

Abstract
Background
β-adrenergic receptor (β-AR) activation can provoke cardiac arrhythmias mediated by cAMP-dependent alterations of Ca2+ signaling. However cAMP can activate both PKA and an Exchange protein directly activated by cAMP (Epac) but their functional interaction is unclear. In heart selective Epac activation can induce potentially arrhythmogenic sarcoplasmic reticulum (SR) Ca2+ release that involves CaMKII effects on the ryanodine receptor (RyR).
Methods and results
We tested whether physiological β-AR activation causes Epac-mediated SR Ca2+ leak and arrhythmias, whether it requires Epac1 vs. Epac2, β1-AR vs. β2-AR and CaMKIIδ-dependent phosphorylation of RyR2-S2814. We used knockout mice for Epac1, Epac2 or both (DKO). All knockouts exhibited unaltered basal cardiac function, Ca2+ handling and hypertrophy in response to pressure overload. However, SR Ca2+ leak induced by the specific Epac activator 8-CPT in wild-type was abolished in Epac2-KO and DKO, but unaltered in Epac1-KO. β-AR-induced arrhythmias were also less inducible in Epac2-KO vs. wild-type. β-AR activation with PKA inhibition, mimicked 8-CPT effects on SR Ca2+ leak, and was prevented by blockade of β1-AR but not β2-AR. CaMKII inhibition (KN93) and genetic ablation of either CaMKIIδ or CaMKII phosphorylation on RyR2-S2814 prevented 8-CPT-induced SR Ca2+ leak.
Conclusions
β1-AR activates Epac2 to induce SR Ca2+ leak via CaMKIIδ-dependent phosphorylation of RyR2-S2814. This pathway contributes to β-AR-induced arrhythmias and reduced cardiac function.
Keywords: arrhythmias, sarcoplasmic reticulum, heart failure, adrenergic receptors, calcium
Introduction
β-adrenergic receptor (β-AR) activation (which is chronic in heart failure, HF) can provoke cardiac arrhythmias due to abnormal diastolic Ca2+ release from the sarcoplasmic reticulum (SR) via phosphorylation of the ryanodine receptor (RyR2).1-3 This also contributes to reduced cardiac contractility caused by a reduced SR Ca2+ load and systolic SR Ca2+ transient.4-7 Early studies of enhanced SR Ca2+ leak in HF or with β-AR activation focused on PKA, a classical cAMP target.1 However, β-AR activation can also induce PKA-independent diastolic SR Ca2+ leak mediated by Ca2+/calmodulin-dependent protein kinase II (CaMKII).8-12 Indeed, Epac (Exchange protein directly activated by cAMP), a cAMP target, has been shown to increase diastolic SR Ca2+ leak via CaMKII.12 Two Epac isoforms have been identified. Epac1 (one cAMP binding domain) seems ubiquitously expressed whereas Epac2 (two cAMP binding sites) seems more prominent in brain, pituitary and adrenal gland13-15.
To date, cardiac Epac activation, using a cAMP analog 8-CPT (8-(4-chlorophenylthio)-2’-O-methyladenosine-3’,5’-cyclic monophosphate) can enhance SR Ca2+ release, via the small-G protein, Rap and phospholipase-Cε11, 16 Although most work agrees on CaMKII as the mediator of Epac-dependent RyR2 activation,11, 12, 17 Epac-induced SR Ca2+ leak can also reduce SR Ca2+ load and release.12, 18, 19 Those effects are similar to those observed in HF 8, 20-22 where Epac is also upregulated.23, 24 Notably, 8-CPT can trigger ventricular arrhythmias in mice and may be involved in cardiac fibrosis through β-AR-dependent production of IL-6 in cardiac fibroblast.17, 25 Thus Epac may be intimately involved in cardiomyopathy such as arrhythmias and HF via alteration of Ca2+ handling.
Despite the emergence of this cAMP- and Epac-induced SR Ca2+ leak pathway, key fundamental issues are still unknown. 1) Most prior work implicating Epac has relied on the pharmacological selective agonist 8-CPT, which could have off-target effects.26 2) It is unknown which Epac isoform mediates RyR and arrhythmogenic effects of Epac activation. 3) It is unknown whether these effects are mediated by type 1 or type 2 β-AR (β1-AR or β2-AR). 4) It has been proposed that Epac-mediated effects might require higher levels of β-AR activation than for PKA activation. 5) It is unknown whether and to what extent Epac contributes to β-AR induced arrhythmias. 6) It is not known if the Epac-dependent effects on SR Ca2+ release require the major cardiac CaMKIIδ isoform or if other isoforms suffice. 7) It is also unclear whether the SR Ca2+ release effects of Epac require the putative RYR CaMKII site S2814. Here we generated Epac knock-out (KO) mice and use other genetically altered mice to unequivocally address all of those points.
We demonstrate that Epac-KO did not alter cardiac function, early pressure overload-induced hypetrophy or basal myocyte Ca2+ handling. Epac-dependent arrhythmogenic effects were prevented by ablation of Epac2, β1-AR, CaMKIIδ and RyR2-S2814 phosphorylation. In conclusion, Epac2 mediates β1-AR-induced cardiac arrhythmias via CaMKIIδ and RyR-S2814 phosphorylation.
Methods
An expanded Method is provided in the Online-only Supplement
Epac Knockout Mouse Lines
Epac1 and Epac2 KO mice were developed via similar strategies (see Online-only Supplemental Methods). The Epac1-KO mice were also used in a parallel initial project.27 Epac1/Epac2 double knockout mice (DKO) were obtained by breeding heterozygous Epac1-KO and Epac2-KO mice to generate double null mice. Littermate Epac2 null mice were used as controls. All procedures were performed in accordance with NIH Guide for the Care and Use of Laboratory Animals and approved by Institutional Animal Care and Use Committee of UCSD.
CaMKIIδC-KO and RyR2-S2814A and S2814D mice
Homozygous CaMKIIδ-KO mice were generated from heterozygous CaMKIIδ mice as previously described.28 RyR2-S2814A and RyR2-S2824D knock-in mice were previously described29 and respectively inhibit or mimic CaMKII phosphorylation at this site on RyR2.
Myocytes isolation
Cardiac ventricular myocytes were isolated using the retrograde Langendorff perfusion technique with Type-2 collagenase (Worthington) perfusion. Cells were kept in 1 mM [Ca] (20-22°C) before starting experiments. Epac-KO cardiomyocytes were isolated from 3 and 10 months old mice. There were no apparent differences between age groups. All procedures were approved by the UC Davis Institutional Animal Care and Use Committee (IACUC).
Echocardiography
Anesthetized mice were analyzed by echocardiography and hemodynamics in 8-month old WT and Epac1-KO, 18-month old WT and Epac2-KO mice and 18-month old control and Epac-DKO mice. Hemodynamic analysis was performed in 3-month old Epac1-KO and WT mice and 5-month old Epac-DKO and control mice. Transverse Aortic Constriction (TAC) was performed in either 3-month old male Epac1 or Epac2 and age-matched WT control mice went either through sham operation (n=3) or TAC surgery (n=6-9).
In Vivo Electrophysiology
Programmed intracardiac stimulation was performed in age-matched control (Ctl) and homozygous Epac2-KO mice, as described.30 The incidence of reproducible sustained VT (episode >10 consecutive beats of VT) was determined ±ISO (ISO, 0.5mg/kg, i.p.).
Line scan confocal microscopy
Ca2+ Transient, SR Ca2+ leak and SR Ca2+ load were assessed in freshly isolated cells loaded with 5 μM Fluo-3AM or Fluo-4AM (Molecular Probes)12 using confocal microscopy (BioRad, Radiance 2100, x40 oil immersion objective). Excitation was at 488nm (Ar Laser) and emission at >505nm. Ad-FRET-based cAMP indicator31 was provided by Dr. Y.K. Xiang (University of California, Davis). Image analysis was performed using ImageJ software and homemade routines in IDL (Interactive Data Language, ITT).
Quantitative PCR
Quantitative PCR was done using GAPDH (QT01658692, Qiagen) as control. Epac1-1 (forward: 5’-CTCTGTCTCTGCCCTGTTCC-3’; reverse: 5’ CGCAAAGAAAGAGTTGAGG-3’), Epac1-2 (forward: 5′-TGTTGGTGAAGGTCAATTCTG-3′; reverse: 5′-CCACACCACGGGCATC-3′), Epac2-1 (forward: 5′-GGCGTACCAGATGACAACCT-3′; reverse: 5′-CCTCCTCAGGAACAAATCCA-3′), Epac2-2(forward: 5′-TGTTAAAGTGTCTGAGACCAGCA-3′; reverse: 5′-AAAGGCTGTCCCAATTCCCAG-3′)
Antibodies
Epac1 antibody was a gift from Dr Xiaodong Cheng (University of Texas Medical Branch). Epac2 and GAPDH antibodies were purchased from Santa Cruz.
Statistical Analysis
Data are expressed as mean ± SEM. For multiple group comparisons, a one way Anova test was followed by Bonferroni post-hoc test. A repeated measures Two way Anova test was followed by a Bonferroni post-hoc test to examine the effect of drug application (before vs. after) between multiple groups. Statistical discrimination between two populations (with or without treatment) was evaluated by Student’s t-test for independent samples and paired t-tests for matched samples. Differences were considered significant for p<0.05.
Results
Generation of Epac1 and Epac2-KO Mouse Lines
Epac1 and Epac2-KO mice were generated using the Cre-loxP system by floxing exons 3-5 and 7, respectively (Online-only Supplemental Data Figure 1A,D). Southern blots show correct recombinant ES cell clones for each case (Online-only Supplemental Data Figure 1B,E). Anti-Epac1 antibody confirmed complete deletion of Epac1 in null mouse hearts (Online-only Supplemental Data Figure 1C). However, commercial anti-Epac2 antibodies could not detect homogenate Epac2 protein. Thus Epac2 was first immunoprecipitated from WT and Epac2-KO hearts before blotting. Flag tagged Epac2 protein expressed in 293T cells were used to confirm antibody efficacy. No Epac2 protein was detected in Epac2-KO hearts (Online-only Supplemental Data Figure 1F). Thus, we created Epac1-KO and Epac2-KO mouse lines.
Cardiomyocytes may express Epac1 and Epac2-mRNA at different levels.23 Quantitative PCR showed higher Epac2-mRNA than Epac1 in both heart homogenates and isolated cardiomyocytes (Online-only Supplemental Data Figure 2A). Epac1 protein level was not altered in Epac2-KO vs. WT control hearts (not shown), nor was Epac2-mRNA altered in Epac1-KO mice (Online-only Supplemental Data Figure 2B), although we could not assess this at the protein level.
Epac is not critical to baseline cardiac function
In vivo baseline cardiac function was normal in all 3 KOs in terms of left ventricular (LV) posterior wall thickness (LVPW), LV internal dimension (LVID) and fractional shortening (FS%), compared to WT for up to 18 months of age (Figure 1, Online-only Supplemental Data Table 1). Moreover, heart rates, LV pressure, ±dP/dtmax and response to dobutamine perfusion were unchanged (Online-only Supplemental Data Table 1). None of the deletions appreciably affected basal cardiac function, heart weight to body weight ratio (Figure 1D). Nor was there any significant difference in the transaortic constriction (TAC; 3-4 weeks) induced hypertrophy, cardiac dysfunction (Figure 1E-F, Online-only Supplemental Data Figure 3) or expression levels of SERCA or NCX (in preliminary Western analysis) in either Epac-KO mouse. The lack of differential phenotype discouraged further study of these TAC mice.
Figure 1. Normal cardiac function in Epac1, Epac2 and DKO assessed by echocardiography.
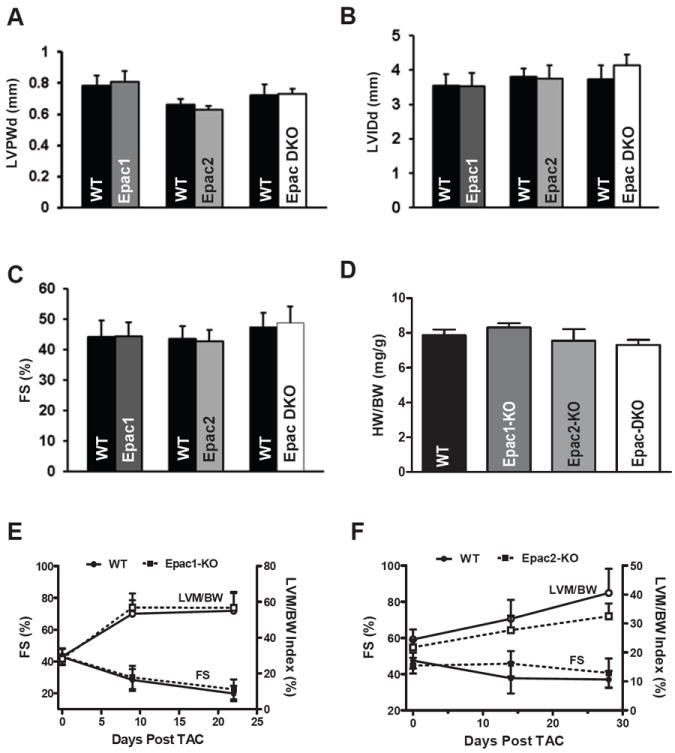
A. 8-months WT (n=6) and Epac1-KO (n=7) mice, 18-months WT (n=7) and Epac2-KO (n=6) mice and 18-months control (n = 7) and Epac-DKO (n=7) mice were measured by echocardiography. Between null mice and controls there was no difference in left ventricular wall thickness (LVPWd), left ventricle internal dimension size (LVIDd) at end of diastole (B), cardiac systolic function (C) or heart weight/Body weight ratio (D; Epac1-KO (n=3), Epac2-KO (n=5) and Epac-DKO (n=3)). E. Cardiac function (fractional shortening, FS) and hypertrophy (left ventricular mass/body weight; LVM/BW) in Epac1-KO (n= 8) and (F) Epac2-KO (n=9) vs. WT measured during 3-4 weeks after TAC.
At the myocyte level, basal Ca2+ transient amplitude and time constant of intracellular Ca2+ concentration ([Ca2+]i)decline (τtwitch) indicative of SR Ca2+-ATPase function (Figure 2A-B) as well as SR Ca2+ content and Na+/Ca2+ exchange function (τNCX; Figure 2A-D) were not significantly different between groups. Diastolic Ca2+ spark frequency (CaSpF, a measure of SR leak) was unaltered among the groups, particularly when normalized to SR Ca2+ load (to account for the intrinsic effect of SR Ca2+ load on CaSpF; Figure 2D-E). So, Epac ablation does not appreciably alter baseline Ca2+ handling. This is consistent with the idea that basal [cAMP] and Epac activation are low in adult ventricular myocytes in the absence of agonists or phosphodiesterase inhibition. It also creates a uniform baseline with which to examine the role of Epac during β-AR activation.
Figure 2. Unaltered basal Ca2+ signaling in Epac-KO mice.
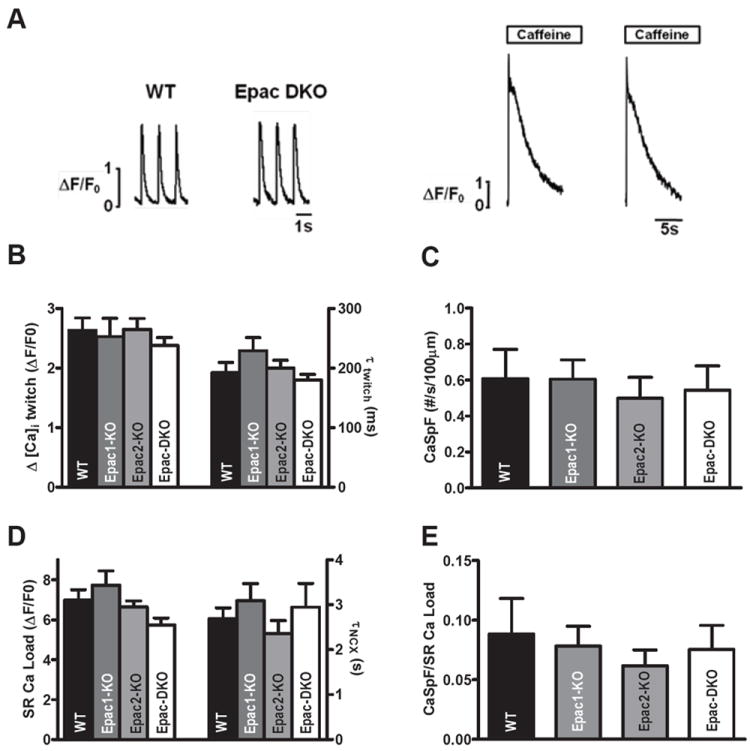
A. Normalized Ca2+ transient (left) and SR Ca2+ load (right) traces, recorded in confocal microscopy from WT and Epac-DKO mice. B. Mean Ca2+ transient amplitude (ΔF/F0, where F0 is diastolic fluorescence). WT (n=31), Epac1-KO (n=17), Epac2-KO (n=39) and Epac-DKO (n=45). C. Mean Ca2+ spark frequency (CaSpF) as number of sparks per 100 μm s-1. WT (n=25), Epac1-KO (n=14), Epac2-KO (n=39) and Epac-DKO (n=34). D. Mean SR Content assessed by 10 mM caffeine-induced fluorescence change (ΔF/F0). WT (n=25), Epac1-KO (n=15), Epac2-KO (n=29) and Epac-DKO (n=14). E. CaSpF normalized to SR Ca2+ load. Number of animals: 7 WT, 6 Epac2-KO, 4 Epac1-KO and 6 Epac DKO (p=NS).
Epac2, not Epac1, is responsible for PKA-independent SR Ca2+ Leak
To test which Epac isoform mediates the previously reported 8-CPT-induced increase of SR Ca2+ release,12 we measured CaSpF in isolated ventricular myocytes. Figure 3A shows that when 10 μM 8-CPT was applied to individual WT myocytes CaSpF was enhanced, as previously described.12 The same was observed in Epac1-KO (especially when normalized to SR Ca load; Figure 3C). In contrast, the 8-CPT-induced increase of CaSpF was totally abolished in Epac2-KO or DKO mice (Figure 3A,C).
Figure 3. Epac2 is critical for Epac-mediated SR Ca2+ leak.
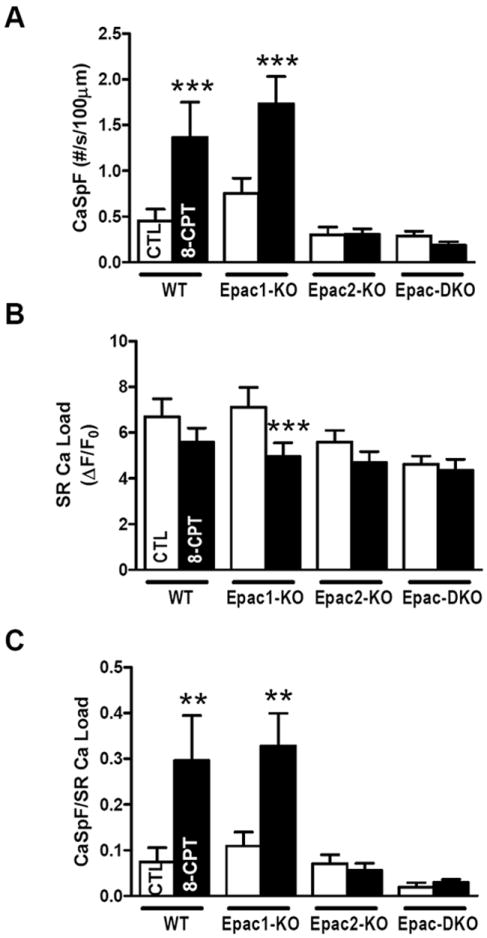
A. Mean CaSpF in intact cardiomyocytes. WT±8-CPT (n=7), Epac1-KO±8-CPT (n=13), Epac2-KO±8-CPT (n=21) and Epac-DKO±8-CPT (n=20). B. SR Ca2+ load. WT±8-CPT (n= 16), Epac1-KO±8-CPT (n=13), Epac2-KO (n=13), Epac2-KO+8-CPT (n=21), Epac-DKO (n=12) and Epac-DKO+8-CPT (n=19). C. CaSpF normalized to SR Ca2+ load. **p<0.01. ***p<0.001
Epac2 contributes to β-AR-induced cardiac arrhythmias
Since Epac2 can enhance diastolic SR Ca release we tested whether Epac2 might contribute to β-AR-induced arrhythmias. This was assessed in vivo as the ability of programmed electrical stimulation to induce sustained ventricular tachycardia (SVT) before or after isoproterenol (ISO) injection (0.5mg/kg, i.p.). This protocol was intentionally aggressive, such that all WT exhibited SVT (p<0.001 vs. control). This allowed the detection of reduced susceptibility and SVT duration in Epac2-KO vs. WT (50%, p<0.05 vs. WT; Figure 4A-C). Furthermore, Epac2-KO exhibited reduced tendency to VT vs. WT following a burst pacing protocol (Online-only Supplemental Data Figure 4). WT and Epac2-KO mice did not exhibit differences in either heart rate, ECG parameters or refractory periods at baseline or in response to ISO (Online-only Supplemental Data Table 2 and Online-only Supplemental Data Table 3). They was also no difference in ventricular effective refractory period between WT and Epac2-KO mice (±ISO; Online-only Supplemental Data Table 4) suggesting that the arrhythmia is unlikely due to altered repolarization, but may indeed reflect reduced triggered activity in Epac2-KO related to less RyR sensitization.
Figure 4. Epac2-KO deletion reduced ISO-dependent increase of pacing-induced ventricular tachycardia.
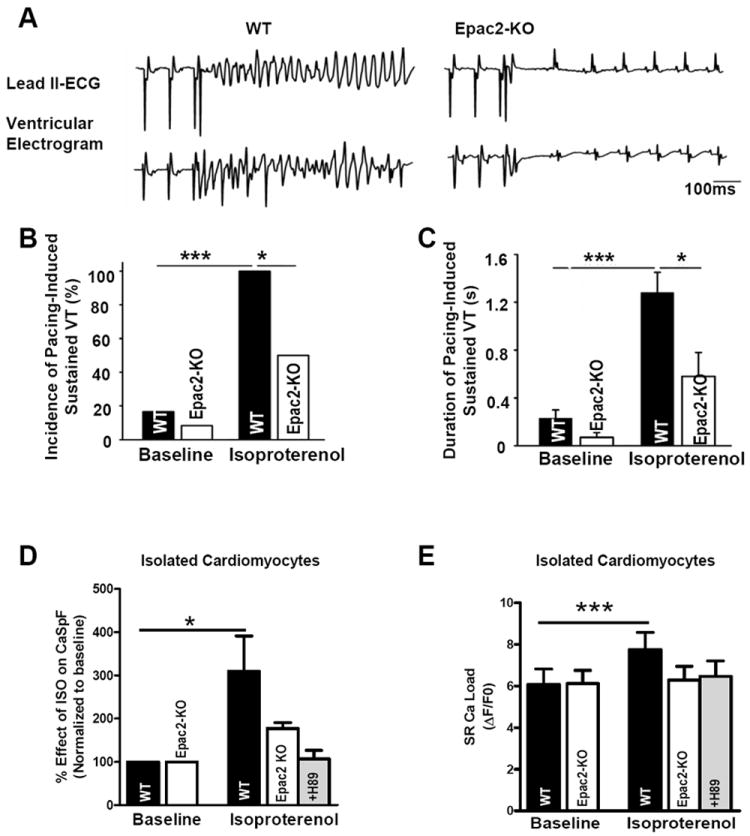
A. Representative simultaneous surface ECG (lead 2) and intra-cardiac ventricular electrogram after ISO (0.5mg/kg) revealed sustained ventricular tachycardia (SVT) in WT and sinus rhythm in Epac2-KO after S1-S2 extra-stimuli at pacing CL of 90ms. B. Mean SVT incidence before and after ISO stimulation. C. Mean duration of SVT under same conditions (n=12 WT and 12 Epac2-KO mice for C and D). D. Percent effect of ISO on CaSpF in isolated cardiomyocytes from WT (n=7) and Epac2-KO±H89 (n=5 and n=12). E. SR Ca load before and after ISO in same cells than D. *P<0.05, ***P<0.01.
To complement those in vivo arrhythmia measurements, we assessed ISO-induced increases in CaSpF in Epac2-KO cardiomyocytes, reasoning that SR Ca release events may be a key underlying mechanism for these triggered arrhythmias (Figure 4D). ISO increased CaSpF in WT, but this effect was dramatically blunted in Epac2-KO. The residual CaSpF enhancement in Epac2-KO mice was abolished by PKA inhibition using H89 (a potent inhibitor of PKA). ISO-induced Ca2+ waves also tended to be less frequent in Epac2-KO vs. WT myocytes (79 ±24% vs. 132 ± 48% increase; NS). In Epac2-KO mice the inhibition of PKA by H89 fully reversed the effects of ISO on Ca transient amplitude (Online-only Supplemental Data Figure 5) and correction for SR Ca2+ load (Figure 4E) did not alter this interpretation. We conclude that Epac2 activation (in parallel with PKA) contributes significantly to β-AR-induced triggered arrhythmias and the SR Ca release events which may underlie them.
Epac is activated by moderate levels of β-AR stimulation
β-AR stimulation activates PKA and Epac, and alters SR Ca2+ leak by phosphorylation of phospholamban (via elevated SR Ca2+ load32) or of RyR2 (which is controversial).1, 5, 32-34 To better understand the physiological significance of Epac signaling, we tested direct β-AR stimulation ±PKA inhibition. First, we assessed the isoproterenol concentration ([ISO]) needed for maximal effects on inotropy, lusitropy and Ca2+ sparks (Figure 5A-B). ISO induced a concentration-dependent increase of Ca2+ transient amplitude, CaSpF and τtwitch (indicative of phospholamban phosphorylation effects). The lusitropic effect was maximal at ~100 nM ISO, while Ca2+ transient amplitude and CaSpF were maximal at 100-300 nM ISO. Thus, we focused on the 30-300 nM range of [ISO] with PKA blocked by pretreatment with 2 μM H89. H89 can influence [cAMP] achieved by ISO application because of PKA effects on phosphodiesterases, which was tested in control experiments using a FRET-based cAMP sensor35 (Online-only Supplemental Data Figure 6). H89 slightly increased basal [cAMP], but 30 nM ISO caused a similar cAMP level to be reached in the absence or presence of H89.
Figure 5. Low β-AR PKA-independent stimulation activates Epac-induced SR Ca2+ leak in rat cardiomyocytes.
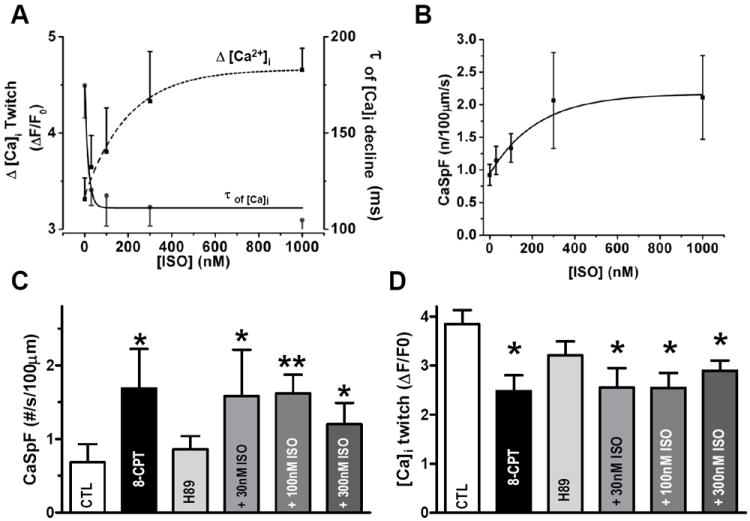
A. Average Ca2+ transient amplitude and Ca2+ transient decay time constant (τ) of SR Ca2+ uptake in intact cardiomyocytes without (n=35) or with 30nM (n=9), 100nM (n=10), 300nM (n=8) or 1μM (n=12) ISO. B. ISO concentration-response curve of CaSpF for conditions obtained as in A. C. CaSpF ±8-CPT and after physiological activation of Epac (ISO+2μM H89). Control (n=8), 8-CPT (n=8), 30nM ISO+H89 (n=4), 100nM ISO+H89 (n=6) and 300nM ISO+H89 (n=6)). D. Average Ca2+ transient amplitude in same conditions. *p<0.05.
H89 pretreatment blocked the ISO-induced increase of Ca2+ transient amplitude, SR Ca2+ load and τtwitch (Online-only Supplemental Data Figure 7), consistent with potent PKA inhibition. In contrast, ISO still greatly increased CaSpF with PKA blocked, at all [ISO] (Figure 5C) and also decreased Ca2+ transient amplitude (Figure 5D). Both of these effects are similar to those observed upon Epac activation by 8-CPT (Figure 5C-D). This implies that most of the PKA-independent CaSpF enhancement is mediated by Epac. The ISO-mediated decrease in Ca2+ transient amplitude when PKA is blocked is consistent with the idea that PKA is required for the β-AR-dependent positive inotropic effect, but that other β-AR-mediated effects negatively influence Ca2+ transients (e.g. SR Ca2+ unloading via Epac). Notably, 30nM ISO+H89 was sufficient to fully induce the PKA-independent enhancement of CaSpF, comparable to that induced by 8-CPT. This suggests that physiologically relevant β-AR activation may activate the Epac2 pathway in parallel to PKA, and at similar [cAMP], and both pathways contribute to arrhythmogenesis.
Epac-induced SR leak is activated by β1-AR
Next we sought to determine whether β1-AR or β2-AR is responsible for mediating the Epac-dependent activation of Ca2+ sparks and arrhythmias11, 12, 17. Figure 6 was done in rat myocytes for comparison with mouse data obtained in other figures as well as previous reports. Both 8-CPT and ISO increased CaSpF (Figure 6A) and reduced steady state SR Ca2+ load (Figure 6C) as seen in mouse experiments (Figure 3). The lower SR Ca2+ load in rat myocytes treated with 8-CPT may be secondary to the increase in CaSpF. This would actually feedback to limit the rise in CaSpF (due to lower luminal SR Ca2+). As in mouse myocytes, in studies with rat myocytes ISO+H89 produce similar effects to that of 8CPT alone (Figure 6A-C). Thus, ISO+H89 is a good treatment to assess β-AR-induced Epac effects independent of PKA.
Figure 6. β1-AR is the upstream activator of Epac-induced SR Ca2+ leak.
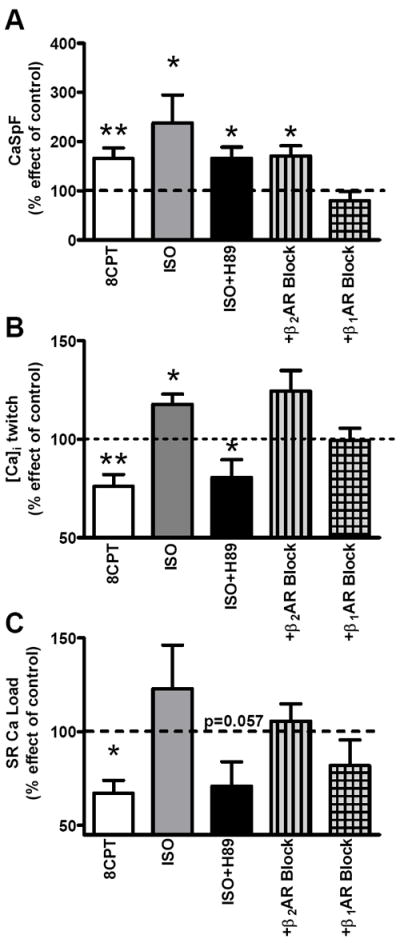
A. CaSpF average under 8-CPT (white, n=7), 100nM ISO (n=5), 100nM ISO+2mM H89 (n=7), ISO+H89+300nM CGP-20712A (β2-AR block; n=8) and ISO+H89+50nM ICI-118.551 (β1-AR block; n=7). Data are percent with respect to control. B. Average Ca2+ transient amplitude under 8-CPT (n=10), 100nM ISO (n=8), ISO+H89 (n=9), ISO+H89+CGP-20712A (n=11) and ISO+H89+ICI-118.551 (n=5). C. Mean of percent effect of SR Ca2+ content vs. control. *p<0.05, **p<0.01.
To determine the β-AR subtype involved, we treated cells with specific β1- and β2-AR inhibitors (along with H89) prior to ISO exposure. β1-AR blockade (CGP-20712A) was able to fully prevent the ISO-induced increase of CaSpF, whereas the response following β2-AR blockade (ICI-118.551) was not different from ISO+H89 (Figure 6A). Consistent with this result, CGP-20712A also restored Ca2+ transient amplitude (99.4±6.2% of control) vs. 8-CPT or ISO+H89 (166±23 and 165±22%) (Figure 6B). The 8-CPT or ISO+H89-dependent rise of SR Ca2+ leak, results in a decrease in SR Ca2+ load in both cases (Figure 6B), which is partially prevented by β1-AR block.
Epac mediates RyR2-S2814 phosphorylation via CaMKIIδ
Our previous work suggested that the Epac-induced CaSpF enhancement involved CaMKII activation and RyR2 phosphorylation.12 However the phosphorylation site and CaMKII isoform required are unknown. RyR2-S2814 has been suggested as an important CaMKII phosphorylation target.30, 36 To test its requirement for Epac-dependent effects, we used RyR2-S2814 knock-in mice. In those miceS2814 was rendered either non-phosphorylatable (RyR2-S2814A)29 or phosphomimetic (RyR2-S2814D).30
In WT mice, CaMKII inhibition (1 μM KN93) blocked the 8-CPT-induced increase of CaSpF and the reduction of SR Ca2+ load and Ca transient (Figure 7A-D). The same effects were observed in both knock-in mice, suggesting that this CaMKII target (and presumably the leak induced), is sufficient to depress twitch Ca2+ transients. These results prove that phosphorylation of this CaMKII target site is essential for Epac-dependent activation of SR Ca2+ leak.
Figure 7. Nonphosphorylatable RyR2-S2814 and CaMKIIδ deletion abolish Epac-induced SR Ca2+ leak.
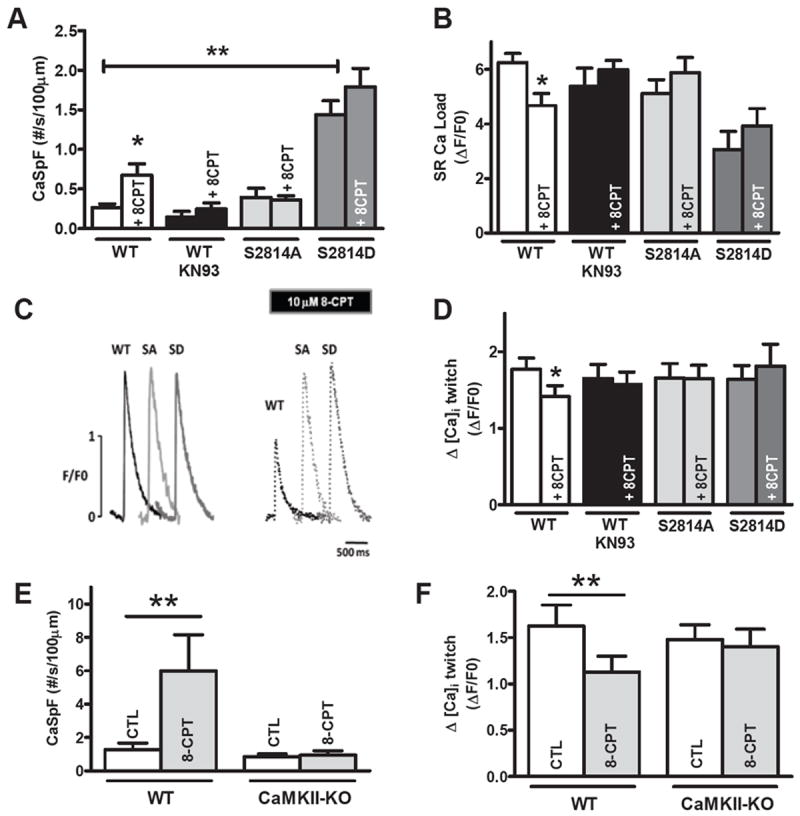
A. CaSpF average from WT (n=16), S2814D (n=9) and S2814A mice (n=10) before and after 8-CPT and ±KN93 (n=9) for WT. B. Mean SR Ca2+ content before 8-CPT in WT (n=18), WT-KN93 (n=9), S2814D (n=15) and S2814A mice (n=11) and +8-CPT in WT (n=16), WT-KN93 (n=9), S2814D (n=5) and S2814A mice (n=13). C. Ca2+ transient traces (F/F0) in WT, S2814A and S2814D. Dotted traces represents same cells with 10mM 8-CPT. D. Average Ca2+ transient amplitude for WT (n=20), WT-KN93 (n=9), S2814A mice (n=11) and S2814D (n=13). E. CaSpF average from WT (n=10), CaMKIIδ KO (n=10) before and after 8-CPT. F. Average Ca2+ transient amplitude from WT (n=10), CaMKIIδ-KO (n=10) under same conditions. * p<0.05, ** p<0.01.
Notably, the S2814A mice behaved most like WT+KN-93, whereas the S2814D mice exhibited much higher basal CaSpF and lower SR Ca2+ load (as recently reported30) but were not significantly modulated by 8-CPT. This is consistent with S2814D being phosphomimetic for CaMKII effects at that site.37 Moreover, the unaltered baseline Ca2+ transient amplitude, despite reduced SR Ca2+ load in S2814D mice is consistent with the CaMKII-dependent enhancement of fractional SR Ca2+ release at a constant SR Ca2+ load and Ca2+ current trigger,38 and hence allows relatively normal Ca2+ transients.
CaMKIIδ is the dominant CaMKII isoforms in cardiomyocytes (70-90%), but other isoforms (e.g. CaMKIIγ) could phosphorylate certain targets and contribute to the inhibitory effects of KN93.39 To test whether the Epac2-dependent effect on Ca2+ sparks is mediated via CaMKIIδ specifically, we assessed Ca2+ transients and sparks in WT vs. CaMKIIδ-KO mice (Figure 7E-F). The 8-CPT-induced increase of CaSpF and decrease of Ca2+ transient amplitude were abolished in CaMKIIδ-KO mice. Thus, the Epac-induced SR Ca2+ leak effect seems to require CaMKIIδ activity (vs. other isoforms).
Discussion
The relatively specific Epac activator 8-CPT has been shown to induce diastolic SR Ca leak and arrhythmias, and in human HF Epac expression is increased. This makes it important to understand how Epac-dependent arrhythmogenic signaling works. 8-CPT could have off-target effects, 26 but these seem negligible with respect to Ca2+ handling because 10 μM 8-CPT had no acute effect on Ca2+ transients or Ca sparks in Epac-2 KO and DKO myocytes. We conclude that myocyte Epac2 is essential for the 8-CPT-induced RyR activation, and that Epac1 cannot substitute. The Epac2-mediated effects on RyR can be induced via β1-AR activation (not by β2-AR) and contribute significantly (along with PKA) to β-AR activation dependent arrhythmogenesis in hearts and myocytes. The fact that the ISO-induced Ca spark enhancement was partially prevented in Epac2-KO mice and further suppressed by PKA inhibition (Fig 4D) suggests that Epac2 and PKA are the main pathways here. Other modulators like nitrosylation or oxidation do not seem critical (but could become relevant under some conditions).
CaMKIIδ is required for these Epac2-mediated effects, and other CaMKII isoforms (which constitute ~20% of myocyte CaMKII activity35) cannot substitute. One specific site on RyR2 (S2814) also seems essential for Epac2-dependent RyR effects, despite the presence of other phosphorylation sites on RyR2.40 This provides novel insight into how Epac works in parallel with the classical cAMP/PKA pathway in cardiomyocytes with respect to Ca2+ handling and arrhythmias in heart.
Epac2, a key protein for β-AR-mediated SR Ca leak and arrhythmia
At baseline, Epac has little impact on cardiac function and Ca2+ signaling, because none of the Epac-KO mice exhibited changes in baseline heart weight/body rate ratio, cardiac function or myocyte Ca2+ handling (including diastolic SR Ca2+ leak, SR Ca2+ load or global SR Ca2+ release). However, Epac is called into play during acute physiological β-AR activation and this could be exacerbated in pathological contexts, such as chronic β-AR stimulation during HF. Indeed, Epac2-KO mice were less susceptible to triggered arrhythmias upon β-AR activation and their myocytes produced less arrhythmogenic Ca sparks (Figure 4). In contrast, our data here showed that neither Epac-KO significantly altered TAC-induced hypertrophy or the associated cardiac function changes (Fig 1E-F, Online-only Supplemental Data Fig 3A-D). These data are consistent with the idea that Epac activation plays a more significant role in β-AR-induced myocyte Ca2+ mishandling and arrhythmias than in the initial stages of hypertrophy. Indeed, Epac activation by 8-CPT in whole hearts increased diastolic SR Ca2+ leak and ventricular arrhythmogenesis.17 We cannot rule out that Epac might be involved in signaling related to HF,22, 41 and to the extent that this CaMKII/RyR2 pathway might be critical in the transition from compensated hypertrophy to HF as seen in CaMKIIδ-KO mice,39 functional upregulation of this Epac pathway during cardiomyopathy1, 23 may contribute to this transition to HF.
We discovered that only Epac2 mediates enhanced SR Ca2+ leak and thus might have different signaling pathways than Epac1 in heart. We found greater Epac2 vs. Epac1 mRNA expression in cardiomyocytes, and while Epac1 mRNA is clearly expressed in heart,14, 15, 23 the Epac1/Epac2 mRNA ratio decreases in adulthood compared to neonates23 making it plausible that Epac2 may have increasing functional effects with age. We anticipate that Epac1 mediates different functions in cardiomyocytes. Indeed, Epac1 is part of a nuclear muscle-specific A-Kinase anchoring protein complex, reinforcing the idea of its role in nuclear signaling.42
Role of Epac vs. PKA during β-AR-mediated SR Ca leak
Acute Epac activation with 8-CPT has been reported to either increase11, 16or decrease (present study)12, 19 Ca transient amplitude, but this may only be an apparent discrepancy related to experimental protocols. We and others12, 19 focused on 8-CPT effects during steady state where Ca transients were decreased. In contrast, larger initial Ca2+ transient during the first 20s are also seen,11, 16 consistent with RyR activation (that we describe). This is similar to the effect of low concentrations of the RyR sensitizer caffeine,43 where initial Ca transients are larger (sensitized release), but those drive greater Ca efflux and progressively lower SR Ca load. If diastolic SR Ca leak is also enhanced, this can result in lower steady state Ca transients than control, as observed with CaMKII activation.44, 45 All agree that Epac activation causes CaMKII-dependent RyR sensitization.
Epac activation increases CaMKII-dependent RyR2 sensitivity to Ca2+ which paralleled alteration described during HF. Indeed in HF cardiac contraction is reduced, in part because reduced SR Ca2+ content attributed to SR Ca2+ leak increase via CaMKII, despite a relative sensitization of the RyR.6, 8, 21, 46, 47 Thus, Epac may, along with PKA, be an important a downstream effector of β-AR-cAMP mediated effects in HF.
It is thought that Epac activation requires higher levels of cAMP compared to activation of PKA,48-50 which would imply that low levels of β-AR activation might preferentially activate PKA, while higher levels would recruit both PKA and Epac. Our data indicate that 30 nM ISO was sufficient to induce maximal Epac-dependent RyR2 effects, a lower [ISO] than required for maximal PKA-dependent effects (Figure 5). Thus, during β-AR stimulation in myocytes, Epac activation likely overlaps with PKA. Indeed, both ISO-induced arrhythmias and SR Ca2+ leak effects were strongly attenuated in Epac2-KO mice, reinforcing the idea that Epac and PKA are co-activated in β-AR-induced SR Ca leak and arrhythmias. Global β-AR activation (PKA+Epac) enhances SR Ca2+ leak, Ca2+ transients and SR Ca2+ load. In contrast under Epac activation alone, Ca2+ transient and SR Ca2+ load decrease because of higher SR Ca2+ leak. The enhanced SR Ca2+ load and Ca2+ transients with global β-AR activation can be explained by strong PKA effects on Ca2+ current and phospholamban which increase SR Ca2+-ATPase activity and SR Ca2+ load. Epac-dependent effects may limit the rise of SR Ca2+ load driven by PKA-dependent effects. Consequently, we expected that β-AR-induced inotropy might be slightly enhanced in Epac-KO mice. This effect was not detected (Online-only Supplemental Data Table 1) possibly because the very strong positive inotropic effects of PKA overcome the modest Epac-mediated negative inotropy. Indeed, the Epac-induced RyR sensitization coupled with increased SR Ca2+ load and Ca2+ current could ablate the moderate negative inotropic effect of Epac.
Epac2 signaling pathway in β-AR-mediated SR Ca leak
In cardiomyocytes, β1-AR and β2-AR coexist. Here, only β1-AR inhibition blocked Epac-mediated Ca2+ signaling alterations. This agrees with previous work where β1-AR favors the formation of a Epac/CaMKII/β-arrestin complex51 and where β1-AR deletion attenuates CaMKII activation.52 Since Epac also activates CaMKII,11, 12, 17 these findings reinforce Epac and CaMKII signaling downstream of β1-ARs (at least at the RyR) and the link between CaMKII-dependent SR Ca2+ leak and arrhythmias.17, 24 The genetic block of RyR S2814 phosphorylation, CaMKIIδ-KO and KN93 fully abolished 8-CPT-induced SR Ca2+ leak in WT (Figure 7). These data extend previous studies11, 12, 17 suggesting that Epac-induced RyR2-S2814 phosphorylation depends on the CaMKIIδ and not other CaMKII isoforms. However, in RyR2-S2814D mice, we observed a larger SR Ca2+ leak at baseline compared to 8-CPT suggesting that Epac may not achieve the maximum RyR2 activation seen in phosphomimetic RyR2-S2814D mice.
In conclusion, our work indicates that β1-AR activates Epac2-dependent SR Ca2+ leak and arrhythmia via CaMKIIδ-dependent phosphorylation of RyR2-S2814. These findings give new insights into Epac physiology and pathology. Indeed, Epac seems non-essential for baseline cardiac function, and does not significantly attenuate the acute inotropic response to potent β-AR activation or the early hypertrophic response to pressure overload. However, we demonstrate a specific role for the Epac2 isoform in mediating PKA-independent SR Ca2+ leak via β1-AR causing RyR-S2814 phosphorylation by CaMKIIδ and SR Ca2+ leak. Further investigations will be needed to understand why both Epac and PKA are activated by β1-AR stimulation and the role of Epac1 (nuclear signaling vs. RyR function or transition to HF).
Supplementary Material
Clinical Perspective Summary.
In heart failure (HF), β-adrenergic receptor (AR) activation can cause arrhythmias, mediated in part by abnormal diastolic Ca2+ release from the sarcoplasmic reticulum (SR). This is generally attributed to β-AR-induced increase in cAMP which activates protein kinase A (PKA) to phosphorylate key targets that increase Ca2+ current influx, SR Ca2+ uptake and possibly the ryanodine receptor (RyR2). Recent studies have identified another direct cAMP target, Epac (Exchange protein directly activated by cAMP) which is upregulated in HF and can be activated in parallel to PKA, to regulate independent targets. Indeed, Epac activation by a cAMP analog (8-CPT) can recapitulate the HF-associated enhancement of pro-arrhythmic SR Ca2+ and the reduced SR Ca2+ load that causes systolic dysfunction. However, cardiac Epac signaling is incompletely understood. In this study, using genetic manipulation of Epac1, Epac2, RyR2 and CaMKII, we have clarified how Epac activation contributes to β-AR-induced arrhythmias. We show that Epac and PKA activity may contribute nearly equally to β-AR induced arrhythmias and diastolic SR Ca leak. Moreover, this Epac pathway specifically requires β1-AR-dependent activation of the Epac2 isoform, which activates the CaMKIIδ isoform to phosphorylate the S2814 site on RyR2 to enhance arrhythmogenic Ca2+ release. These mechanistic findings clarify β-AR-mediated Ca2+ leak-induced arrhythmia signaling and highlight that Epac2 could be an important novel therapeutic target in the treatment of arrhythmias.
Acknowledgments
We thank Khanha Dao for technical assistance.
Funding Sources: This study was funding by grants NIH P01-HL080101 (DMB, JHB and JC), R37-HL30077 (DMB), R01-HL089598 (XTW) and Fondation Leducq (DMB and XTW). XTW is a W.M. Keck Foundation Distinguished Young Scholar in Medical Research, and is funded by NIH grants (R01-HL089598; R01-HL091947). Dr. van Oort was supported by an American Physiology Society postdoctoral Fellowship.
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 2.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 3.Ogrodnik J, Niggli E. Increased Ca2+ leak and spatiotemporal coherence of Ca2+ release in cardiomyocytes during beta-adrenergic stimulation. J Physiol. 2010;588:225–242. doi: 10.1113/jphysiol.2009.181800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belevych A, Kubalova Z, Terentyev D, Hamlin RL, Carnes CA, Gyorke S. Enhanced ryanodine receptor-mediated calcium leak determines reduced sarcoplasmic reticulum calcium content in chronic canine heart failure. Biophys J. 2007;93:4083–4092. doi: 10.1529/biophysj.107.114546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bers DM, Eisner DA, Valdivia HH. Sarcoplasmic reticulum Ca2+ and heart failure: Roles of diastolic leak and Ca2+ transport. Circ Res. 2003;93:487–490. doi: 10.1161/01.RES.0000091871.54907.6B. [DOI] [PubMed] [Google Scholar]
- 6.Shannon TR, Pogwizd SM, Bers DM. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ Res. 2003;93:592–594. doi: 10.1161/01.RES.0000093399.11734.B3. [DOI] [PubMed] [Google Scholar]
- 7.Wehrens XH, Lehnart SE, Marks AR. Intracellular calcium release and cardiac disease. Annu Rev Physiol. 2005;67:69–98. doi: 10.1146/annurev.physiol.67.040403.114521. [DOI] [PubMed] [Google Scholar]
- 8.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 9.Curran J, Hinton MJ, Rios E, Bers DM, Shannon TR. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res. 2007;100:391–398. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- 10.Grimm M, Brown JH. Beta-adrenergic receptor signaling in the heart: Role of CaMKII. J Mol Cell Cardiol. 2010;48:322–330. doi: 10.1016/j.yjmcc.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oestreich EA, Malik S, Goonasekera SA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV. Epac and phospholipase cepsilon regulate Ca2+ release in the heart by activation of protein kinase cepsilon and calcium-calmodulin kinase ii. J Biol Chem. 2009;284:1514–1522. doi: 10.1074/jbc.M806994200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pereira L, Metrich M, Fernandez-Velasco M, Lucas A, Leroy J, Perrier R, Morel E, Fischmeister R, Richard S, Benitah JP, Lezoualc’h F, Gomez AM. The camp binding protein epac modulates Ca2+ sparks by a Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes. J Physiol. 2007;583:685–694. doi: 10.1113/jphysiol.2007.133066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bos JL. Epac: A new camp target and new avenues in camp research. Nat Rev Mol Cell Biol. 2003;4:733–738. doi: 10.1038/nrm1197. [DOI] [PubMed] [Google Scholar]
- 14.de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a rap1 guanine-nucleotide-exchange factor directly activated by cyclic amp. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 15.Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. A family of camp-binding proteins that directly activate rap1. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- 16.Oestreich EA, Wang H, Malik S, Kaproth-Joslin KA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV. Epac-mediated activation of phospholipase c(epsilon) plays a critical role in beta-adrenergic receptor-dependent enhancement of Ca2+mobilization in cardiac myocytes. J Biol Chem. 2007;282:5488–5495. doi: 10.1074/jbc.M608495200. [DOI] [PubMed] [Google Scholar]
- 17.Hothi SS, Gurung IS, Heathcote JC, Zhang Y, Booth SW, Skepper JN, Grace AA, Huang CL. Epac activation, altered calcium homeostasis and ventricular arrhythmogenesis in the murine heart. Pflugers Arch. 2008;457:253–270. doi: 10.1007/s00424-008-0508-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cazorla O, Lucas A, Poirier F, Lacampagne A, Lezoualc’h F. The camp binding protein Epac regulates cardiac myofilament function. Proc Natl Acad Sci U S A. 2009;106:14144–14149. doi: 10.1073/pnas.0812536106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pereira L, Ruiz-Hurtado G, Morel E, Laurent AC, Metrich M, Dominguez-Rodriguez A, Lauton-Santos S, Lucas A, Benitah JP, Bers DM, Lezoualc’h F, Gomez AM. Epac enhances excitation-transcription coupling in cardiac myocytes. J Mol Cell Cardiol. 2012;52:283–291. doi: 10.1016/j.yjmcc.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pieske B, Maier LS, Schmidt-Schweda S. Sarcoplasmic reticulum Ca2+ load in human heart failure. Basic Res Cardiol. 2002;97(Suppl 1):I63–71. doi: 10.1007/s003950200032. [DOI] [PubMed] [Google Scholar]
- 21.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;88:1159–1167. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- 22.Curran J, Brown KH, Santiago DJ, Pogwizd S, Bers DM, Shannon TR. Spontaneous ca waves in ventricular myocytes from failing hearts depend on Ca2+-calmodulin-dependent protein kinase ii. J Mol Cell Cardiol. 2010;49:25–32. doi: 10.1016/j.yjmcc.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ulucan C, Wang X, Baljinnyam E, Bai Y, Okumura S, Sato M, Minamisawa S, Hirotani S, Ishikawa Y. Developmental changes in gene expression of Epac and its upregulation in myocardial hypertrophy. Am J Physiol Heart Circ Physiol. 2007;293:H1662–1672. doi: 10.1152/ajpheart.00159.2007. [DOI] [PubMed] [Google Scholar]
- 24.Metrich M, Lucas A, Gastineau M, Samuel JL, Heymes C, Morel E, Lezoualc’h F. Epac mediates beta-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ Res. 2008;102:959–965. doi: 10.1161/CIRCRESAHA.107.164947. [DOI] [PubMed] [Google Scholar]
- 25.Chen C, Du J, Feng W, Song Y, Lu Z, Xu M, Li Z, Zhang Y. Beta-adrenergic receptors stimulate interleukin-6 production through epac-dependent activation of PKCdelta/p38 MAPK signalling in neonatal mouse cardiac fibroblasts. Br J Pharmacol. 2012;166:676–688. doi: 10.1111/j.1476-5381.2011.01785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poppe H, Rybalkin SD, Rehmann H, Hinds TR, Tang XB, Christensen AE, Schwede F, Genieser HG, Bos JL, Doskeland SO, Beavo JA, Butt E. Cyclic nucleotide analogs as probes of signaling pathways. Nature methods. 2008;5:277–278. doi: 10.1038/nmeth0408-277. [DOI] [PubMed] [Google Scholar]
- 27.Yan J, Mei FC, Cheng H, Lao DH, Hu Y, Wei J, Patrikeev I, Hao D, Stutz SJ, Dineley KT, Motamedi M, Hommel JD, Cunningham KA, Chen J, Cheng X. Enhanced leptin sensitivity, reduced adiposity and improved glucose homeostasis in mice lacking of exchange protein directly activated by cAMP isoform 1. Mol Cell Biol. 2013 Dec 21; doi: 10.1128/MCB.01227-12. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND, Peterson KL, Chen J, Bers D, Heller Brown J. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119:1230–1240. doi: 10.1172/JCI38022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Muller FU, Schmitz W, Schotten U, Anderson ME, Valderrabano M, Dobrev D, Wehrens XH. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119:1940–1951. doi: 10.1172/JCI37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM, Wehrens XH. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinaseII promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669–2679. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DiPilato LM, Cheng X, Zhang J. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of camp signaling within discrete subcellular compartments. Proc Natl Acad Sci U S A. 2004;101:16513–16518. doi: 10.1073/pnas.0405973101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Kranias EG, Mignery GA, Bers DM. Protein kinase a phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ Res. 2002;90:309–316. doi: 10.1161/hh0302.105660. [DOI] [PubMed] [Google Scholar]
- 33.Bers DM. CamkII inhibition in heart failure makes jump to human. Circ Res. 2010;107:1044–1046. doi: 10.1161/CIRCRESAHA.110.231902. [DOI] [PubMed] [Google Scholar]
- 34.Guo T, Cornea RL, Huke S, Camors E, Yang Y, Picht E, Fruen BR, Bers DM. Kinetics of FKBP12.6 binding to ryanodine receptors in permeabilized cardiac myocytes and effects on ca sparks. Circ Res. 2010;106:1743–1752. doi: 10.1161/CIRCRESAHA.110.219816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Allen MD, DiPilato LM, Rahdar M, Ren YR, Chong C, Liu JO, Zhang J. Reading dynamic kinase activity in living cells for high-throughput screening. ACS chemical biology. 2006;1:371–376. doi: 10.1021/cb600202f. [DOI] [PubMed] [Google Scholar]
- 36.Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004;94:e61–70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- 37.Guo T, Zhang T, Mestril R, Bers DM. Ca2+/calmodulin-dependent protein kinase ii phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ Res. 2006;99:398–406. doi: 10.1161/01.RES.0000236756.06252.13. [DOI] [PubMed] [Google Scholar]
- 38.Li L, Satoh H, Ginsburg KS, Bers DM. The effect of Ca2+-calmodulin-dependent protein kinase II on cardiac excitation-contraction coupling in ferret ventricular myocytes. J Physiol. 1997;501(Pt 1):17–31. doi: 10.1111/j.1469-7793.1997.017bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND, Peterson KL, Chen J, Bers D, Brown JH. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119:1230–1240. doi: 10.1172/JCI38022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rodriguez P, Bhogal MS, Colyer J. Stoichiometric phosphorylation of cardiac ryanodine receptor on serine 2809 by calmodulin-dependent kinase II and protein kinase A. J Biol Chem. 2003;278:38593–38600. doi: 10.1074/jbc.C301180200. [DOI] [PubMed] [Google Scholar]
- 41.Sag CM, Wadsack DP, Khabbazzadeh S, Abesser M, Grefe C, Neumann K, Opiela MK, Backs J, Olson EN, Brown JH, Neef S, Maier SK, Maier LS. Calcium/calmodulin-dependent protein kinase II contributes to cardiac arrhythmogenesis in heart failure. Circ Heart Fail. 2009;2:664–675. doi: 10.1161/CIRCHEARTFAILURE.109.865279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McConnachie G, Langeberg LK, Scott JD. Akap signaling complexes: Getting to the heart of the matter. Trends Mol Med. 2006;12:317–323. doi: 10.1016/j.molmed.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 43.Trafford AW, Sibbring GC, Diaz ME, Eisner DA. The effects of low concentrations of caffeine on spontaneous ca release in isolated rat ventricular myocytes. Cell Calcium. 2000;28:269–276. doi: 10.1054/ceca.2000.0156. [DOI] [PubMed] [Google Scholar]
- 44.Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH, Bers DM. Transgenic camkiideltac overexpression uniquely alters cardiac myocyte Ca2+ handling: Reduced sr ca2+ load and activated SR Ca2+ release. Circ Res. 2003;92:904–911. doi: 10.1161/01.RES.0000069685.20258.F1. [DOI] [PubMed] [Google Scholar]
- 45.Kohlhaas M, Zhang T, Seidler T, Zibrova D, Dybkova N, Steen A, Wagner S, Chen L, Brown JH, Bers DM, Maier LS. Increased sarcoplasmic reticulum calcium leak but unaltered contractility by acute CaMKII overexpression in isolated rabbit cardiac myocytes. Circ Res. 2006;98:235–244. doi: 10.1161/01.RES.0000200739.90811.9f. [DOI] [PubMed] [Google Scholar]
- 46.Lindner M, Erdmann E, Beuckelmann DJ. Calcium content of the sarcoplasmic reticulum in isolated ventricular myocytes from patients with terminal heart failure. J Mol Cell Cardiol. 1998;30:743–749. doi: 10.1006/jmcc.1997.0626. [DOI] [PubMed] [Google Scholar]
- 47.Pogwizd SM, Qi M, Yuan W, Samarel AM, Bers DM. Upregulation of Na+/Ca2+ exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ Res. 1999;85:1009–1019. doi: 10.1161/01.res.85.11.1009. [DOI] [PubMed] [Google Scholar]
- 48.Bos JL. Epac proteins: Multi-purpose camp targets. Trends Biochem Sci. 2006;31:680–686. doi: 10.1016/j.tibs.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 49.Rehmann H, Schwede F, Doskeland SO, Wittinghofer A, Bos JL. Ligand-mediated activation of the cAMP-responsive guanine nucleotide exchange factor Epac. J Biol Chem. 2003;278:38548–38556. doi: 10.1074/jbc.M306292200. [DOI] [PubMed] [Google Scholar]
- 50.de Rooij J, Rehmann H, van Triest M, Cool RH, Wittinghofer A, Bos JL. Mechanism of regulation of the Epac family of cAMP-dependent rapgefs. J Biol Chem. 2000;275:20829–20836. doi: 10.1074/jbc.M001113200. [DOI] [PubMed] [Google Scholar]
- 51.Mangmool S, Shukla AK, Rockman HA. Beta-arrestin-dependent activation of Ca2+/calmodulin kinaseII after beta(1)-adrenergic receptor stimulation. J Cell Biol. 2010;189:573–587. doi: 10.1083/jcb.200911047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yoo B, Lemaire A, Mangmool S, Wolf MJ, Curcio A, Mao L, Rockman HA. Beta1-adrenergic receptors stimulate cardiac contractility and CaMKII activation in vivo and enhance cardiac dysfunction following myocardial infarction. Am J Physiol Heart Circ Physiol. 2009;297:H1377–1386. doi: 10.1152/ajpheart.00504.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.