clear increases in pupil size and subjective changes of visual perception were noted after 20 and 30 mg. The effects resolved rapidly and were no more than mild to moderate, but were initially disconcerting and were not anticipated. Therefore, a more specific study on the effect of the drug on visual system function was initiated before multiple dosing was started.
Visual effects study
Based on the conjecture that the visual symptoms were more likely to reflect a retinal rather than central origin, consultation with ophthalmology experts and a review of the literature revealed that the retina is rich in glycine receptors. It appeared that although CNS studies of other glycinergic or glutamatergic agents did not specifically describe visual disturbances 18, 24, 25, such effects have been reported with high dose intravesicular glycine administration in men, following prostate surgery 26, 27. Based on the combination of drug-induced pupil dilation and abnormal vision, direct retinal effects were considered more probable than autonomically induced mydriasis or central visual impairment. Detailed assessments of retinal architecture and function in rats and dogs exposed to SCH 900435 did not demonstrate any electrophysiological or structural abnormalities of the retina or other components of the visual system after repeated dosing (data on file). In the first-in-human study, all symptoms were mild and rapidly reversible without any sequelae, both subjectively and during follow-up. Moreover, the resolution of visual symptoms was faster than the reduction of plasma concentrations, suggesting that the visual effects may not have functional consequences. This was investigated in a dedicated ophthalmologic study with SCH 900435 at Moorfields Eye Hospital in London, UK.
Subjects and methods
The visual effects study was a double-blind, placebo-controlled, four period crossover study involving the administration of three ascending single oral doses of SCH 900435 (5, 13 and 20 mg) in which the placebo was inserted randomly to 24 healthy male subjects. The exclusion and inclusion criteria and study restrictions were similar to the single ascending dose study. Inclusion criteria for the study included a normal eye examination and good visual acuity (6/9 or better in each eye). Each treatment period lasted 1 day and was separated by a wash-out of at least 3 days. Subjects were further randomized into one of three groups of eight subjects in which PK, PD and several PD eye assessments were performed. In group 1 (fully dark adapted with dilated pupils, duration approximately 12 min, in cycles of 25 min) the following PD tests were performed: dark adaptation, measuring the threshold of light detection without electrophysiology, rod specific single flash, red flash, intermediate, International Society for Clinical Electrophysiology of Vision (ISCEV) standard and ISCEV + 0.6 LV (see appendix II online). In group 2 (light conditions with dilated pupils, duration approximately 20 min, in cycles of 25 min) the following tests were performed: single flash cone response - 2 Hz, flicker response - 30 Hz, on/off response, S-cone response and photopic negative response (see appendix II online). In group 3 (light conditions with undilated pupils, duration approximately 45 min) the following tests were performed: VEP - pattern reversal/flash, pattern ERG, colour contrast sensitivity, visual acuity, pupillometry (see appendix II online). PK, PD and statistics are described in more detail in appendix II (online). Endocrinologic investigations (performed only in groups 1 and 2 due to very intensive testing in group 3) included repeated measurements of LH, FSH and testosterone. AEs, laboratory safety parameters, ECG, blood pressure and heart rate measurements were performed regularly during the study. SCH 900435 blood samples were obtained predose and frequently up to 24 h post-dose (for PK methods and statistics see SAD study appendix I – online).
Results and discussion
Twenty-four (three groups of eight) subjects were included. There were no SAEs or clinically relevant changes in laboratory safety parameters, vital signs and ECG data. The majority of the AEs were considered to be of mild intensity (102 out of 109 AEs), four AEs were of moderate intensity and three were of severe intensity. One visual AE after 13 mg SCH 900435 and two following 20 mg and were similar to the events that were also described in the SAD study. The symptoms were transient and resolved without intervention.
Pharmacokinetics and hormone results were similar to those observed in the SAD study (data not shown), with the exception of the FSH profile which showed no consistent decrease in this study (in contrast to the SAD study). Overall, there was no consistent change in any ophthalmologic test result in any subject, other than those that may be expected from normal variation. The visual symptoms questionnaire provided the most sensitive and specific assessment of visual disturbance (Table 5). The questionnaire 1.5 h post-dose seemed to indicate more frequent eye effects with the higher doses. After 8 h post-dose most effects had disappeared. Extraneous factors (e.g. subject tiredness and dizziness, adaptation/intolerance to the testing) and the awareness of the subjects of possible eye symptoms could possibly be an explanation for the relatively frequent reporting of blurred vision pre-dose and following administration of placebo. Even though there might the influence of abovementioned extraneous factors, confidence was raised by the observation of significant dose–response relationships and clear consistency among the studies (SAD and visual effects studies) for the most important (visual and neuroendocrine) pharmacodynamic effects and the data provided gave no indication for clinical concerns preventing the conduct of a multiple (ascending) dose study.
Table 5.
Results of visual analogue scales in visual effects study – number of subjects responding ‘yes’ to the question [number of subjects considering the effect as severe]
| Question/Study time | Placebo | SCH 900435 | ||
|---|---|---|---|---|
| 5 mg | 13 mg | 20 mg | ||
| n | 24 | 24 | 24 | 24 |
| Your vision is blurred | ||||
| Pre-dose | 9 [2] | 11 [3] | 12 [2] | 12 [2] |
| 1.5 h post-dose | 7 | 10 [1] | 16 [1] | 14 [1] |
| 8 h post-dose | 0 | 1 | 0 | 1 |
| There appears to be a film over your eyes | ||||
| Pre-dose | 1 | 2 | 2 | 1 |
| 1.5 h post-dose | 1 | 2 | 3 | 3 [1] |
| 8 h post-dose | 0 | 0 | 0 | 0 |
| You have difficulty assessing how far away objects are | ||||
| Pre-dose | 2 | 1 | 2 | 2 |
| 1.5 h post-dose | 2 | 2 | 3 [1] | 8 [1] |
| 8 h post-dose | 0 | 0 | 0 | 0 |
| You are seeing flashes of light | ||||
| Pre-dose | 0 | 0 | 0 | 0 |
| 1.5 h post-dose | 0 | 0 | 5 [1] | 7 [1] |
| 8 h post-dose | 0 | 0 | 0 | 0 |
| You are seeing dark patches | ||||
| Pre-dose | 0 | 0 | 0 | 0 |
| 1.5 h post-dose | 0 | 0 | 2 [1] | 3 [1] |
| 8 h post-dose | 0 | 0 | 0 | 0 |
| You are having more difficulty than usual focusing | ||||
| Pre-dose | 7 [1] | 11 [2] | 8 | 7 [1] |
| 1.5 h post-dose | 6 | 7 [1] | 14 [1] | 10 [1] |
| 8 h post-dose | 1 | 2 | 1 | 1 |
| You would not feel safe driving a car with your vision as it is | ||||
| Pre-dose | 5 [1] | 4 | 3 | 5 |
| 1.5 h post-dose | 4 | 6 | 11 [1] | 12 [1] |
| 8 h post-dose | 3 | 1 | 0 | 1 |
| Do you have any other visual symptoms or disturbance? | ||||
| Pre-dose | 0 | 0 | 0 | 0 |
| 1.5 h post-dose | 0 | 0 | 2 [1] | 3 [1] |
| 8 h post-dose | 1 | 1 | 0 | 0 |
Multiple (ascending) dose (MAD) study
Subjects and methods
The MAD study was performed at Guy's Drug Research Unit, London, UK. Selection criteria and study restrictions were similar to the visual effects study. The study design of the MAD study was double-blind, placebo-controlled and parallel group. Multiple (ascending) oral doses between 4 and 30 mg were given once or twice daily to 40 healthy male subjects (five groups of eight – six active ingredient and two placebo) for 13 days (Table S1). Dose groups 1, 2 and 3 received 4, 8 and 16 mg once daily (respectively), while dose group 4 received 12 mg twice daily (and started with 12 mg once daily on day 1 and ended with 16 mg once daily on day 13). Subjects in dose group 5 (dose titration) were given 8 mg, 12 mg, 16 mg, 20 mg all for a period of 3 days subsequently and then received 30 mg once daily for 1 day.
Similar to the SAD and visual effects study, PK samples were obtained frequently for up to 72 h after the last SCH 900435 dose and AEs, laboratory safety parameters, ECG, oral body temperature, blood pressure and heart rate measurements were followed-up regularly (see for the study design Table S2). PD and safety assessments in the MAD study were based on the results of the SAD and visual effects study: visual analogue scales (VAS), pupillometry and neuroendocrine effects (LH, FSH, testosterone) (Table S2). In addition, the cognitive effects of multiple dose treatment with SCH 900435 were examined with the following tests from the Cognitive Drug Research (CDR) test battery: simple reaction time, digit vigilance, choice reaction time, spatial working memory, numeric working memory, immediate word recall, word recognition, postural stability and tracking. At screening and follow-up, visual examinations were performed at Moorfields Eye Hospital, London, and subjects were given a questionnaire consisting of eight questions to ascertain visual symptoms. The primary aim of this MAD study was to examine the clinical course of the events that had been observed during single dosing. Descriptive and summary statistics for the VAS and AEs were used, as statistical models were considered to have a chance of obstructing the identification of isolated but relevant clinical observations. The statistical analyses performed on PK and the methods of the CDR test battery are described in more detail in appendix III (online).
Results and discussion
Forty healthy male subjects, with a mean (range) age of 25.9 (18–42) years, were included in the study. One subject was withdrawn from the study on day 12 (before the morning administration of SCH 900435) due to a persistent focusing difficulty; these symptoms disappeared by the evening of day 12. All other subjects completed the study. There were no clinically relevant abnormal findings in the laboratory safety parameters, vital signs or ECG data. The most common AEs reported by subjects receiving SCH 900435 were eye disorders and CNS disorders (Table 6). These symptoms were mostly noted between 0.5 and 1 h post-dose after both single and multiple doses and did not appear to be prolonged. None of the AEs was severe and no SAEs were reported during the study. Notably, the total number of eye disorders following SCH 900435 12 mg twice daily (6) (or after dose titration (8)) were lower than following 16 mg once daily (12) (Table 6). Although the ocular effects were less frequent after dosing twice daily or titration, there were comparable incidences of general CNS AEs (such as dizziness). However, it should be kept in mind that these remarks are based on a very low number of subjects and no statistical support is given.
Table 6.
Number of subjects (%) with AEs possible or probably related to SCH 900435 for ‘eye disorders’ and ‘CNS disorders’ in the multiple (ascending) dose study
| Treatment | ||||||
|---|---|---|---|---|---|---|
| Placebo | 4 mg once daily | 8 mg once daily | 16 mg once daily | 12 mg twice daily. | Dose titration | |
| n = 10 | n = 6 | n = 6 | n = 6 | n = 6 | n = 6 | |
| Eye disorder | ||||||
| Accommodation disorder | 0 | 0 | 0 | 0 | 1 (17) | 0 |
| Eye pain | 0 | 0 | 0 | 0 | 0 | 2 (33) |
| Eyelid irritation | 0 | 0 | 0 | 1 (17) | 0 | 0 |
| Ocular discomfort | 0 | 0 | 0 | 0 | 1 (17) | 0 |
| Photopsia | 0 | 0 | 0 | 3 (50) | 0 | 1 (17) |
| Vision blurred | 0 | 0 | 1 (17) | 5 (83) | 2 (33) | 4 (67) |
| Visual disturbance | 0 | 0 | 1 (17) | 3 (50) | 2 (33) | 1 (17) |
| Central nervous system disorders | ||||||
| Balance disorders | 0 | 0 | 0 | 0 | 1 (17) | 0 |
| Coordination abnormal | 0 | 0 | 0 | 2 (33) | 0 | 0 |
| Dizziness | 1 (10) | 0 | 1 (17) | 3 (50) | 2 (33) | 5 (83) |
| Dizziness postural | 0 | 0 | 1 (17) | 3 (50) | 3 (50) | 2 (33) |
| Headache | 2 (20) | 0 | 0 | 1 (17) | 3 (50) | 2 (33) |
| Lethargy | 0 | 0 | 0 | 2 (33) | 2 (33) | 6 (100) |
| Memory impairment | 0 | 0 | 0 | 0 | 0 | 1 (17) |
| Somnolence | 1 (10) | 0 | 0 | 4 (67) | 1 (17) | 3 (50) |
| Tremor | 1 (10) | 0 | 0 | 0 | 0 | 0 |
| Tunnel vision | 0 | 0 | 0 | 1 (17) | 0 | 0 |
Although a decrease in pupil diameter was seen (as opposed to the dilation seen in the single dose study), this change was not sustained, not evident under the other conditions or in the other dose groups and it cannot be excluded that this finding was coincidental in this small group of subjects. No other clinically relevant deviations in PD effects (VAS and cognitive CDR tests) were found.
Similarly to the SAD study, FSH, LH and testosterone significantly decreased during multiple dosing (see Figure 7 for LH; the figures for the other hormones were similar and were therefore not shown). There was no difference in hormone reductions between the first and last days of dosing. The effect was generally largest following administration of 16 mg SCH 900435.
Figure 7.
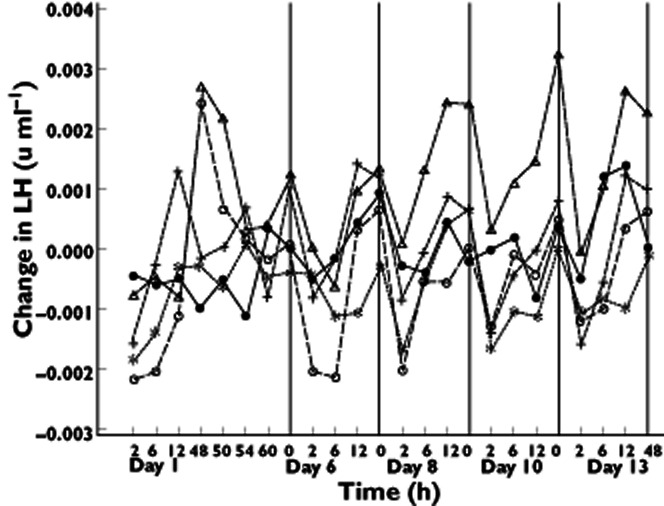
Mean changes from baseline LH values against time following administration of SCH 900435 or placebo in multiple ascending dose study. Treatment:  , placebo;
, placebo;  , 4 mg once daily;
, 4 mg once daily;  , 8 mg once daily;
, 8 mg once daily;  , 16 mg once daily;
, 16 mg once daily;  , 12 mg twice daily daily
, 12 mg twice daily daily
The PK parameters of the multiple (ascending) dose study were as expected from the single dose and the visual effects study (data therefore not shown). Steady-state concentrations were reached after between 2 and 6 days of daily dosing. Mean accumulation of SCH 900435 exposure (AUC) at steady-state after once daily dosing ranged between 5% and 11% and was 22% after 12 mg twice daily treatment. The elimination half-life was increased 20% after 10 days of daily dosing. The PK of SCH 900435 were dose-proportional at steady-state in the dose range 4–16 mg once daily and 12 mg twice daily.
In summary, the incidence of central nervous and visual symptoms measured by AE reporting increased when the dose of SCH 900435 was higher than 8 mg once daily. The eye symptoms were similar to those observed in the SAD and visual effects study. The other PD measurements did not show relevant or consistent changes in the doses tested in this study, except limited decreases in gonadotrophic hormones. Clinically, the results might indicate that dose titration and twice daily (compared with once daily) dosing of SCH 900435 improve tolerance to the subjective visual disturbances that primarily occur shortly after the initiation of treatment. However, this should be re-evaluated in a larger number of patients.
Discussion
The changes in visual VAS scales (mainly in the SAD study), the reported visual symptoms and the pupil dilation that were observed in the SCH 900435 studies are all compatible with a direct retinal effect of SCH 900435. Visual spots can be a manifestation of cortical spreading depression such as that which occurs during a migraine aura, and it cannot be excluded that a similar phenomenon caused the visual symptoms that occurred with SCH 900435. This would be consistent with a potential role of enhanced glutamatergic activity, which among other biochemical derangements has been postulated to play a role in cortical spreading depression 28. On the other hand, visual auras typically migrate and this was not reported by participants in our studies. Also, no other migrating neurological deficits or headaches occurred. Furthermore, pupillary abnormalities are usually not observed in occipital visual disturbance. A transient change in retinal function may then explain the visual symptoms, even in the absence of any detectable changes in retinal function as measured electrophysiologically.
Glycine is one of the essential neurotransmitters modulating visual signals in the retina 29. The retinal amacrine cells appear to possess a specific uptake mechanism for glycine 30 and contribute to the generation of the oscillatory potentials (OPs) in the electroretinogram. In different animal models these potentials were found to be blocked by glycine 30, 31. In a recent study with a glycine re-uptake inhibitor in healthy humans a high incidence of visual effects was described 32. At the higher dose ranges, visual effects were reported that may have been comparable with the symptoms observed in our studies, including visual disturbance, blurred vision, photophobia, diplopia and photopsia. The time course of the effects in the current study was also compatible with an acute pharmacological effect. In the SAD study, the reported visual symptoms and pupil dilation were both clearly related to dose (and consequently plasma concentration). Although no formal concentration–effect analyses were performed, it seemed that the subjective visual changes (assessed with the Bowdle VAS colours in the SAD study) resolved more rapidly than the plasma concentrations. As this could point to tolerance for this effect, this was further explored in the multiple ascending dose study by administrating the compound twice daily or by dose titration. The visual system is highly adaptive, so the question remained whether symptoms faded because of (retinal) adaptation to ongoing functional impairments. Preclinical multiple dose studies showed no morphological abnormalities in the eyes of experimental animals and the dedicated ophthalmologic study found no electrophysiological or functional indications of significant retinal impairment. In the MAD study, the incidence of AEs related to eye disorders increased with ascending doses of SCH 900435. However, the total reported number of eye disturbances was less frequent after 12 mg twice daily dosing and dose titration than with 16 mg once daily. Also, the 20 mg twice daily dosing (in the dose titration group) is more than two times higher than the maximum dose tolerated after once daily dosing (i.e. 16 mg) (see Table 6). This provided further confidence that the rapid resolution of the visual symptoms after a single dose or during dose titration reflects the development of pharmacological tolerance without additional functional consequences. However, the numbers of subjects on which this is based are small and studies with larger numbers of subjects should be performed before a definitive conclusion can be drawn. Also, all eye disturbances should be investigated in patients. Theoretically, it is not expected that tolerance for the desired effects will occur due to a different hypothesized mechanism of action, but this also should be studied in patients before firm conclusions can be drawn about response persistence. Although no clear retinal changes were found during the described healthy volunteer studies, it may be prudent to perform ophthalmologic investigations periodically (e.g. visual acuity, visual fields and VAS) at least during clinical trials.
The SAD and MAD studies showed consistent decreases in sex hormones after SCH 900435, with a linear dose–response relation. The results for FSH did not reach statistical significance in the visual effects study, for reasons that are not entirely clear, but testosterone concentrations in this study decreased similarly to the SAD study. Literature on the relationship between NMDA receptor stimulation and FSH, LH or testosterone production in humans is sparse, and most studies show no clear association 33, 34 or contradict the present findings 35. In preclinical experiments, an increase of FSH was found 36, 37. The testosterone reductions found in this study are compatible with the diminished gonadotrophic hormone concentrations, particularly of LH. The differences in the response of LH and FSH after SCH 900435 administration are partly related to the differences in half-life. LH has an elimination half-life of around 1 to 2 h 38, while that of FSH is in the region of 5 h 39. Although a direct effect of SCH 900435 on the expression of any of these hormones individually cannot be excluded, the time courses of the hormonal effects and the linear relation between dose and effect agree better with a common effect on LH and FSH release, most likely due to reduction of GnRH release 40, and suggesting the site of action of SCH 900435 to be the hypothalamus. These gonadotrophic responses are dose dependent and quite consistent, which makes these hormones (and LH in particular), the most reliable and sensitive pharmacodynamic indicators of a central SCH 900435 effect that were identified in these studies.
In general, animal studies of glycine re-uptake inhibitors and glycine agonists showed central nervous system depression (i.e. reduction in motor activity) 7, 41, 42. In apparent contrast, SCH 900435 produced slight improvements of smooth pursuit eye movements in the SAD study, which seemed to be dose dependent, and hence to be related to the pharmacological activity of the drug. Improved smooth pursuit could point to a mild stimulant effect of SCH 900435. On the other hand, the EEG demonstrated small statistically significant reductions in alpha2 power (10.5–12.5 Hz), which are more in line with CNS depression. However, this alpha reduction could be secondary to the visual symptoms caused by SCH 900435, since EEG alpha rhythm is particularly sensitive to visual input (e.g. eye opening or closure) and subjective well being (e.g. suppression by nausea and dizziness). These findings should be replicated before firm conclusions can be drawn.
Since experience with indirect NMDA-receptor activators in humans was limited, a cautious and adjustable approach to development of these drugs in humans is required. The present paper describes how a novel glycine re-uptake inhibitor, which had a steep dose–response curve for adverse events in test animals and showed unanticipated and initially disconcerting visual symptoms in humans, was safely introduced in healthy volunteers using a research strategy consisting of frequent interim analyses of PK characteristics and of data intensive PD effects, and a return to dedicated animal studies of the visual system, before the drug was reintroduced in humans. Unanticipated visual effects were detected from adverse event reports, and could be interpreted and pursued more reliably using frequent measurements of visual analogue scales for a broad range of subjective symptoms, and pharmacodynamic measures of multiple systemic and CNS effects, including pupil size. This strategy is particularly useful for chemical entities with novel mechanisms of action, which have a higher chance of showing unanticipated effects.
Intensive pharmacodynamic measurements can also provide indications for CNS penetration and dose-related pharmacological activity, especially since the early studies in healthy volunteers almost always cover a large dosage range. The establishment of dose-related changes provides strong evidence for a drug-related effect. This is clearly demonstrated by the consistency of almost all effects that were observed during the first study, even though they were unexpected and may even have initially seemed spurious. It cannot be ascertained whether the indications for CNS penetration and pharmacological activity of SCH 900435 are due to GlyT1 re-uptake inhibition, although this is suggested by the high potency and selectivity of SCH 900435 at this site. The retina, which is particularly rich in glycine and glutamatecontaining cells 43, has a structure similar to the blood–brain barrier. The observed visual spots in the SAD study together with the dose-responsive hormone changes and neurophysiological effects could indicate that SCH 900435 passes the blood retinal barrier and penetrates into the CNS. Long term studies and investigations with other glycine re-uptake inhibitors are needed to confirm whether retinal and gonadotropic changes are pharmacological effects of this novel drug class. Although these studies provided indications that SCH 900435 has a pharmacological effect in the CNS at doses that are well tolerated after titration, the antipsychotic effects and the therapeutically active doses of SCH 900435 and of glycine enhancement strategies in schizophrenia in general 44, 45 remain to be established.
Acknowledgments
The authors would like to thank Ge Ruigt for his help with the EEG
Conflict of Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare ML had support from MSD (formerly Organon) for the submitted work; PP, JUH and HJK were employees at MSD (formerly Organon) in the previous 3 years andno other relationships or activities that could appear to have influenced the submitted work.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Table S1
Treatment groups, multiple (ascending) dose study
Table S2
Study design, pharmacodynamic measurements, multiple (ascending) dose study
Appendix S1
Description of pharmacodynamic tests and statistics, single ascending dose study
References
- 1.Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976;192:481–483. doi: 10.1126/science.3854. [DOI] [PubMed] [Google Scholar]
- 2.Seeman P, Chau-Wong M, Tedesco J, Wong K. Brain receptors for antipsychotic drugs and dopamine: direct binding assays. Proc Natl Acad Sci USA. 1975;72:4376–4380. doi: 10.1073/pnas.72.11.4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Javitt DC. Is the glycine site half saturated or half unsaturated? Effects of glutamatergic drugs in schizophrenia patients. Curr Opin Psychiatry. 2006;19:151–157. doi: 10.1097/01.yco.0000214340.14131.bd. [DOI] [PubMed] [Google Scholar]
- 4.Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Jr, Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- 5.Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- 6.Krystal JH, D'Souza DC, Petrakis IL, Belger A, Berman RM, Charney DS, Abi-Saab W, Madonick S. NMDA agonists and antagonists as probes of glutamatergic dysfunction and pharmacotherapies in neuropsychiatric disorders. Harv Rev Psychiatry. 1999;7:125–143. [PubMed] [Google Scholar]
- 7.N-Methyl MJ. N-Methyl-D-aspartate receptors as a target for improved antipsychotic agents: novel insights and clinical perspectives. Psychopharmacology (Berl) 2005;179:30–53. doi: 10.1007/s00213-005-2199-1. [DOI] [PubMed] [Google Scholar]
- 8.Schmidt WJ, Kretschmer BD. Behavioural pharmacology of glutamate receptors in the basal ganglia. Neurosci Biobehav Rev. 1997;21:381–392. doi: 10.1016/s0149-7634(96)00044-9. [DOI] [PubMed] [Google Scholar]
- 9.Javitt DC, Hashim A, Sershen H. Modulation of striatal dopamine release by glycine transport inhibitors. Neuropsychopharmacology. 2005;30:649–656. doi: 10.1038/sj.npp.1300589. [DOI] [PubMed] [Google Scholar]
- 10.Olney JW, Farber NB. Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry. 1995;52:998–1007. doi: 10.1001/archpsyc.1995.03950240016004. [DOI] [PubMed] [Google Scholar]
- 11.Adams B, Moghaddam B. Corticolimbic dopamine neurotransmission is temporally dissociated from the cognitive and locomotor effects of phencyclidine. J Neurosci. 1998;18:5545–5554. doi: 10.1523/JNEUROSCI.18-14-05545.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Javitt DC. Glycine modulators in schizophrenia. Curr Opin Investig Drugs. 2002;3:1067–1072. [PubMed] [Google Scholar]
- 13.Smith KE, Borden LA, Hartig PR, Branchek T, Weinshank RL. Cloning and expression of a glycine transporter reveal colocalization with NMDA receptors. Neuron. 1992;8:927–935. doi: 10.1016/0896-6273(92)90207-t. [DOI] [PubMed] [Google Scholar]
- 14.Aragon C, Lopez-Corcuera B. Glycine transporters: crucial roles of pharmacological interest revealed by gene deletion. Trends Pharmacol Sci. 2005;26:283–286. doi: 10.1016/j.tips.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 15.Chen L, Muhlhauser M, Yang CR. Glycine tranporter-1 blockade potentiates NMDA-mediated responses in rat prefrontal cortical neurons in vitro and in vivo. J Neurophysiol. 2003;89:691–703. doi: 10.1152/jn.00680.2002. [DOI] [PubMed] [Google Scholar]
- 16.Depoortere R, Dargazanli G, Estenne-Bouhtou G, Coste A, Lanneau C, Desvignes C, Poncelet M, Heaulme M, Santucci V, Decobert M, Cudennec A, Voltz C, Boulay D, Terranova JP, Stemmelin J, Roger P, Marabout B, Sevrin M, Vige X, Biton B, Steinberg R, Francon D, Alonso R, Avenet P, Oury-Donat F, Perrault G, Griebel G, George P, Soubrie P, Scatton B. Neurochemical, electrophysiological and pharmacological profiles of the selective inhibitor of the glycine transporter-1 SSR504734, a potential new type of antipsychotic. Neuropsychopharmacology. 2005;30:1963–1985. doi: 10.1038/sj.npp.1300772. [DOI] [PubMed] [Google Scholar]
- 17.Harsing LG, Jr, Gacsalyi I, Szabo G, Schmidt E, Sziray N, Sebban C, Tesolin-Decros B, Matyus P, Egyed A, Spedding M, Levay G. The glycine transporter-1 inhibitors NFPS and Org 24461: a pharmacological study. Pharmacol Biochem Behav. 2003;74:811–825. doi: 10.1016/s0091-3057(02)01078-x. [DOI] [PubMed] [Google Scholar]
- 18.Javitt DC, Balla A, Sershen H, Lajtha A. A.E. Bennett Research Award. Reversal of phencyclidine-induced effects by glycine and glycine transport inhibitors. Biol Psychiatry. 1999;45:668–679. doi: 10.1016/s0006-3223(98)00237-6. [DOI] [PubMed] [Google Scholar]
- 19.Le Pen G, Kew J, Alberati D, Borroni E, Heitz MP, Moreau JL. Prepulse inhibition deficits of the startle reflex in neonatal ventral hippocampal-lesioned rats: reversal by glycine and a glycine transporter inhibitor. Biol Psychiatry. 2003;54:1162–1170. doi: 10.1016/s0006-3223(03)00374-3. [DOI] [PubMed] [Google Scholar]
- 20.Tsai G, Lane HY, Yang P, Chong MY, Lange N. Glycine transporter I inhibitor, N-methylglycine (sarcosine), added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry. 2004;55:452–456. doi: 10.1016/j.biopsych.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 21.Lane HY, Chang YC, Liu YC, Chiu CC, Tsai GE. Sarcosine or d-serine add-on treatment for acute exacerbation of schizophrenia: a randomized, double-blind, placebo-controlled study. Arch Gen Psychiatry. 2005;62:1196–1204. doi: 10.1001/archpsyc.62.11.1196. [DOI] [PubMed] [Google Scholar]
- 22.Tsai GE, Yang P, Chang YC, Chong MY. d-alanine added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry. 2006;59:230–234. doi: 10.1016/j.biopsych.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 23.D'Souza DC, Singh N, Elander J, Carbuto M, Pittman B, de Haes JU, Sjogren M, Peeters P, Ranganathan M, Schipper J. Glycine transporter inhibitor attenuates the psychotomimetic effects of ketamine in healthy males: preliminary evidence. Neuropsychopharmacology. 2012;37:1036–1046. doi: 10.1038/npp.2011.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goff DC, Tsai G, Levitt J, Amico E, Manoach D, Schoenfeld DA, Hayden DL, McCarley R, Coyle JT. A placebo-controlled trial of d-cycloserine added to conventional neuroleptics in patients with schizophrenia. Arch Gen Psychiatry. 1999;56:21–27. doi: 10.1001/archpsyc.56.1.21. [DOI] [PubMed] [Google Scholar]
- 25.Goff DC, Coyle JT. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry. 2001;158:1367–1377. doi: 10.1176/appi.ajp.158.9.1367. [DOI] [PubMed] [Google Scholar]
- 26.Mizutani AR, Parker J, Katz J, Schmidt J. Visual disturbances, serum glycine levels and transurethral resection of the prostate. J Urol. 1990;144:697–699. doi: 10.1016/s0022-5347(17)39558-7. [DOI] [PubMed] [Google Scholar]
- 27.Hahn RG, Andersson T, Sikk M. Eye symptoms, visual evoked potentials and EEG during intravenous infusion of glycine. Acta Anaesthesiol Scand. 1995;39:214–219. doi: 10.1111/j.1399-6576.1995.tb04046.x. [DOI] [PubMed] [Google Scholar]
- 28.Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med. 2011;17:439–447. doi: 10.1038/nm.2333. [DOI] [PubMed] [Google Scholar]
- 29.Shen W, Jiang Z. Characterization of glycinergic synapses in vertebrate retinas. J Biomed Sci. 2007;14:5–13. doi: 10.1007/s11373-006-9118-2. [DOI] [PubMed] [Google Scholar]
- 30.Korol S, Leuenberger PM, Englert U, Babel J. In vivo effects of glycine on retinal ultrastructure and averaged electroretinogram. Brain Res. 1975;97:235–251. doi: 10.1016/0006-8993(75)90447-3. [DOI] [PubMed] [Google Scholar]
- 31.Wachtmeister L, Dowling JE. The oscillatory potentials of the mudpuppy retina. Invest Ophthalmol Vis Sci. 1978;17:1176–1188. [PubMed] [Google Scholar]
- 32.Ouellet D, Sutherland S, Wang T, Griffini P, Murthy V. First-time-in-human study with GSK1018921, a selective GlyT1 inhibitor: relationship between exposure and dizziness. Clin Pharmacol Ther. 2011;90:597–604. doi: 10.1038/clpt.2011.154. [DOI] [PubMed] [Google Scholar]
- 33.van Berckel BN, Lipsch C, Timp S, Gispen-de Wied C, Wynne H, van Ree JM, Kahn RS. Behavioral and neuroendocrine effects of the partial NMDA agonist d-cycloserine in healthy subjects. Neuropsychopharmacology. 1997;16:317–324. doi: 10.1016/S0893-133X(96)00196-0. [DOI] [PubMed] [Google Scholar]
- 34.van Berckel BN, Oranje B, van Ree JM, Verbaten MN, Kahn RS. The effects of low dose ketamine on sensory gating, neuroendocrine secretion and behavior in healthy human subjects. Psychopharmacology (Berl) 1998;137:271–281. doi: 10.1007/s002130050620. [DOI] [PubMed] [Google Scholar]
- 35.van Berckel BN, Lipsch C, Gispen-de Wied C, Wynne HJ, Blankenstein MA, van Ree JM, Kahn RS. The partial NMDA agonist d-cycloserine stimulates LH secretion in healthy volunteers. Psychopharmacology. 1998;138:190–197. doi: 10.1007/s002130050662. [DOI] [PubMed] [Google Scholar]
- 36.Bonavera JJ, Swerdloff RS, Sinha Hakim AP, Lue YH, Wang C. Aging results in attenuated gonadotropin releasing hormone-luteinizing hormone axis responsiveness to glutamate receptor agonist N-methyl-D-aspartate. J Neuroendocrinol. 1998;10:93–99. doi: 10.1046/j.1365-2826.1998.00177.x. [DOI] [PubMed] [Google Scholar]
- 37.Bettendorf M, de Zegher F, Albers N, Hart CS, Kaplan SL, Grumbach MM. Acute N-methyl-D,L-aspartate administration stimulates the luteinizing hormone releasing hormone pulse generator in the ovine fetus. Horm Res. 1999;51:25–30. doi: 10.1159/000023309. [DOI] [PubMed] [Google Scholar]
- 38.Tornoe CW, Agerso H, Senderovitz T, Nielsen HA, Madsen H, Karlsson MO, Jonsson EN. Population pharmacokinetic/pharmacodynamic (PK/PD) modelling of the hypothalamic-pituitary-gonadal axis following treatment with GnRH analogues. Br J Clin Pharmacol. 2007;63:648–664. doi: 10.1111/j.1365-2125.2006.02820.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.le Cotonnec JY, Loumaye E, Porchet HC, Beltrami V, Munafo A. Pharmacokinetic and pharmacodynamic interactions between recombinant human luteinizing hormone and recombinant human follicle-stimulating hormone. Fertil Steril. 1998;69:201–209. doi: 10.1016/s0015-0282(97)00503-7. [DOI] [PubMed] [Google Scholar]
- 40.Brann DW, Mahesh VB. Glutamate: a major neuroendocrine excitatory signal mediating steroid effects on gonadotropin secretion. J Steroid Biochem Mol Biol. 1995;53:325–329. doi: 10.1016/0960-0760(95)00070-g. [DOI] [PubMed] [Google Scholar]
- 41.Lechner SM. Glutamate-based therapeutic approaches: inhibitors of glycine transport. Curr Opin Pharmacol. 2006;6:75–81. doi: 10.1016/j.coph.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 42.Eulenburg V, Armsen W, Betz H, Gomeza J. Glycine transporters: essential regulators of neurotransmission. Trends Biochem Sci. 2005;30:325–333. doi: 10.1016/j.tibs.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 43.Davanger S, Storm-Mathisen J, Ottersen OP. Colocalization of glutamate and glycine in bipolar cell terminals of the human retina. Exp Brain Res. 1994;98:342–354. doi: 10.1007/BF00228422. [DOI] [PubMed] [Google Scholar]
- 44.Buchanan RW, Javitt DC, Marder SR, Schooler NR, Gold JM, McMahon RP, Heresco-Levy U, Carpenter WT. The Cognitive and Negative Symptoms in Schizophrenia Trial (CONSIST): the efficacy of glutamatergic agents for negative symptoms and cognitive impairments. Am J Psychiatry. 2007;164:1593–1602. doi: 10.1176/appi.ajp.2007.06081358. [DOI] [PubMed] [Google Scholar]
- 45.Duncan EJ, Szilagyi S, Schwartz MP, Bugarski-Kirola D, Kunzova A, Negi S, Stephanides M, Efferen TR, Angrist B, Peselow E, Corwin J, Gonzenbach S, Rotrosen JP. Effects of d-cycloserine on negative symptoms in schizophrenia. Schizophr Res. 2004;71:239–248. doi: 10.1016/j.schres.2004.03.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.