

Abstract
Aim
To investigate the effects of the cytochrome P450 3A4 (CYP3A4) inhibitor ketoconazole on the pharmacokinetics (PK) and pharmacodynamics of fluticasone furoate (FF) and vilanterol trifenatate (VI).
Methods
Two double-blind, randomized, placebo-controlled, two-way crossover studies in healthy subjects. In study 1, subjects received single doses of ketoconazole (400 mg) or placebo on days 1–6, with a single dose of inhaled VI (25 μg) on day 5. Pharmacodynamic and PK data were obtained up to 48 h following the VI dose. In study 2, subjects received once daily ketoconazole (400 mg) or placebo for 11 days, with FF/VI (200/25 μg) for the final 7 days. Pharmacodynamic and PK data were obtained up to 48 h following the day 11 dose.
Results
In study 1, there was no effect of co-administration of ketoconazole and VI on pharmacodynamic or PK parameters. In study 2, co-administration of ketoconazole and FF/VI had no effect on 0–4 h maximal heart rate or minimal blood potassium {treatment difference [90% confidence interval (CI)] –0.6 beats min–1 (−5.8, 4.5) and 0.04 mmol l−1 (−0.03, 0.11), respectively}, whilst there was a 27% decrease in 24 h weighted mean serum cortisol [treatment ratio (90% CI) 0.73 (0.62, 0.86)]. Co-administration of ketoconazole increased [percentage change (90% CI)] FF area under the curve (0-24) and maximal plasma concentration by 36% (16, 59) and 33% (12, 58), respectively, and VI area under the curve (0–t′) and maximal plasma concentration by 65% (38, 97) and 22% (8, 38), respectively.
Conclusion
Co-administration of FF/VI or VI with ketoconazole resulted in a less than twofold increase in systemic exposure to FF and VI. There was no increase in β-agonist systemic pharmacodynamic effects, while serum cortisol was decreased by 27%. Co-administration of FF/VI with strong CYP3A4 inhibitors has the potential to increase systemic exposure to both fluticasone furoate and vilanterol, which could lead to an increase in the potential for adverse reactions.
Keywords: drug interactions, fluticasone furoate, ketoconazole, vilanterol
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Fluticasone furoate, an inhaled corticosteroid, and vilanterol trifenatate, an inhaled β2-agonist, both with 24 h duration, are in development for the treatment of asthma and chronic obstructive pulmonary disease.
Preclinical data suggest that both are predominantly metabolized by cytochrome P450 3A4 (CYP3A4).
WHAT THIS STUDY ADDS
In the presence of the potent CYP3A4 inhibitor, ketoconazole, the systemic exposure to fluticasone furoate and vilanterol increased by less than twofold, indicating that they are substrates of CYP3A and are susceptible to interactions with potent CYP3A inhibitors. Co-administration of fluticasone furoate/vilanterol with strong CYP3A4 inhibitors has the potential to increase systemic exposure to both fluticasone furoate and vilanterol which could lead to an increase in the potential for adverse reactions.
Introduction
It is widely accepted that today's practice of polypharmacy inevitably increases the incidence of drug–drug interactions. A key cause for drug–drug interactions is the inhibition of cytochrome P450 enzymes, which are responsible for the metabolic clearance of many drugs 1. Vilanterol trifenatate (VI) is a novel long-acting β2-agonist, which has been demonstrated to provide 24 h bronchodilatation in subjects with asthma and chronic obstructive pulmonary disease (COPD) 2–4. It is being developed as a once daily clinical treatment for asthma and COPD in combination with fluticasone furoate (FF), a novel inhaled corticosteroid also active for 24 h 5. Both FF and VI are predominantly metabolized via cytochrome P450 3A4 (CYP3A4) and are also substrates for P-glycoprotein (PgP). Clinical studies have demonstrated that the oral bioavailability and/or systemic clearance of many CYP3A4 and/or PgP substrate drugs can be altered by concomitant administration of CYP3A4 inhibitors and/or PgP inhibitors 6–9, such as ketoconazole 10. Ketoconazole is a recommended and commonly used probe to assess the impact of potential metabolic inhibition of CYP3A4 11 and has also been shown to inhibit the PgP efflux 12 and to inhibit PgP-mediated drug transport in human cell lines 13.
The unwanted systemic effects of FF and VI are limited by topical administration in the lung, low therapeutic doses and low oral bioavailability of the swallowed portion of the dose. At high, supratherapeutic doses, however, both compounds have the potential to produce predictable class-related, systemic pharmacodynamic (PD) effects; FF, as an inhaled corticosteroid, can be associated with decreases in serum cortisol, while VI, like other long-acting β-agonists, could cause hypokalaemia, hyperglycaemia and tachycardia. Although effects on these parameters are not seen at the therapeutic doses of FF and VI under evaluation in Phase III studies in asthma and COPD 2, 3, increased PD responses could occur if CYP3A4 and/or PgP inhibition by administration of concomitant drugs increased FF and/or VI systemic exposure. Two studies were therefore conducted to investigate whether co-administration of ketoconazole affects the pharmacokinetics (PK) and PD of either VI or the combination of FF/VI in healthy subjects. As systemic exposure to both FF and VI is low at therapeutic doses, systemic PD end-points [serum cortisol (for FF) and heart rate and blood potassium levels (for VI)] were used as primary end-points, with FF and VI pharmacokinetics (PK) determined as secondary end-points.
Methods
Study design and subjects
All subjects gave written informed consent prior to any study-related procedures, and the protocols were approved by the appropriate institutional review boards and conducted in accordance with good clinical practice guidelines and the Declaration of Helsinki. Two studies have been described in this manuscript; GSK protocol number HZA105548 (clinicaltrials.gov NCT01165125) and GSK protocol number B2C112205 (clinicaltrials.gov NCT00866515). Both studies were single-centre, randomized, double-blind with respect to ketoconazole, two-way crossover studies in 18–20 healthy male and female nonsmoking subjects. Both VI and FF/VI were administered via a dual-strip dry powder inhaler, which contained VI in one strip and either placebo or FF in the second strip 14.
Study 1 (B2C112205)
The primary objective of this pilot study was to determine whether co-administration of repeat dose ketoconazole at a dose of 400 mg once daily with single dose inhaled VI (25 μg) had an effect on supine heart rate and blood potassium levels. During two treatment periods of 7 days, subjects received single doses of oral ketoconazole or placebo on the morning of days 1–6, with a single dose of inhaled VI co-administered on the morning of day 5. After a minimal 7 day washout, subjects crossed over to receive the other treatment. Healthy subjects between 18 and 65 years of age with an forced expiratory volume in one second (FEV1) ≥ 80% predicted, FEV1/forced vital capacity ratio ≥ 0.7 who were not currently smoking nor had smoked for 12 months and had a pack history of ≤10 pack years were included.
Study 2 (HZA105548)
The primary objective was to determine whether repeat dose co-administration of ketoconazole with FF/VI had an effect on heart rate, blood potassium levels and serum cortisol. During the two treatment periods, subjects received repeat dose oral ketoconazole at a dose of 400 mg or matching placebo once daily in the morning for 11 days. On days 5–11, FF/VI at a dose of 200/25 μg was co-administered once a day for 7 days. The washout between treatment periods was 7–14 days. After the screening visit, each subject was admitted to the unit on day −1 and remained resident until 1 h postdosing with ketoconazole or placebo on day 1. Subjects returned to the unit for two further in-house periods on the evening of day 4 until the morning of day 6 and from the evening of day 10 to the morning of day 12. On days 2–4 and 7–10, the subjects returned to the unit each morning to be dosed and remained on the unit until at least 1 h postdose. Healthy subjects between 18 and 64 years of age with an FEV1 ≥ 85% of predicted who were current nonsmokers who had not smoked for 12 months and had a pack history of ≤5 pack years were included.
All females were required to be either of nonchild-bearing potential or taking adequate contraception throughout the study period in both studies.
Outcome measurements
Pharmacodynamic end-points
In study 1, the primary end-points (0–4 h) were the maximal and weighted mean heart rate and minimal blood potassium level on the morning of day 5. Secondary PD end-points included maximal QT interval calculated using Fridericia's correction factor (QTcF) and weighted mean blood potassium on the morning of day 5.
In study 2, the primary end-points were the maximal heart rate and minimal blood potassium level (0–4 h) and the weighted mean serum cortisol (0–24 h) on day 11. Secondary PD end-points (0–4 h) included maximal heart rate, maximal QTcF and minimal blood potassium on the morning of day 5 and minimal diastolic blood pressure, maximal systolic blood pressure and maximal QTcF on the morning of day 11. Pharmacodynamic data were collected predose (day −1) and after 7 days repeat dose co-administration of FF/VI and ketoconazole or placebo. Blood glucose levels were also measured, primarily for safety, on day 5.
Whole blood samples (approximately 0.5 ml) were analysed for potassium and glucose using the i-STAT1 portable chemical analyser (Abbott Laboratories, Forest Lake, IL, USA). In study 1, on days 4 and 5, samples were taken predose and at 20, 40, 60, 80 and 100 min, 2, 3, 4 and 24 h postdose. In study 2, samples were taken predose and at 30 min, 1, 2 and 4 h on in-patient days 5 and 11 of the study for measurement of blood potassium and on day 5 for blood glucose. Samples for measurement of serum cortisol were taken predose and at 1, 2, 3, 4, 6, 8, 10, 12, 16 and 24 h on day 11 of the study.
Pharmacokinetic end-points
Secondary PK end-points included plasma concentrations and derived PK parameters for VI on days 5−7 in study 1 and for FF and VI on days 5 and 11 in study 2. In study 1, samples were taken predose and at 2, 5, 10, 15, 20, 30, 40, 60, 80 and 100 min and 2, 3, 4, 6, 8, 12, 16, 24, 30, 36 and 48 h postdose during each treatment period. In study 2, samples were taken predose and at 5, 15, 30 and 45 min and 1, 1.5, 2, 3, 4, 6, 8, 10, 12 and 24 h postdose on in-patient days 5 and 11 of the study. Additional samples were taken 36 and 48 h after the day 11 FF/VI dose.
Blood samples (approximately 3 ml) for the determination of FF and VI concentrations were collected into tubes containing EDTA. Plasma samples were analysed separately for FF (150 μl of plasma) and VI (200 μl of plasma) using validated analytical methods based on solid-phase extraction with isotopically labelled internal standards, followed by high-performance liquid chromatography/mass spectrometry analysis using a turbo ion-spray interface and multiple reaction monitoring. The protonated ion masses monitored for FF were 539 for the parent ion and 313 for the daughter ion and for VI were 486 for the parent ion and 159 for the daughter ion. The lower limits of quantification for FF and VI were both 10 pg ml−1 and the higher limits of quantification were 1000 pg ml−1 and 10 000 pg ml−1, respectively. For FF, the within-run precision, between-run precision and bias were all ≤13.1%. For VI, the within-run precision, between-run precision and bias were all ≤14.4%. Quality controls prepared at three different concentrations analysed with each batch of samples met the acceptance criteria. Pharmacokinetic parameters were calculated by standard noncompartmental analysis using WinNonLin Pro v5.2. In study 1, the following PK parameters were derived: area under the concentration–time curve from time zero to last quantifiable concentration [AUC(0–t)], maximal observed concentration (Cmax), first occurrence of maximal observed concentration (tmax) and time of the last quantifiable concentration (tlast) on days 5–7. In study 2, Cmax, tmax and AUC(0–t) were derived on days 5 and 11, and terminal plasma elimination rate constant (λz), apparent terminal elimination half-life (t1/2), AUC(0–24), AUC(0–t) and apparent clearance (CL/F) were derived on day 11. The AUC(0–24) could not be derived for FF on day 5 or VI on both days and therefore AUC(0–t′) was derived, where t′ was the common time of the last quantifiable concentration within a subject for each day across treatments.
Safety end-points
Secondary safety end-points included adverse events (AEs), laboratory safety tests (haematology, clinical chemistry and urinalysis), vital signs, 12-lead electrocardiography (ECG) and Holter monitoring. Clinical laboratory tests were analysed locally by the site for each study.
Statistical analysis
In both studies, VI or FF/VI and ketoconazole were declared to be equivalent to VI or FF/VI and placebo if the 90% confidence interval (CIs) for the estimated treatment difference for maximal heart rate were within ±10 beats min–1 and the 90% CIs for the estimated treatment difference for minimal blood potassium treatment difference were within ±0.22 mmol l−1. In study 2, equivalence was also declared if the weighted mean serum cortisol 90% CIs for the estimated treatment ratio were within 0.8–1.25. The equivalence limits for heart rate and potassium were selected based on treatment effects that would be considered to be of clinical concern. The equivalence limit for heart rate (90% CIs for mean difference contained within ±10 beats min–1) was selected based on a treatment effect on mean maximal heart rate of approximately 5 beats min–1. This would typically represent an increase in heart rate of <10% and is considerably less than that produced by normal daily activities, diurnal variation or by established β-agonists, such as salbutamol 15.
The sample size calculations were based on equivalence of VI or FF/VI with ketoconazole and VI or FF/VI with placebo for the primary end-points. For study 1, a sample size of 12 subjects was chosen, which assumed that there would be no difference between the two treatments. With eight subjects, the study would have in excess of 90% power to show this if the standard deviation for maximal heart rate was 5.1 beats min–1, and with 12 subjects the study would have in excess of 90% power to show this if the standard deviation for minimal potassium were 0.15 mmol l−1. For study 2, a sample size of 15 subjects was chosen, driven by variability estimates for 24 h weighted mean serum cortisol, where with a standard deviation of 0.17 nmol l−1 the study would have an excess of 90% power to demonstrate equivalence. In study 1, each derived PD parameter for heart rate, potassium and QTc(F) was analysed using a repeated measures, mixed effect model, with fixed effects terms for period, treatment, day, treatment by day, subject baseline, period baseline and period baseline by day interaction. Subject was treated as a random effect in the model. A formal statistical analysis was carried out to compare VI AUC(0–t) and Cmax between treatments. A mixed effects model analysis of variance was performed, with period and treatment fitted as fixed effects and subject as a random effect.
In study 2, each derived day 11 PD parameter was analysed using a mixed effects model with fixed effect terms for treatment, period, subject baseline and period baseline, and subject fitted as a random effect. The serum cortisol 0–24 h weighted means were loge transformed prior to data analysis. Point estimates and their associated 95% confidence intervals were constructed for each treatment. The difference between the adjusted means and corresponding two-sided 90% confidence interval for FF/VI with ketoconazole relative to FF/VI with placebo was calculated, then back transformed to provide the corresponding estimates for the ratio. On day 11, following loge transformation, AUC of FF and VI were analysed separately using a mixed effects model with fixed effect terms for treatment and period. Subject was treated as a random effect in the model. The AUC(0–24) and AUC (0–t′) were analysed for FF and VI, respectively. Data for Cmax were analysed by fitting a mixed effects model with fixed effect terms for treatment, period, day and day by treatment. Subject was treated as a random effect in the model.
Accumulation ratios were determined from AUC and Cmax on days 5 and 11. Mixed effects models were fitted with fixed effect terms for treatment, period, day and day by treatment. Subject was treated as a random effect in the model. For FF, AUC(0–t) and AUC(0–24) were analysed for days 5 and 11, respectively, and for VI, AUC(0–t′) was analysed.
The noncompartmental PK analysis was conducted by ICON Development Solutions (Ellicott City, MD, USA) and the statistical analyses of PK data were performed by Synergy, Statistics and Programming (Slough, UK).
Results
Study population
All subjects who received at least one dose of study treatment were included in the ‘all subjects’ population. The baseline characteristics of the subjects in the two studies are summarized in Table 1.
Table 1.
Baseline characteristics (healthy subjects)
| Characteristic | Study 1 | Study 2 |
|---|---|---|
| Subjects randomized (n) | 20 | 18 |
| Subjects completed (n) | 18 | 18 |
| Mean age [years (range)] | 30.1 (18–52) | 29.3 (22–39) |
| Males [n (%)] | 9 (45) | 11 (61) |
| Mean height [cm (range)] | 171.1 (150–184) | 171.2 (153–183) |
| Mean weight [kg (range)] | 71.6 (50.6–101.3) | 69.95 (48.4–85.6) |
| Mean body mass index [kg m−2 (range)] | 24.3 (19.0–29.9) | 23.68 (20.2–27.3) |
| Race, Caucasian [n (%)] | 17 (85) | 11 (61) |
Pharmacodynamic end-points
Primary
In both studies, equivalence was concluded for maximal heart rate (0–4 h) and the weighted mean heart rate (0–4 h; study 1) because the 90% CIs for the mean difference between the two treatments were contained within ±10 beats min–1 (Table 2). Equivalence was also concluded in both studies for minimal blood potassium (0–4 h) and weighted mean blood potassium (study 1) because the 90% CIs for the mean difference between the two treatments were contained within ±0.22 mmol l−1 (Table 2). In study 2, the weighted mean serum cortisol (0–24 h) was on average 27% lower with ketoconazole co-administration and equivalence was not concluded because the 90% CIs for the ratio were not completely within the range of 0.80–1.25 (90% CI 0.62, 0.86; Figure 1 and Table 2); however, the majority of subjects showed minimal change between individual weighted mean serum cortisol values between the two treatments (Figure 1).
Table 2.
Statistical analysis of pharmacodynamic parameters after co-administration of repeat dose ketoconazole or placebo with single dose vilanterol (study 1) and repeat dose FF/VI (study 2)
| Parameter | Treatment | n | Adjusted means | Treatment difference (90% CI) |
|---|---|---|---|---|
| Study 1 (differences from baseline) | ||||
| Heart rate (beats min–1; maximum 0–4 h) | VI + ketoconazole | 19 | 10.90 | 2.54 (−0.75, 5.83) |
| VI + placebo | 18 | 8.36 | ||
| Heart rate (beats min–1; weighted mean 0–4 h) | VI + ketoconazole | 19 | 3.57 | 2.40 (0.10, 4.69) |
| VI + placebo | 18 | 1.17 | ||
| Blood potassium (mmol l−1; minimum 0–4 h) | VI + ketoconazole | 19 | −0.133 | 0.041 (−0.065, 0.148) |
| VI + placebo | 18 | −0.175 | ||
| Blood potassium (mmol l−1; weighted mean 0–4 h) | VI + ketoconazole | 19 | 0.056 | 0.041 (−0.059, 0.140) |
| VI + placebo | 18 | 0.016 | ||
| QTcF (ms; maximum 0–4 h) | VI + ketoconazole | 19 | 17.9 | 3.1 (−2.8, 8.9) |
| VI + placebo | 18 | 14.8 | ||
| QTcF (ms; weighted mean 0–4 h) | VI + ketoconazole | 19 | 4.7 | 4.7 (0.4, 8.9) |
| VI + placebo | 18 | 0.0 | ||
| Study 2 (absolute values) | ||||
| Heart rate (beats min–1; maximum 0–4 h) | FF/VI + ketoconazole | 18 | 73.5 | –0.6 (–5.8, 4.5) |
| FF/VI + placebo | 18 | 74.1 | ||
| Blood potassium (mmol l−1; minimum 0–4 h) | FF/VI + ketoconazole | 18 | 3.72 | 0.04 (–0.03, 0.11) |
| FF/VI + placebo | 18 | 3.68 | ||
| Serum cortisol (mmol l−1; weighted mean 0–24 h) | FF/VI + ketoconazole | 18 | 104.6 | 0.73* (0.62, 0.86) |
| FF/VI + placebo | 18 | 142.5 | ||
| Systolic blood pressure (mmHg; maximum 0–4 h) | FF/VI + ketoconazole | 18 | 114.9 | –0.1 (–2.7, 2.5) |
| FF/VI + placebo | 18 | 115.0 | ||
| Diastolic blood pressure (mmHg; minimum 0–4 h) | FF/VI + ketoconazole | 18 | 57.8 | –2.0 (–4.4, 0.3) |
| FF/VI + placebo | 18 | 59.8 | ||
| QTcF (ms; maximum 0–4 h) | FF/VI + ketoconazole | 18 | 426.0 | 7.6 (4.5, 10.6) |
| FF/VI + placebo | 18 | 418.4 |
Abbreviations: CI, confidence interval; FF, fluticasone furoate; QTcF, QT interval calculated using Fridericia's correction factor; VI, vilanterol trifenatate.
Ratio.
Figure 1.
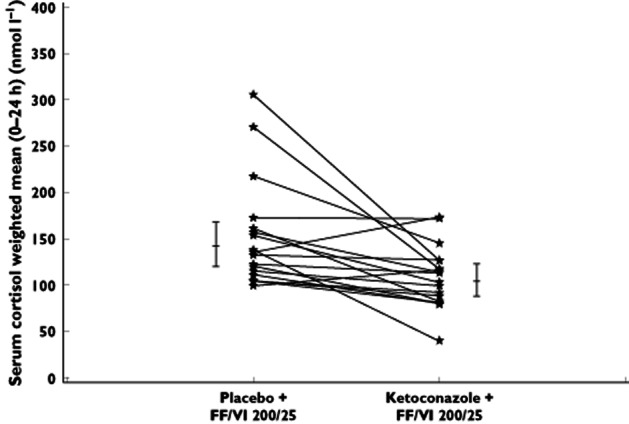
Individual subject and adjusted mean (95% confidence interval) weighted mean serum cortisol (0–24 h) after co-administration of repeat dose ketoconazole or placebo and repeat dose fluticasone furoate/vilanterol trifenatate (FF/VI; study 2)
Secondary
In study 1, there was no evidence of an effect of ketoconazole alone on 0–4 h maximal and weighted mean QTcF on day 4. On day 5, the 90% CIs for ketoconazole co-administration vs. VI with placebo for the 0–4 h maximal QTcF included zero and the mean treatment difference was 3.1 ms (90% CI −2.8, 8.9). For 0–4 h weighted mean QTcF, the 90% CIs for ketoconazole co-administration vs. VI with placebo did not include zero and the mean difference was 4.7 ms (90% CI 0.4, 8.9; Table 2). In study 2, there was no evidence of an effect of ketoconazole co-administration on minimal diastolic blood pressure or maximal systolic blood pressure (0–4 h; Table 2). The maximal QTcF (0–4 h) was, on average, 7.6 ms (90% CI 4.5, 10.6) higher following repeat dosing with FF/VI with ketoconazole compared with FF/VI with placebo (Table 2). Blood glucose values were comparable between the two treatments. The mean maximal blood glucose (0–4 h) on day 5 was 5.06 mmol l−1 (95% CI 4.95, 5.18) following administration of FF/VI with placebo and 5.21 mmol l−1 (90% CI 5.09, 5.33) following FF/VI with ketoconazole.
Pharmacokinetic end-points
The PK population contained all subjects from whom a PK sample was obtained and analysed. In study 1, the ratio of the adjusted means and corresponding 90% CIs for AUC(0–t) showed evidence of a higher VI systemic exposure (on average 1.9-fold) when given with ketoconazole compared with VI and placebo. For Cmax, there was no clear evidence of a difference between VI and ketoconazole and VI and placebo. The ratio of the adjusted means was 0.89 and the corresponding 90% CI contained 1.00 (Table 3).
Table 3.
Statistical analysis of FF and VI pharmacokinetic parameters after co-administration of repeat dose ketoconazole or placebo with repeat dose FF/VI (study 2) and with single dose VI (study 1)
| Parameter | Comparison | Ratio of geometric means | 90% CI of the ratio |
|---|---|---|---|
| Fluticasone furoate (study 2) | |||
| AUC(0–24) | FF/VI + ketoconazole/FF/VI + placebo | 1.36 | (1.16, 1.59) |
| Cmax | FF/VI + ketoconazole/FF/VI + placebo | 1.33 | (1.12, 1.58) |
| Vilanterol (study 2) | |||
| AUC(0–t′) | FF/VI + ketoconazole/FF/VI + placebo | 1.65 | (1.38, 1.97) |
| Cmax | FF/VI + ketoconazole/FF/VI + placebo | 1.22 | (1.08, 1.38) |
| Vilanterol (study 1) | |||
| AUC(0–t) | VI + ketoconazole/VI + placebo | 1.90 | (1.37, 2.64) |
| Cmax | VI + ketoconazole/VI + placebo | 0.89 | (0.67, 1.18) |
Abbreviations: AUC, area under the curve; CI, confidence interval; Cmax, maximum plasma concentration; FF, fluticasone furoate; VI, vilanterol trifenatate.
In study 2, co-administration with ketoconazole resulted in greater FF and VI exposure (Table 3). The mean FF AUC(0–24) and Cmax were increased by 36 and 33%, respectively, and the mean VI AUC(0–t′) and Cmax were increased by 65 and 22%, respectively (Figure 2 and Table 3).
Figure 2.
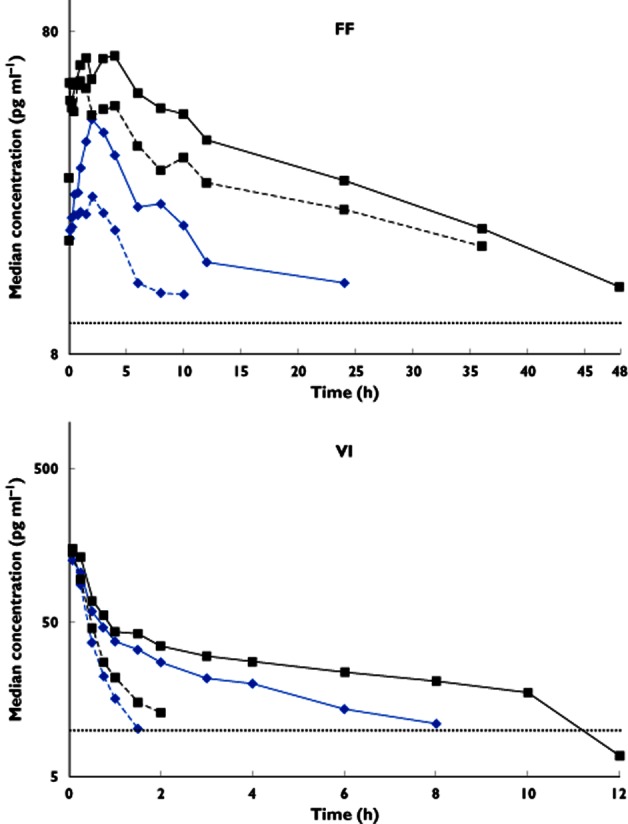
Single and repeat dose median plasma semi-log concentration–time profiles for fluticasone furoate (FF; 0–48 h) and vilanterol trifenatate (VI; 0–12 h) after co-administration of ketoconazole or placebo and FF/VI (study 2).  , single dose FF/VI + placebo;
, single dose FF/VI + placebo;  , single dose FF/VI + ketoconazole;
, single dose FF/VI + ketoconazole;  , repeat dose FF/VI + placebo;
, repeat dose FF/VI + placebo;  , repeat dose FF/VI + ketoconazole; and
, repeat dose FF/VI + ketoconazole; and  , lower limit of quantification (LLQ) 10 pg ml–1
, lower limit of quantification (LLQ) 10 pg ml–1
In study 2, accumulation of FF or VI was similar following both treatments. From the first to the seventh day of dosing, the mean FF Cmax increased by 79% (90% CI 51, 113) and by 105% (90% CI 72, 143) for FF/VI and ketoconazole and FF/VI and placebo, respectively, whilst the mean VI Cmax increased by 28% (90% CI 13, 44) and 18% (90% CI 4, 34), respectively. Assessment of accumulation using AUC was confounded by the fact that single dose data were truncated due to concentrations falling below the lower limit of quantification at earlier time points than for repeat dosing and hence are not reported. Although there was reduced CL/F for FF and VI following FF/VI with ketoconazole compared with FF/VI with placebo the FF apparent half-life was similar for the two treatments (on average 22 h; Table 4). The apparent half-life of VI was longer following administration of FF/VI with ketoconazole (on average 7.5 h) compared with FF/VI with placebo (on average 1.1 h; Table 4).
Table 4.
Summary of pharmacokinetic parameters following repeat dose co-administration of FF/VI with ketoconazole or placebo for 7 days (study 2)
| Parameter | Treatment | Fluticasone furoate | Vilanterol | ||
|---|---|---|---|---|---|
| Geometric mean (CV%) | 95% CI | Geometric mean (CV%) | 95% CI | ||
| AUC* (pg h ml−1) | FF/VI + placebo | 715 (24.2) | (635, 806) | 76.1 (124) | (47.1, 123) |
| FF/VI + ketoconazole | 970 (53.5) | (756, 1245) | 125 (186) | (68.1, 230) | |
| Cmax (pg ml−1) | FF/VI + placebo | 62 (14.6) | (57.7, 66.7) | 120 (59.6) | (91.5, 158) |
| FF/VI + ketoconazole | 82.5 (34.6) | (69.8, 97.5) | 147 (46.5) | (118, 183) | |
| t1/2 (h) | FF/VI + placebo | 22.0 (34.6) | (16.1, 30.1) | 1.11 (103) | (0.45, 2.70) |
| FF/VI + ketoconazole | 22.2 (15.3) | (19.9, 24.8) | 7.51 (72.2) | (4.56, 12.3) | |
| tmax† (h) | FF/VI + placebo | 0.77 (0.00–2.00) | NA | 0.08 (0.08–0.25) | NA |
| FF/VI + ketoconazole | 2.00 (0.00–4.02) | NA | 0.08 (0.00–0.25) | NA | |
| CL/F (l h−1) | FF/VI + placebo | 280 (24.2) | (248, 315) | 257 (82.4) | 178, 372 |
| FF/VI + ketoconazole | 206 (53.5) | (161, 265) | 80.9 (129) | 49.4, 132 | |
Abbreviations: AUC, area under the curve; CI, confidence interval; CL/F, apparent clearance; Cmax, maximum plasma concentration; CV, coefficient of variation; FF, fluticasone furoate; NA, not applicable; t1/2, half-life; tmax, time to maximum plasma concentration; VI, vilanterol trifenatate.
FF = AUC(0–24); VI = AUC(0–t′).
Median (range).
Safety
In both studies, treatment was well tolerated alone and when co-administered with ketoconazole. No SAEs were reported and no subject withdrew from the studies due to an AE. The most frequently reported AE in both studies was headache; seven of 19 subjects reported headache with VI and ketoconazole compared with two of 18 with VI and placebo in study 1, and seven of 18 subjects reported headache with FF/VI and ketoconazole compared with three of 18 with FF/VI and placebo in study 2. There was no clinical laboratory, vital sign or ECG abnormality of clinical significance in any subject in either study.
Discussion
These two studies were performed to determine whether co-administration of repeat dose ketoconazole, a strong CYP3A4 and potent PgP inhibitor 10, had an effect on the pharmacodynamics and pharmacokinetics of either single dose inhaled VI (25 μg) or repeat dose inhaled FF/VI (200/25 μg) in healthy subjects. Vilanterol and FF are substrates for PgP and are primarily metabolized by the CYP3A4 isoenzyme. Both FF and VI have high plasma clearance, indicative of extensive first-pass metabolism, resulting in low oral bioavailability from the swallowed portion of the dose (1.26 and <2%, respectively 16). Consequently, systemic exposure for both inhaled FF and VI is primarily due to absorption of the inhaled portion of the dose delivered to the lung. Co-administration of an inhibitor of PgP and the CYP3A4 isoenzyme, such as ketoconazole, may potentially increase the systemic exposure to VI and/or FF when co-administered by inhibition of first-pass metabolism and/or reduced metabolic clearance, which can result in an increase in systemic effects. In the pilot study (study 1), co-administration of a single inhaled dose of VI with repeat dose ketoconazole did not result in a significant increase in β2-adrenoceptor-mediated systemic PD effects. Co-administration of VI with ketoconazole resulted in a small, approximately twofold increase in VI systemic exposure (AUC) compared with VI and placebo, although there was no evidence for an increase in the VI Cmax. Previous repeat dose studies in healthy subjects have shown that VI doses up to 100 μg have little effect on β2-adrenoceptor-mediated systemic PD parameters, are well tolerated and are not associated with any clinically significant unwanted systemic effects 4. The general lack of VI pharmacodynamic effects on heart rate, blood potassium and QTcF seen with ketoconazole co-administration is consistent with these effects being predominantly attributable to the VI Cmax, which was unchanged, while the relatively limited increase in the VI AUC was probably insufficient to result in an increase in these systemic effects.
Co-administration of repeat dose ketoconazole and FF/VI (study 2) had no effect on heart rate or blood potassium compared with FF/VI and placebo, because predefined equivalence criteria were met. Likewise, there was no effect on either minimal diastolic or maximal systolic blood pressure. This lack of effect was consistent with the results of study 1. Co-administration of repeat dose ketoconazole and FF/VI did have a small effect on maximal QTcF (0–4 h), which was on average 7.6 ms greater compared with FF/VI and placebo. This small difference in QTcF was not associated with any absolute QTcF values of clinical concern. In this study, ketoconazole was only co-administered with FF/VI, and so any effects of ketoconazole alone could not be determined with this study design. Administration of ketoconazole alone (at a dose of 200 mg twice daily) has been reported to be associated with QTcF increases of ∼5–6 ms in one study 17, while increases of up to ∼6–12 ms have been reported, which are not considered to be clinically relevant 18. Based on these data, it is considered likely that the small difference in maximal QTcF seen with ketoconazole and FF/VI co-administration could be entirely attributable to ketoconazole rather than due to the minimal increase in VI exposure, particularly given that no increases were observed in any other systemic β2-adrenoceptor-mediated PD effects. Co-administration of repeat dose ketoconazole and FF/VI resulted in an increase in corticosteroid-mediated systemic effects. Equivalence was not concluded, because serum cortisol was on average 27% lower for FF/VI and ketoconazole when compared with FF/VI with placebo. This effect was in line with the increase in FF systemic exposure seen with FF/VI and ketoconazole co-administration on day 11, with the FF AUC(0–24) being on average 36% higher and the FF Cmax on average 33% higher compared with FF/VI with placebo. The increased FF AUC(0–24) following FF/VI with ketoconazole compared with FF/VI with placebo was reflected in a lower CL/F. However, given that the FF half-life was similar for the two treatments (on average 22 h), and the increase in Cmax was similar to that for AUC, the data would suggest that the increased FF exposure following co-administration of FF/VI with ketoconazole was due to an increase in oral bioavailability of FF rather than a decrease in the rate of plasma clearance. The systemic exposure of VI on day 11 was also significantly higher following administration of FF/VI and ketoconazole compared with FF/VI with placebo. There was an average increase of 65% in VI AUC(0–t′) and an average increase of 22% for VI Cmax following repeat dosing of inhaled FF/VI with ketoconazole compared with inhaled FF/VI with placebo. Although the apparent t½ of VI was longer following administration of FF/VI with ketoconazole (on average 7.5 h) compared with FF/VI with placebo (on average 1.1 h), this difference may be due to the estimate of VI apparent t½ being confounded by data following FF/VI with placebo being censored as a result of data falling below the lower limits of quantification at a notably earlier time point than those following FF/VI with ketoconazole. In this study, it was therefore not possible to determine whether the reduced VI CL/F was due to increased oral bioavailability of VI or a decrease in the rate of plasma clearance. However, the finding in study 1, where AUC was increased but Cmax was unchanged, would suggest that the effect for VI is more likely to be due to a decreased rate of plasma clearance of drug absorbed from the inhaled portion of the dose rather than increased oral bioavailability as a consequence of inhibition of first-pass metabolism. Although this study was conducted in healthy subjects, the results can be extrapolated to target patient populations. Moreover, as has been reported with fluticasone propionate in asthma 19 and COPD 20, systemic exposure to both FF and VI is anticipated to be lower in subjects with asthma or COPD compared with healthy subjects. On this basis, greater pharmacodynamic effects with FF/VI and ketoconazole co-administration would not be anticipated in clinical use. Administration of VI or FF/VI with ketoconazole or placebo was generally well tolerated. There were no SAEs and no AEs leading to withdrawal. There were also no clinically significant safety laboratory, vital signs or 12-lead ECG findings in either study.
Other long-acting beta agonists and inhaled corticosteroids are also metabolized by CYP3A4, and co-administration with CYP3A4 inhibitors can lead to increased systemic exposure and pharmacodynamic effects. Co-administration of ketoconazole with the LABA salmeterol resulted in increased salmeterol exposure (16-fold increase in AUC and 1.4-fold increase in Cmax 21), although the systemic effects were less marked than might have been expected. Likewise, co-adminstration of intranasal fluticasone propionate with ritonavir significantly increased fluticasone propionate exposure, resulting in significantly reduced serum cortisol concentrations 22. Overall, the increases in VI and FF exposure seen with ketoconazole co-administration in the present study were of a similar or lesser magnitude that those reported with salmeterol or fluticasone propionate.
In this study, ketoconazole was selected as a representative strong CYP3A4 and potent PgP inhibitor 10. Although not specifically studied, co-administration of other strong CYP3A4 and potent PgP inhibitors (e.g. clarithromycin, itraconazole and ritonavir 10) with FF/VI would be anticipated to produce similar increases in FF and VI exposure and effects on serum cortisol.
Conclusion
Co-administration of VI or FF/VI with ketoconazole resulted in a less than twofold increase in systemic exposure to FF and VI. There was no increase in β2-agonist systemic pharmacodynamic effects, while serum cortisol was decreased by 27%. Co-administration of FF/VI with strong CYP3A4 inhibitors has the potential to increase systemic exposure to both fluticasone furoate and vilanterol, which could lead to an increase in the potential for adverse reactions.
Acknowledgments
The authors wish to acknowledge ICON Development Solutions (Ellicott City, MD, USA) for conducting the derivation of PK parameters under the direction of Clinical Pharmacology Modelling and Simulation, GSK and Worldwide Bioanalysis DMPK, Ware, UK for analysing the PK samples. The authors also wish to acknowledge Diana Jones of Cambrian Clinical Associates Ltd for development of manuscript first draft, editorial suggestions to draft versions of this paper, assembling tables and figures, collating author comments and referencing. This assistance was funded by GSK.
Competing Interests
The following studies funded by GSK have been incorporated into this manuscript: protocol number HZA105548 (clinicaltrials.gov NCT01165125) and protocol number B2C112205 (clinicaltrials.gov NCT00866515). All listed authors meet the criteria for authorship set forth by the International Committee for Medical Journal Editors. All the authors (with the exception of Lee Tombs) are employees of GSK (and hold shares in the firm). There are no other competing interests to declare.
References
- 1.Clarke SE, Jones BC. Human cytochromes P450 and their role in metabolism-based drug-drug interactions. In: Rodrigues AD, editor. Drug-Drug Interactions. New York: Marcel Dekker; 2002. pp. 55–88. [Google Scholar]
- 2.Lötvall J, Bateman ED, Bleecker ER, Busse W, Woodcock A, Follows R, Lim J, Stone S, Jacques L, Haumann B. Dose-related efficacy of vilanterol trifenatate (VI), a long-acting beta2 agonist (LABA) with inherent 24-hour activity, in patients with persistent asthma. Eur Respir J. 2010;36(Suppl. 54):1013s. [Google Scholar]
- 3.Hanania NA, Feldman G, Zachgo W, Shim JJ, Crim C, Sanford L, Lettis S, Barnhart F, Haumann B. Safety of vilanterol trifenatate (VI) in a COPD dose-ranging study. Chest. 2012;142:119–127. doi: 10.1378/chest.11-2231. [DOI] [PubMed] [Google Scholar]
- 4.Kempsford RD, Norris V, Siederer SK. The pharmacodynamics, pharmacokinetics and tolerability of repeat doses of the novel inhaled long-acting beta2 adrenoceptor agonist (LABA) GW642444 (25, 50 and 100mcg) in healthy subjects. Am J Respir Crit Care Med. 2010;181:A4461. [Google Scholar]
- 5.van den Berge M, Luijk B, Bareille P, Dallow N, Postma DS, Lammers J-WJ. Prolonged protection of the new inhaled corticosteroid fluticasone furoate against AMP hyperresponsiveness in patients with asthma. Allergy. 2010;65:1531–1535. doi: 10.1111/j.1398-9995.2010.02414.x. [DOI] [PubMed] [Google Scholar]
- 6.Dollery C, editor. Therapeutic Drugs. Vol. 2. Edinburgh: Churchill Livingstone; 1991. Ketoconazole; pp. 21–24. [Google Scholar]
- 7.Brindley C, Falcoz C, Makie AE, Bye A. Absorption kinetics after inhalation of fluticasone propionate via the Diskhaler, Diskus and metered-dose inhaler in healthy volunteers. Clin Pharmacokinet. 2000;39:1–8. doi: 10.2165/00003088-200039001-00001. [DOI] [PubMed] [Google Scholar]
- 8.Tsunoda SM, Velez RL, von Moltke LL, Greenblatt DJ. Differentiation of intestinal and hepatic cytochrome P450 3A activity with use of midazolam as an in vivo probe: effect of ketoconazole. Clin Pharmacol Ther. 1999;66:461–471. doi: 10.1016/S0009-9236(99)70009-3. [DOI] [PubMed] [Google Scholar]
- 9.Gomez DY, Wacher VJ, Tomlanovich SJ, Herbert MF, Benet LZ. The effects of ketoconazole on the intestinal metabolism and bioavailability of cyclosporine. Int J Clin Pharmacol Ther. 1995;58:15–19. doi: 10.1016/0009-9236(95)90067-5. [DOI] [PubMed] [Google Scholar]
- 10.Zhang L, Zhang Y, Huang SM. Scientific and regulatory perspectives on metabolizing enzyme-transporter interplay and its role in drug interactions: challenges in predicting drug interactions. Mol Pharm. 2009;6:1766–1774. doi: 10.1021/mp900132e. [DOI] [PubMed] [Google Scholar]
- 11.FDA. 1996. FDA. Guidance for Industry: Drug interaction studies – study design, data analysis and implications for dosing and labelling. Available at http://www.fda.gov/cder/guidance/index.htm (last accessed 14 November 2011)
- 12.Ward KW, Stelman GJ, Morgan JA, Zeigler KS, Azzarano LM, Kehler JR, McSurdy-Freed JE, Proksch JW, Smith BR. Development of an in vivo preclinical screen model to estimate absorption and first-pass hepatic extraction of xenobiotics. II. Use of ketoconazole to identify P-glycoprotein/CYP3A-limited bioavailability in the monkey. Drug Metab Dispos. 2004;32:172–177. doi: 10.1124/dmd.32.2.172. [DOI] [PubMed] [Google Scholar]
- 13.Kim RB, Wandel C, Leake B, Cvetkovic M, Fromm MF, Dempsey PJ, Roden MM, Belas F, Chaudhary AK, Roden DM, Wood AJ, Wilkinson GR. Interrelationship between substrates and inhibitors of human CYP3A and P-glycoprotein. Pharm Res. 1999;16:408–414. doi: 10.1023/a:1018877803319. [DOI] [PubMed] [Google Scholar]
- 14.Prime D, De Backer W, Hamilton M, Cahn A, Preece A, Kelleher D, Lindo E. Comparison of inhalation profiles through a novel dry powder inhaler (nDPI) and lung function measurements for healthy subjects, asthma and chronic obstructive pulmonary disease (COPD) patients. Am J Respir Crit Care Med. 2012;185:A2940. [Google Scholar]
- 15.Cekici L, Arschang V, Kohansal R, Burghuber OC. Short-term effects of 2009 Short-term effects of inhaled salbutamol on autonomic cardiovascular control in healthy subjects: a placebo-controlled study. Br J Clin Pharmacol. 2009;67:394–402. doi: 10.1111/j.1365-2125.2009.03377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allen A, Bianco J, Bal J, Tombs L, Kempsford R. The absolute bioavailability of fluticasone furoate (FF) and vilanterol (VI) following inhaled administration in combination in healthy subjects. Eur Respir J. 2011;38(Suppl. 55):724s. [Google Scholar]
- 17.Robert M, Salvà M, Segarra R, Pavesi M, Esbri R, Roberts D, Golor G. The prokinetic cinitapride has no clinically relevant pharmacokinetic interaction and effect on QT during coadministration with ketoconazole. Drug Metab Dispos. 2007;35:1149–1156. doi: 10.1124/dmd.106.010835. [DOI] [PubMed] [Google Scholar]
- 18.Nizoral (ketoconazole) Product Information. 2010. Available at http://www.medicines.org.uk/EMC/medicine/912/SPC/Nizoral%20Tablets (last accessed 7 December 2010)
- 19.Brutsche MH, Brutsche IC, Munavvar M, Langley SJ, Masterson CM, Daley-Yates PT, Brown R, Custovic A, Woodcock A. Comparison of pharmacokinetics and systemic effects of inhaled fluticasone propionate in patients with asthma and healthy volunteers: a randomised crossover study. Lancet. 2000;356:556–561. doi: 10.1016/S0140-6736(00)02581-2. [DOI] [PubMed] [Google Scholar]
- 20.Singh DS, Whale C, Houghton N, Daley-Yates P, Kirby SM, Woodcock AA. Pharmacokinetics and systemic effects of inhaled fluticasone propionate in chronic obstructive pulmonary disease. Br J Clin Pharmacol. 2003;55:375–381. doi: 10.1046/j.1365-2125.2003.01758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seretide Product Information. 2011. Available at http://hcp.gsk.com/therapy_areas/respiratory-and-allergy/seretide/542852/ (last accessed 20 September 2012)
- 22. Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2003/020121s028lbl.pdf (last accessed 19 August 2012)