

Abstract
Fear memories are acquired through neuronal plasticity, an orchestrated sequence of events regulated at circuit and cellular levels. The conventional model of fear acquisition assumes unimodal (e.g. excitatory or inhibitory) roles of modulatory receptors in controlling neuronal activity and learning. Contrary to this view, we show that protease-activated receptor-1 (PAR1) promotes contrasting neuronal responses depending on the emotional status of an animal by a dynamic shift between distinct G protein coupling partners. In the basolateral amygdala of fear-naïve mice PAR1 couples to Gαq/11 and Gαo proteins, while after fear conditioning coupling to Gαo increases. Concurrently, stimulation of PAR1 before conditioning enhanced, but afterwards it inhibited firing of basal amygdala neurons. An initial impairment of the long-term potentiation (LTP) in PAR1-deficient mice was transformed into an increase in LTP and enhancement of fear after conditioning. These effects correlated with more frequent AMPA receptor-mediated miniature post synaptic events and increased neuronal excitability. Our findings point to experience-specific shifts in PAR1-G protein-coupling in the amygdala as a novel mechanism regulating neuronal excitability and fear.
Introduction
Seven transmembrane G protein-coupled receptors (GPCRs) constitute the largest family of signaling proteins and are activated by a variety of natural ligands (1). On agonist binding GPCRs transduce cellular signals through a wide range of G proteins, linked to diverse biochemical pathways. Over the past decade a concept of invariable, static association of distinct GPCRs with predetermined G proteins, has been challenged. It has been demonstrated that different synthetic ligands acting at the same receptor can elicit qualitatively different biochemical and cellular responses (2) by stabilizing GPCRs in different conformational states (3-5) and activating alternative transduction cascades.
Optogenetic studies show that spatiotemporally precise control over intracellular signaling processes in discrete brain regions could serve to control higher behavioral functions in mammals (6). Directing neuronal signaling through distinct transduction pathways in the nucleus accumbens of mice using light-activated, genetically-engineered GPCRs elicits contrasting reward-related behaviors in the conditioned place preference paradigm (6). These experiments raise a fascinating possibility that GPCRs exhibit input-specific intracellular signaling by endogenous agonists, which could serve as a mechanism for experience-induced plasticity, emotion and learning in the mammalian brain.
Protease-activated receptors (PARs) are a family of GPCRs activated through proteolysis within their extracellular N-terminal domain, exposing a tethered ligand that acts as an agonist at the receptor (7). The best characterized member of this class, PAR1, can be cleaved by several native proteases, such as thrombin, tPA, plasmin, factor X or activated protein C (8) that have distinct substrate specificities. Thus, on PAR1 cleavage, a different N-terminal of the receptor is generated, consistent with distinct proteases eliciting contrasting cellular responses (8). The above findings indicate that PAR1 might be subject to agonist-directed signaling by the distinct protease-generated endogenous ligands.
Extracellular proteases in the amygdala and hippocampus are key regulators of neuronal activity leading to fear and anxiety (9-12). Spatially and temporally controlled release of proteases at excitatory synapses (9-15) modulates neuronal activity by cleaving membrane receptors, latent growth factors, or adhesion molecules (16, 17). These proteases also activate PAR1 (18, 19), a GPCR previously linked to excitatory synaptic transmission and learning (13, 19).
Here we show that PAR1 elicits contrasting neuronal responses and determines state-dependent neuronal network properties depending on the fear status of the animal by a dynamic control of PAR1 coupling to distinct G protein subtypes. Thus, biased GPCR signaling can serve as a fundamental mechanism of experience-induced neuronal plasticity in the mammalian brain, controlling neuronal responses, emotion and learning.
Results
PAR1 is enriched in mouse amygdala neurons
PARs are abundant in the brain, but their region-, circuit- and cell type-specific expression varies among species (20, 21) suggesting species-specific functions. To better understand the role of PAR1 in the mouse central nervous system we examined the presence of PAR1 in different brain regions by Western blotting and found the highest levels in the basolateral amygdala (381 ± 68% enrichment relative to the hypothalamus; n = 3 per brain region; Figure 1A-C). Cortical expression was almost equally prominent (351 ± 14%), while the thalamic and medulla levels were moderate (244 ± 23% and 252 ± 37%). PAR1 protein expression in the hippocampus, cerebellum and hypothalamus were the lowest (172 ± 68%; 162 ± 26%; 100 ± 11%, respectively).
Figure 1. PAR1 is expressed by neurons in the basal nucleus of the amygdala.

Brain regions (AMY - amygdala; CER - cerebellum; HIP - hippocampus; HYP - hypothalamus; THA - thalamus; COR - cortex; MED – medulla) were dissected from naïve PAR1+/+ mice and PAR1 levels measured by Western blotting (representative blots in A). The PAR1 band was not observed in PAR1−/− animals (B). The highest expression of PAR1 (normalized to β-actin levels) was detected in the amygdala, and the lowest in the hypothalamus (quantification in C; n = 3 per brain region). Quadruple staining for PAR1 (D, H, J; green), neuronal marker NeuN (E, I; red), astrocyte marker glial fibrillary acidic protein (GFAP, F, I, J; far red pseudocolored blue) and DAPI (G) was performed using coronal brain sections containing the amygdala. Immunohistochemistry revealed the majority of PAR1-positive cells within the basal nucleus co-localizing with NeuN, indicative of their neuronal phenotype (H, quantified in L, n = 5 mice). A weaker expression was occasionally observed in astrocytes (J, quantified in L). PAR1 signal was not observed when the primary antibody was omitted (K). Schematic representation of the study timeline and design (M). Various experimental combinations of the above were used as indicated in the text. Data shown as mean ± SEM.
We next examined the pattern of PAR1 expression by immunohistochemistry. We found PAR1-positive cells in most regions of a mouse brain, with strong staining in the basolateral complex of the amygdala (n = 5; Figure 1D). To determine the phenotype of PAR1-positive cells in this brain region we performed multi-label immunohistochemistry using astrocyte-specific (anti-GFAP) or neuron-specific (anti-NeuN) antibodies together with an anti-PAR1 immunoglobulin. We found that 92.9 ± 3.6% of PAR1-positive cells co-localized with NeuN, indicative of their neuronal phenotype (n = 5; Figure 1D-L). PAR1 expression was most prominent in cell bodies and neuronal processes. Immunohistochemistry revealed weak expression of PAR1 in a small sub-population of astrocytes (Figure 1F, J).
PAR1 is cleaved during conditioning and bidirectionally regulates LTP and fear
We first examined whether PAR1 is cleaved during fear conditioning in the basolateral amygdala. Western blotting showed that the density of a band recognised by an antibody raised against the extracellular portion of PAR1 decreased shortly after conditioning (but not after the unpaired protocol; n = 4 per group, p < 0.05) and returned to normal within 48 hours (Figure 2A), indicative of PAR1 cleavage and its subsequent replacement.
Figure 2. Bidirectional, fear-dependent regulation of L-LTP by PAR1.

(A) Western blotting demonstrated a 40% decrease in the amygdala PAR1 levels 5 min after fear conditioning consistent with its cleavage by proteases (n = 4 per group). PAR1 levels were back to normal 48 h later. Field potential amplitude (normalized to the maximum FP for each slice) recorded in the basal nucleus as a function of stimulus intensity applied to the lateral nucleus (input-output relationship; electrode placement shown in B) was similar in PAR1+/+ and PAR1−/− mice (C). Tetanic stimulation (asterisks in D, E, F, H) resulted in a stable LTP in naïve PAR1+/+ (D; black symbols) which lasted more than two hours and was depressed in PAR1−/− mice (red symbols). Fear conditioning caused a reversal of LTP in PAR1−/− animals which was approximately 2-fold higher than in PAR1+/+ mice during the course of the experiment (E). When the tone and footshock were delivered in a random manner the increase in the early phase of LTP (E-LTP) in PAR1−/− mice persisted, while the increase in the late phase (L-LTP) was no longer observed (F). Individual representative traces at the baseline (1), 30 seconds (2) and 2 hours post –tetanus (3) are shown as inserts in D, E and F. Amygdala slices from PAR1+/+ and PAR1−/− mice were perfused with picrotoxin to block GABAA receptors. The picrotoxin-induced increase in the field potential amplitude recorded in the basal nucleus of the amygdala was similar in both genotypes (G). Moreover, blocking the inhibitory input did not affect the impairment in the late phase of LTP observed in PAR1−/− mice (H) indicating that GABA-ergic mechanisms in the amygdala are not affected by the deletion of the PAR1 gene. Please note an inhibitory effect of picrotoxin on post-tetanic potentiation equally pronounced in both genotypes. n = 6-8 per group. Data shown as mean ± SEM. LA – lateral amygdala, BLA – basal amygdala, MeA – medial amygdala, CA – central amygdala.
The basal nucleus of the amygdala serves as an important site of plasticity in fear conditioning and is involved in the acquisition of conditioned stimuli (22-24). To study the role of PAR1 in amygdalar plasticity we induced LTP in the lateral-basal amygdala pathway (25) of fear-naïve PAR1+/+ and PAR1−/− mice (Figure 2B). Basal synaptic transmission was indistinguishable between genotypes (Figure 2C). However, the absence of PAR1 impaired LTP in this pathway in fear-naïve animals (Figure 2D; N = 6 animals, n = 8 slices per group, p < 0.01 at 120 min). This reduction was not due to an enhancement of inhibitory mechanisms (26), because neither basal excitability (Figure 2G) nor the decrease in LTP (Figure 2H; N = 6, n= 6-8, p < 0.01 at 120 min) were affected by the GABAA receptor antagonist picrotoxin in PAR1−/− compared to PAR1+/+ animals. Miniature inhibitory post-synaptic currents (mIPSCs) in whole-cell voltage clamp experiments showed similar frequency and amplitude between PAR1+/+ and PAR1−/− mice in the basal amygdala neurons (Figures S1 and 2) confirming that GABAergic inhibition was unaffected in PAR1−/− animals.
Fear learning liberates extracellular proteases (11, 12) and promotes cleavage of PAR1 in the amygdala (Figure 2A) so we next investigated the contribution of PAR1 to fear-related neuronal plasticity. We subjected PAR1+/+ and PAR1−/− mice to a mild fear conditioning protocol and induced LTP in the lateral-basal pathway. In contrast to fear-naïve conditions, we found a 2-fold increase in LTP in PAR1−/− mice as compared to PAR1+/+ animals (Figure 2E; N = 6, n = 8 per group, p < 0.001 and p < 0.01 for 60 and 120 min, respectively).
To examine whether the above changes could be attributed to learning or rather stress-related alterations in neuronal physiology, we subjected mice to an unpaired conditioning protocol and induced LTP. Late phase of LTP was defined based on its protein synthesis-dependence (25) (Figure S3). The increase in the late phase of LTP (L-LTP) in PAR1−/− mice was due to fear learning (no change observed following the unpaired protocol; N = 6, n = 7 per group, p > 0.05 between PAR1+/+ and PAR1−/− mice at 120 min), while the rise in the early phase (E-LTP) was footshock-dependent (p < 0.0001 at 30 sec, Figure 2F).
Electrically evoked LTP is often occluded by the “LTP-like changes” induced by the conditioning procedure (see Figure S4 and ref. (27)). To investigate the relationship between L-LTP in the lateral-basal pathway and fear memory strength in PAR1−/− mice we measured tone-associated freezing following fear conditioning. In contrast to earlier studies (13) we used a mild conditioning protocol resulting in ~30% freezing in wild-type mice to detect either enhancement or reduction in fear learning in PAR1−/− mice. Two-way ANOVA revealed a strong effect of conditioning (F(1, 48)=43.38; p<0.0001) and genotype (F(1, 48)=9.5; p<0.01), but most importantly a significant genotype x conditioning interaction (F(1, 48)=4.69; p<0.05) indicating that the increase in L-LTP in PAR1−/− mice coincided with the enhancement of amygdala-dependent fear memory (n = 11-14 per group, p < 0.001; Figure 3A-C) when compared to wild-type animals that underwent the same treatment. Pain threshold, general motor activity, anxiety and hippocampal functions were indistinguishable between the genotypes (Figure 3D and S5) indicating that these parameters did not contribute to differences in fear learning.
Figure 3. Enhancement of conditioned fear by genetic or pharmacological disruption of PAR1.

PAR1+/+ or PAR1−/− mice were subjected to fear conditioning and amygdala-dependent memory tested two days later. The fear conditioning-related increase in amygdalar L-LTP in PAR1−/− mice (see Figure 2E) was accompanied by the increase in the amygdala-dependent fear measured as a percentage of time spent immobile (average values and representative movement traces recorded during fear retrieval in A-C). When exposed to the tone (time indicated as the red line on the x-axis in A-B) PAR1−/− mice froze significantly more than PAR1+/+ animals (yellow areas indicate immobility). Animals that received unpaired treatment did not show learning. Behavioral reaction of both geneotypes to the aversive stimulus was indistinguishable, indicating that the observed augmentation of fear learning in PAR1−/− mice could not be attributed to altered pain sensitivity (D). The effect of genetic disruption of PAR1 was mimicked by bilateral intra-amygdala injections of PAR1 antagonist SCH79797 during conditioning (E). In A-C n = 11-14 per group; D, E n = 9-10 per group. * p < 0.05, ** p < 0.01, *** p < 0.001. Data shown as mean ± SEM.
We next tested if the effect of genetic disruption of the PAR1 gene could be mimicked by its pharmacological inhibition. To this end we bilaterally infused PAR1 antagonist SCH79797 into the amygdala of PAR1+/+ mice before conditioning. We found that the enhancement of fear observed in PAR1–deficient animals was recapitulated by pharmacological inhibition of PAR1 in the amygdalae of PAR1+/+ mice (Figures 3E and S6; n = 8-9 per group) indicating that the effect of PAR1 on cued fear learning was amygdala-specific.
PAR1-G protein coupling is dynamic, associative and experience-specific
What is the mechanism of the reversal of LTP and enhancement of fear memory following conditioning in PAR1−/− mice? One possibility is that the G protein-coupling profile of PAR1 and its role in regulating the above phenomena could be dynamic and regulated by fear conditioning. To investigate this hypothesis we focused on PAR1 association with Gαq/11 and Gαo proteins because they are abundantly expressed in the brain (28, 29) and elicit contrasting effects in the nervous system (30). To assess whether PAR1-G protein coupling in the amygdala is dynamic and regulated by fear learning we measured agonist-stimulated [35S]GTPγS binding to G proteins in membrane preparations. By combining this with an immunoprecipitation step we quantified the amount of PAR1 signaling through specific G proteins (31, 32). The PAR1 agonist TRag stimulated [35S]GTPγS-for-GDP exchange on both Gαq/11 and Gαo proteins in membranes prepared from PAR1+/+, but not PAR1−/− brain tissue (Figure 4A; n = 3-5), confirming the protocol’s specificity. We found that in the amygdala of fear-naïve mice PAR1 was preferentially coupled to Gαq/11 (increase in [35S]GTPγS binding-over-basal: Gαq/11 = 1326 ± 181 vs Gαo = 884 ± 84 d.p.m., n = 7 per group). 48 h after fear conditioning Gαq/11/Gαo protein expression (Figure 4B), and basal Gαo-/Gαq/11-[35S]GTPγS binding were not altered (see Methods), but a pronounced increase in PAR1 coupling to Gαo was found (Figure 4C). Thus, while TRag-stimulated [35S]GTPγS-for-GDP exchange on Gαq/11 was unaltered (92 ± 24 and 105 ± 7% of fear-naïve values in fear conditioned and the unpaired groups, respectively; n = 5 per group), there was a learning-specific 2-fold increase in receptor-mediated [35S]GTPγS binding to Gαo proteins (185 ± 15 and 107 ± 7% of fear-naïve values in fear conditioned and the unpaired group, respectively; n = 5 per group), demonstrating a selective increase in coupling through this arm of the signaling pathway (Figure 4C). The above experiments indicate that fear conditioning (but not the unpaired protocol) shifted preferential coupling of PAR1 in the basolateral complex of the amygdala from excitatory Gαq/11 in naïve mice towards inhibitory Gαo to suppress fear-induced increases in the activation of principal neurons of basal amygdala (Figure 4D). Consistent with the previously reported inhibition of cyclic AMP generation by Gαo (33), we also found an increase in fear-induced CREB phosphorylation in PAR1−/− mice compared to PAR1+/+ animals (Figure 4E).
Figure 4. PAR1 / Gα-protein coupling in the amygdala is dynamic and regulated by fear learning.

To assess whether fear conditioning alters PAR1 Gα-protein binding in the amygdala we performed [35S]GTPγS binding and Gα-specific immunoprecipitation assay. (A) TRag-induced increases in [35S]GTPγS binding to Gαo and Gαq/11 were abolished in PAR1−/− animals confirming assay specificity. (B) Western blotting demonstrated that fear conditioning did not affect cellular Gαo or Gαq/11 levels. (C) Stimulation of PAR1 in PAR1+/+ mice with TRag revealed a fear conditioning-specific 2-fold increase in PAR1 Gαo coupling while Gαq/11 coupling was unaffected. The unpaired protocol did not change PAR1-G protein association. Fear conditioning-induced shift towards inhibitory Gαo binding of PAR1 was consistent with greater inductions of c-Fos (D) and phospho-CREB (E) in the basal nucleus of the amygdala of PAR1−/− mice after conditioning. *p < 0.05, ** p < 0.01; Data shown as means ± SEM. Bars in D and E equal 100 μm.
PAR1 regulates neuronal excitability in experience-dependent manner
To investigate the electrophysiological consequences of fear-mediated changes in PAR1-G protein coupling that could underlie the reversal of LTP, we subjected PAR1+/+ and PAR1−/− mice to fear-conditioning and studied the excitability of principal basal amygdala neurons in current clamp recordings. Two-way ANOVA revealed a significant genotype x conditioning interaction on input resistance (F(1.28)=5.18; p<0.05; Figures 5D and S7D); inter-group comparison pointed to an increase in neuronal excitability in PAR1−/− mice after fear conditioning (p<0.05; n = 6-8 mice per group; Figures 5A-D and S7 for fear-naïve mice). These findings are consistent with the increase in LTP in PAR1−/− mice (Figure 2E), higher c-Fos activation (Figure 4D), and preferential coupling of PAR1 to the inhibitory Gαo (Figure 4C) in PAR1+/+ animals after fear conditioning.
Figure 5. Fear conditioning-dependent, bidirectional control of firing frequency of basal amygdala neurons by PAR1.

(A-D) Current-clamp experiments revealed higher neuronal firing rate in PAR1−/− mice after fear conditioning. Voltage responses (representative traces in A, B) were recorded by currents steps from −100 to +600 pA in 50 pA increments (C) from principal neurons of the basal nucleus of PAR1+/+ and PAR1−/− mice 48 h after fear conditioning. Number of action potential spikes was counted as a function of depolarizing current injection (D). Fear conditioning significantly increased action potential firing rate in PAR1−/− (p < 0.01 at 150 pA; p < 0.05 at 200 pA). Insert shows a significant increase in the mean input resistance in PAR1−/− mice (n=8) compared to PAR1+/+ (n = 7; p<0.05). (E) Representative traces of current-clamp recordings at −40 mV from neurons of naïve, unpaired or fear conditioned mice before or after treatment with PAR1 antagonist SCH79797 (1 μM), or agonist TRag (5 μM). In fear conditioned (but not naïve or unpaired) PAR1+/+ mice inhibition of PAR1 with SCH79797 caused an increase while stimulation with TRag elicited a decrease in neuronal firing frequency (individual cells in F and means ± SEM for each group in G) *p < 0.05; **p < 0.01.
The neuronal consequences of PAR1-G protein coupling were tested before and after fear conditioning in wild-type amygdala neurons by measuring the change in firing frequency following pharmacological manipulation of PAR1. In naïve or unpaired groups the PAR1 agonist TRag or antagonist SCH79797, elicited mixed inhibitory and excitatory responses suggesting variable PAR1 coupling to both Gαo and Gαq/11 at single neuron level (Figure 5E-G). In contrast, after fear conditioning PAR1 agonist elicited a sharp decrease in firing frequency (F(5, 25) = 4.25; p < 0.01; n= 5-6 cells per group), supporting the idea of PAR1 switching its coupling to inhibitory Gαo proteins (Figure 4C). This finding was further corroborated by the PAR1 antagonist triggering an increase in neuronal firing frequency in fear conditioned basal amygdala neurons (Figure 5E-G).
PAR1 regulation of neuronal firing is AMPA-receptor dependent
What electrophysiological phenomena might explain the increase in excitability and L-LTP in PAR1−/− mice? Changes in L-LTP have previously been attributed to the modulation of AMPA receptors (34, 35). NMDA receptors are not critical for L-LTP in the lateral-basal amygdala pathway, since blocking these receptors failed to prevent both the basal responses and L-LTP (see Figure S8). In contrast, the AMPA receptor antagonist CNQX completely blocked basal transmission within this pathway (Figure S9). Therefore, to further investigate the electrophysiological mechanism downstream of the fear-mediated G protein switch that could underlie the reversal of L-LTP in PAR1−/− mice we examined AMPA-receptor-dependent miniature excitatory post-synaptic currents (mEPSCs), in principal basal amygdala neurons using whole cell recording. While we did not detect any differences between the genotypes in fear-naïve mice, two-way ANOVA revealed a significant genotype (F(1, 44)=4.43; p<0.05) and conditioning (F(1, 44)=6.48; p<0.05) effects on interevent interval with a 2-fold increase in the frequency of mEPSCs (n = 12 cells per group analyzed over 2 min, p < 0.05; Figure 6A-C and Figure S10) after fear-conditioning in PAR1-deficient mice compared to PAR1+/+ animals.
Figure 6. PAR1 regulates fear-dependent changes in mEPSCs in the basal nucleus of the amygdala.
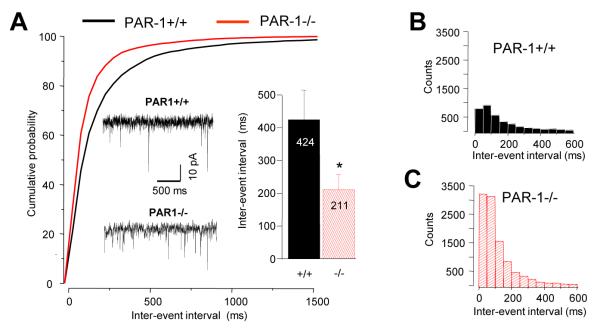
AMPA-receptor-dependent miniature excitatory post-synaptic currents (mEPSCs), in principal basal amygdala neurons after fear conditioning were examined using whole cell recording. Voltage-clamp experiments demonstrated that mEPSCs were ~2-fold more frequent in PAR1−/− animals (p < 0.05; cumulative distribution and mean ± SEM in A, inter-event distribution in B and C, sample traces shown as the insert). *p < 0.05. Data shown as means ± SEM.
Discussion
In this study we propose experience-dependent changes in GPCR coupling to G protein subtypes as a novel form of plasticity in the mammalian brain, which results in a fundamental shift in receptor signaling properties. Using PAR1 in a mouse as a model we demonstrated that during fear conditioning GPCR-G protein interactions are dynamic and regulate neuronal excitability, plasticity and learning.
GPCRs have the ability to signal through different G protein subtypes in a ligand-specific manner, a phenomenon called ligand-directed trafficking of signaling (1, 36). Stimulation of serotonin, opioid, vasopressin, dopamine or β-adrenergic receptors with structurally distinct agonists stabilize the receptor in ligand-specific conformations and shifts receptor signaling towards a different G protein and related transduction pathways (37, 38). Thus, on binding of different pharmacological agents, the same receptor can promote contrasting cellular responses.
In our studies we focused on PAR1 for several reasons. First, protease-activated receptors are unique among GPCRs due to their unusual mode of activation. They can be cleaved by several proteases which produce distinct responses through activation of the same PAR (39). Thus, this phenomenon might involve selective, protease-dependent coupling of PARs to different G protein(s) thereby promoting different sets of signaling events. Second, various forms of neuronal activity promote the release of different proteases into the extracellular space (9); (10-12, 15, 16) suggesting that the type of experience may determine the relative contribution of distinct proteases towards PAR1 cleavage and its association with G protein subtypes. Third, PAR1 is enriched in the amygdala (this paper) suggesting it could be altered during fear conditioning and modulate the expression of fear.
We show here that in response to fear conditioning, PAR1 receptors in the basolateral complex of the amygdala shift their G protein coupling profile to regulate neuronal plasticity, excitability and memory of traumatic experience. Fear conditioning “re-programs” PAR1 receptors, which can activate both Gαq/11 and Gαo proteins, shifting their G protein coupling preference more towards Gαo and inhibiting neuronal excitability, activity and synaptic plasticity. Consequently, stimulation of PAR1 before and after fear conditioning elicits opposite effects on neuronal activity in the basal amygdala – an increase in neuronal firing frequency in fear-naïve and a decrease in fear-conditioned animals. Thus, the PAR1-G protein switch serves as an experience-specific, ultrashort negative-feedback loop operating at a subcellular level to fine-tune neuronal activity underlying formation of traumatic memories. Consequently, PAR1−/− mice show lower L-LTP before and enhanced L-LTP after fear conditioning, consistent with the disruption of the dynamic, PAR1-dependent G protein-coupling regulating neuronal activity and plasticity.
It has been shown in various systems that activation of PAR1 by different proteases (or a different concentration of the same protease) often results in contrasting physiological outcomes (8). Opposite effects of low versus high doses of the PAR1 agonist thrombin have been observed both in the nervous system and other tissues. Liberation of excessive amounts of the protease (or an entirely different set of proteases) from presynaptic terminals in response to strong stimuli may results in the loss of substrate specificity and subsequent cleavage of PAR1 at low-affinity (or, in case of other proteases, entirely new) cleavage sites. Moreover, different sets of environmental stimuli liberating different “protease cocktails” into the amygdala synapses through a variety of neuronal projections would leave their unique, experience-specific “cleavage footprints” on PAR1 (and/or its molecular partners) and may determine its G protein coupling characteristics. Such a mechanism would provide a dynamic and precise control of GPCR/G protein coupling in response to different stimulus potencies and contingencies. This hypothesis is supported by our study (enhancement of fear learning in PAR1−/− mice following a mild conditioning protocol) and that of Almonte et al. (impairment of fear responses in PAR1−/− animals following a strong fear conditioning protocol) (13). Such a hypothesis is also in agreement with previous reports indicating that the strength of fear memories does not always correlate with training intensity, but often follows a non-linear model (40).
In spite of the involvement of G proteins and GPCRs in various forms of learning in a variety of species the dynamic nature of experience-driven GPCR-G protein coupling has, to our knowledge, never been demonstrated. Although it has been shown that another GPCR, corticotropin-releasing factor receptor (CRF-R), differentially regulates conditioned fear responses in C57BL/6N and BALB/c mouse strains by coupling to a distinct sets of G proteins, it was unclear whether receptor-G protein coupling was static or dynamic (41). Other studies have found that perturbing cyclic AMP signaling by expression of constitutively-active form of Gαs in forebrain neurons leads to learning impairment as well as to a number of schizophrenia-related phenotypes (42, 43). Recent studies in mice using genetically-engineered and light-activated GPCRs elegantly showed that selective activation of distinct intracellular signaling cascades in the nucleus accumbens elicits contrasting reward-related behaviours in the conditioned place preference paradigm (6). Altogether, the above experiments indicate that experience-specific shift in the GPCR-G protein coupling could control multiple aspects of synaptic plasticity associated with emotionality and learning in mammals.
Previous studies have shown PAR-1 could regulate the NMDA receptor function (44). However, to our knowledge the mechanism of action of PAR-1 in the amygdala has never been studied and it is likely pathway-specific. The mechanism of action of PAR-1 may depend on regional differences in its modulatory signaling components (e.g. the expression, alternative splicing or posttranslational modifications of PAR-1, other PARs, their putative activating proteases or protease inhibitors).
In summary, our previous and current data support a model where PAR-1 G protein switch serves as a buffering system protecting against the development of traumatic memories in response to weak (i.e. insignificant) aversive encounters. In response to an aversive experience, plasticity-related proteases are liberated into amygdala synapses (9, 11) to cleave PAR1. This cleavage changes PAR1-G protein coupling preference towards Gαo. Subsequently, the shift in the G protein association affects AMPA-mediated responses and neuronal excitability. In PAR1+/+ animals the effects of this modified G protein coupling could be counterbalanced by simultaneous adjustments of other receptor systems. The physiological and behavioral consequences of these adjustments are rapidly exposed in PAR1−/− mice where fear-conditioning affects synaptic transmission and plasticity, whereas these parameters are largely unaffected in PAR1+/+ animals. These changes manifest themselves as enhanced L-LTP, fear learning, neuronal excitability and AMPA-mediated responses in PAR1−/− mice, consistent with the lack of PAR1-evoked, Gαo-mediated inhibition.
The above mechanism has a significant translational potential. First, defective functioning of the PAR1 / G protein buffering system (as a result of mutations in the PAR-1 / G-protein machinery or their molecular partners) may lead to predisposition to the development of stress-related psychiatric disorders in response to mild trauma. Identification of the underlying mutations may open new avenues for treatment/prevention of anxiety disorders (including the posttraumatic stress disorder) or depression. Underlying defects could potentially be corrected by locking GPCR in a desired G-protein coupling state using biased ligands (see (45)).
Given the plethora of GPCRs in the central nervous system and their diverse mechanisms of activation it is likely that distinct sensory or emotional states could create their unique, experience-specific “biochemical footprints” across the brain through exclusive GPCR-G protein effector coupling profiles. Such a mechanism would provide both flexible and precise means of controlling other aspects of animal behaviour.
Material and Methods
Animals
Experiments were performed on male, adult wild-type (C57/BL6) or PAR1−/− mice genotyped as previously described (46). PAR-1−/− mice have been backcrossed to C57/Bl6/J for 10 generations. All animals we housed in a single animal holding room. PAR1−/− and PAR1+/+ mice received similar level of maternal care and husbandry.
Fear Conditioning
PAR1+/+ or PAR1−/− mice were placed in the conditioning chamber and received three conditioned stimulus-unconditioned stimulus (CS-US) pairings. The last 2 sec of the tone (CS, 30 sec, 2.8 kH, 85dB) were paired with the footshock (US, 2 sec, 0.4 mA) delivered through a grid floor and were then mice were moved to their home cage. In the “unpaired” group the tone and footshock were delivered in a random manner. Cued-conditioning was evaluated 48 h after training in a novel context (chamber with flat plastic floor and walls) for 2 min, after which the CS was delivered (2 min, 2.8 kHz, 85 dB) and freezing monitored. Data were analyzed using FreezeView software (Coulbourn Instruments).
Pain threshold
Each behavioural experiment was performed on a separate cohort of mice. PAR1+/+ and −/− mice were subjected to a series of mild footshocks of increasing intensities (in 0.05 mA increments) and behavioral reaction was measured as previously described (47).
Novel object recognition
Mice were placed in a 50×50×50 cm plexiglas box, left free to explore two objects for 5 min. The time of exploration of each object was recorded (head facing the object within 3 cm). At the end of the session mice were put back to their home cage. 1.5 hours and 24 hours later one of the objects was replaced with an unfamiliar object and mice were retested and time spent exploring each object recorded.
Elevated-plus maze
The elevated-plus maze test was performed as previously described (9). The apparatus consisted of four non-transparent white Plexiglas arms: two enclosed arms (50 × 10 × 30 cm) that formed a cross shape with the two open arms (50 × 10 cm) opposite each other. The maze was 55 cm above the floor and dimly illuminated. Mice were placed individually on the central platform, facing an open arm, and allowed to explore the apparatus for 5 min. Behavior was recorded by an overhead camera. The number of entries of the animal from the central platform to closed or open arms was counted.
Western blotting
Their brains were removed and brain regions dissected from a coronal slice approx −0.58 to −2.3 mm relative to Bregma. Samples from the hypothalamus medulla and cerebellar cortex were taken from the remaining tissue. Samples were homogenized (10) (see Supplementary Material for details) and the protein concentration adjusted. Reduced and denatured samples were subjected to SDS-PAGE electrophoresis, transferred onto nitrocellulose membrane and blocked in TBST-milk (TBS, 0.1% Tween 20, 5% skim milk) for 1 h at RT. The membrane was probed with a rabbit anti-PAR1 (gift of Dr. M. Runge, 1:1000, 4°C, overnight), rabbit anti-Gαq/11 antiserum (1:500 in TBST 5% milk)(48) or mouse anti-Gαo antibody (Santa Cruz Biotechnology, 1:500 in TBST 5% milk) followed by compatible HRP-labeled secondary antibodies (both at 1:1000, 1 h at RT). The signal was normalized to actin. Luminescence was detected using Western Blot luminal reagent and photographic film. Band intensities were measured using Scion Image.
Immunohistochemistry
The brains were fixed in 4% PFA overnight. 70 μm coronal free-floating sections were blocked with goat serum in PBS (1:500 for 4 h, RT) and incubated with rabbit anti-PAR1 (1:500, 4°C for 4 h), followed by the addition of mouse anti-NeuN (Chemicon, 1:200, 4°C, overnight) and chicken anti-GFAP (Abcam, 1:1000, 4°C, overnight) to the same wells. To examine the level of c-Fos and P-CREB separate sections were incubated with anti-c-Fos (Cell Signaling, 1:200) or anti-phospho-CREB antibodies (Cell Signaling, 1:200), respectively. After several washes, the sections were incubated with the appropriate AlexaFluor 488, 546, 647 or Cy3-labeled secondary antibodies (Invitrogen or Abcam, all at 1:500, 4°C, overnight). DAPI (pseudocolored grey) was used to visualize cell nuclei in conjunction with PAR1, NeuN and GFAP labeling. Sections in which the primary antibodies were omitted served as controls. The images were collected using LSM5 Exciter confocal microscope.
[35S]GTPγS binding and Gα-specific immunoprecipitation assay
Mouse amygdalae were dissected from coronal vibratome slices under a microscope and homogenized. [35S]GTPγS-Gαq/11 immuno-specific binding was performed as previously described (31, 48) with minor modifications (see Supplementary Material for details). Non-specific binding was determined in the presence of unlabelled GTPγS (10 μM). Basal [35S]GTPγS binding to Gαo: naïve, 3108 ± 508; unpaired, 2816 ± 377; fear conditioned, 2929 ± 501 d.p.m.; binding to Gαq/11: naïve, 1118 ± 185; unpaired, 1411 ± 242; fear conditioned, 1097 ± 69 d.p.m.
Stereotaxic injections
Mice were bilaterally implanted with stainless steel guide cannulae (26-gauge; Plastics One, Roanoke, VA) aimed above the basolateral complex of the amygdala (1.5 mm posterior to bregma, 3.5 lateral and 4.0 ventral). After one week the mice were injected with SCH79797 (1 μM, 0.5 μL over 15 minutes) or vehicle (1:4 DMSO in artificial cerebro-spinal fluid composed of 10 mM HEPES, 140 mM NaCl, 2.8 mM KCl, 1 mM MgCl2*6H2O, 2 mM CaCl2*2H2O, 10 mM glucose; pH 7.3) and using the injection cannulae; 33-gauge, projecting 0.75 mm) followed by fear conditioning. After the experiment a small amount of bromophenol blue was injected to visualize the guide and injection cannulae tracks, the brains were sectioned and the cannulae placement was determined histologically.
Electrophysiology
Coronal slices containing the amygdala (400 μm for extracellular recordings or 300 μm for whole cell recordings) were prepared from 8-12 weeks-old PAR1−/− or PAR+/+ mice (see Supplementary Material for details). The mice were either left undisturbed (naïve), subjected to the unpaired protocol or fear conditioning 48 hours prior to recordings.
Extracellular recordings in the lateral-basal pathway were made using a bipolar tungsten electrode (WPI) and glass microelectrodes (1-2 mΩ) filled with ACSF. Several field potential (FP) traces were averaged to create a template and only the responses matching the template were analyzed. Long-term potentiation (LTP) was elicited by two trains of high-frequency tetanic stimulation (100 Hz, 1 sec, 10 sec interval) repeated 4 times at 3 min interval. The recordings were amplified, filtered (10 kHz) and digitized (50 kHz).
Whole-cell recordings (see Supplementary Material for details) were made from somata of principal neurons of the basal nucleus of the amygdala. Principal neurons and interneurons were distinguished by their morphological and electrophysiological properties (49). Maximum series resistance of 10-15 MΩ was tolerated and neurons with a resting membrane potential below −50 mV were used. In current-clamp recordings the membrane potential was kept at −80 mV (or −40mV where indicated). Input resistance and instantaneous spike frequency were derived from traces in which cells were injected with 200 msec current pulses (−100 to +600 pA; 50 pA increments). In order to record mEPSCs TTX (1 μM; Latoxan) and picrotoxin (100 μM; Sigma) were routinely included in the extracellular solution. The cell membrane was clamped at −70 mV and at the end of each experiment mEPSCs were blocked with CNQX (10 μM) to confirm they were mediated by AMPA receptors. To study mIPSCs cell membrane was clamped at −70 mV. TTX (1 μM), AP-V (50 μM) and CNQX (10 μM; Tocris Bioscience) were routinely added to the extracellular solution. At the end of each experiment mIPSCs were blocked with picrotoxin (100 μM) to confirm they were GABAA -mediated.
Statistical analysis
Statistical analysis was performed using Statistica and GraphPad Prism. All values are expressed as means ± SEM. Student t-test (for two groups) or analysis of variance (ANOVA) for multiple comparisons followed by Tukey’s post test were used as appropriate. P values of less than 0.05 were considered significant.
Supplementary Material
Acknowledgements
This work was supported by a Medical Research Council project grant (G0500231/73852), and a Marie Curie Excellence grant (MEXT-CT-2006-042265 from European Commission) to Robert Pawlak. We acknowledge support of ECMNet (COST BM1001). We are obliged to Dr. Marshall Runge for his gift of the anti-PAR1 antibody, to Drs. Shaun Coughlin and Rachel Chambers for PAR1−/− mice.
References
- 1.Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilchrist A. Modulating G-protein-coupled receptors: from traditional pharmacology to allosterics. Trends Pharmacol Sci. 2007;28:431–437. doi: 10.1016/j.tips.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 3.Bokoch MP, Zou Y, Rasmussen SG, Liu CW, Nygaard R, Rosenbaum DM, Fung JJ, Choi HJ, Thian FS, Kobilka TS, et al. Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature. 2010;463:108–112. doi: 10.1038/nature08650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SG, Choi HJ, Devree BT, Sunahara RK, et al. Structure and function of an irreversible agonist-beta(2) adrenoceptor complex. Nature. 2011;469:236–240. doi: 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Airan RD, Thompson KR, Fenno LE, Bernstein H, Deisseroth K. Temporally precise in vivo control of intracellular signalling. Nature. 2009;458:1025–1029. doi: 10.1038/nature07926. [DOI] [PubMed] [Google Scholar]
- 7.Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- 8.Soh UJ, Dores MR, Chen B, Trejo J. Signal transduction by protease-activated receptors. Br J Pharmacol. 2010;160:191–203. doi: 10.1111/j.1476-5381.2010.00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Attwood BK, Bourgognon JM, Patel S, Mucha M, Schiavon E, Skrzypiec AE, Young KW, Shiosaka S, Korostynski M, Piechota M, et al. Neuropsin cleaves EphB2 in the amygdala to control anxiety. Nature. 2011;473:372–375. doi: 10.1038/nature09938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matys T, Pawlak R, Matys E, Pavlides C, McEwen BS, Strickland S. Tissue plasminogen activator promotes the effects of corticotropin-releasing factor on the amygdala and anxiety-like behavior. Proc Natl Acad Sci U S A. 2004;101:16345–16350. doi: 10.1073/pnas.0407355101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pawlak R, Magarinos AM, Melchor J, McEwen B, Strickland S. Tissue plasminogen activator in the amygdala is critical for stress-induced anxiety-like behavior. Nat Neurosci. 2003;6:168–174. doi: 10.1038/nn998. [DOI] [PubMed] [Google Scholar]
- 12.Pawlak R, Rao BS, Melchor JP, Chattarji S, McEwen B, Strickland S. Tissue plasminogen activator and plasminogen mediate stress-induced decline of neuronal and cognitive functions in the mouse hippocampus. Proc Natl Acad Sci U S A. 2005;102:18201–18206. doi: 10.1073/pnas.0509232102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Almonte AG, Hamill CE, Chhatwal JP, Wingo TS, Barber JA, Lyuboslavsky PN, David Sweatt J, Ressler KJ, White DA, Traynelis SF. Learning and memory deficits in mice lacking protease activated receptor-1. Neurobiol Learn Mem. 2007;88:295–304. doi: 10.1016/j.nlm.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horii Y, Yamasaki N, Miyakawa T, Shiosaka S. Increased anxiety-like behavior in neuropsin (kallikrein-related peptidase 8) gene-deficient mice. Behav Neurosci. 2008;122:498–504. doi: 10.1037/0735-7044.122.3.498. [DOI] [PubMed] [Google Scholar]
- 15.Maggio N, Shavit E, Chapman J, Segal M. Thrombin induces long-term potentiation of reactivity to afferent stimulation and facilitates epileptic seizures in rat hippocampal slices: toward understanding the functional consequences of cerebrovascular insults. J Neurosci. 2008;28:732–736. doi: 10.1523/JNEUROSCI.3665-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsumoto-Miyai K, Ninomiya A, Yamasaki H, Tamura H, Nakamura Y, Shiosaka S. NMDA-dependent proteolysis of presynaptic adhesion molecule L1 in the hippocampus by neuropsin. J Neurosci. 2003;23:7727–7736. doi: 10.1523/JNEUROSCI.23-21-07727.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Samson AL, Medcalf RL. Tissue-type plasminogen activator: a multifaceted modulator of neurotransmission and synaptic plasticity. Neuron. 2006;50:673–678. doi: 10.1016/j.neuron.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 18.Luo W, Wang Y, Reiser G. Protease-activated receptors in the brain: receptor expression, activation, and functions in neurodegeneration and neuroprotection. Brain Res Rev. 2007;56:331–345. doi: 10.1016/j.brainresrev.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 19.Traynelis SF, Trejo J. Protease-activated receptor signaling: new roles and regulatory mechanisms. Curr Opin Hematol. 2007;14:230–235. doi: 10.1097/MOH.0b013e3280dce568. [DOI] [PubMed] [Google Scholar]
- 20.Junge CE, Lee CJ, Hubbard KB, Zhang Z, Olson JJ, Hepler JR, Brat DJ, Traynelis SF. Protease-activated receptor-1 in human brain: localization and functional expression in astrocytes. Exp Neurol. 2004;188:94–103. doi: 10.1016/j.expneurol.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 21.Striggow F, Riek-Burchardt M, Kiesel A, Schmidt W, Henrich-Noack P, Breder J, Krug M, Reymann KG, Reiser G. Four different types of protease-activated receptors are widely expressed in the brain and are up-regulated in hippocampus by severe ischemia. Eur J Neurosci. 2001;14:595–608. doi: 10.1046/j.0953-816x.2001.01676.x. [DOI] [PubMed] [Google Scholar]
- 22.Amorapanth P, LeDoux JE, Nader K. Different lateral amygdala outputs mediate reactions and actions elicited by a fear-arousing stimulus. Nat Neurosci. 2000;3:74–79. doi: 10.1038/71145. [DOI] [PubMed] [Google Scholar]
- 23.Doyere V, Schafe GE, Sigurdsson T, LeDoux JE. Long-term potentiation in freely moving rats reveals asymmetries in thalamic and cortical inputs to the lateral amygdala. Eur J Neurosci. 2003;17:2703–2715. doi: 10.1046/j.1460-9568.2003.02707.x. [DOI] [PubMed] [Google Scholar]
- 24.Goosens KA, Maren S. Contextual and auditory fear conditioning are mediated by the lateral, basal, and central amygdaloid nuclei in rats. Learn Mem. 2001;8:148–155. doi: 10.1101/lm.37601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang YY, Martin KC, Kandel ER. Both protein kinase A and mitogen-activated protein kinase are required in the amygdala for the macromolecular synthesis-dependent late phase of long-term potentiation. J Neurosci. 2000;20:6317–6325. doi: 10.1523/JNEUROSCI.20-17-06317.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Luthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. doi: 10.1016/j.neuron.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 27.Hong I, Kim J, Lee J, Park S, Song B, Kim J, An B, Park K, Lee HW, Lee S, et al. Reversible plasticity of fear memory-encoding amygdala synaptic circuits even after fear memory consolidation. PLoS One. 2011;6:e24260. doi: 10.1371/journal.pone.0024260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neves SR, Ram PT, Iyengar R. G protein pathways. Science. 2002;296:1636–1639. doi: 10.1126/science.1071550. [DOI] [PubMed] [Google Scholar]
- 29.Strittmatter SM, Valenzuela D, Kennedy TE, Neer EJ, Fishman MC. G0 is a major growth cone protein subject to regulation by GAP-43. Nature. 1990;344:836–841. doi: 10.1038/344836a0. [DOI] [PubMed] [Google Scholar]
- 30.Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 31.Akam EC, Challiss RA, Nahorski SR. G(q/11) and G(i/o) activation profiles in CHO cells expressing human muscarinic acetylcholine receptors: dependence on agonist as well as receptor-subtype. Br J Pharmacol. 2001;132:950–958. doi: 10.1038/sj.bjp.0703892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milligan G. Principles: extending the utility of [35S]GTP gamma S binding assays. Trends Pharmacol Sci. 2003;24:87–90. doi: 10.1016/s0165-6147(02)00027-5. [DOI] [PubMed] [Google Scholar]
- 33.Taussig R, Tang WJ, Hepler JR, Gilman AG. Distinct patterns of bidirectional regulation of mammalian adenylyl cyclases. J Biol Chem. 1994;269:6093–6100. [PubMed] [Google Scholar]
- 34.Man HY, Wang Q, Lu WY, Ju W, Ahmadian G, Liu L, D’Souza S, Wong TP, Taghibiglou C, Lu J, et al. Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron. 2003;38:611–624. doi: 10.1016/s0896-6273(03)00228-9. [DOI] [PubMed] [Google Scholar]
- 35.Watt AJ, Sjostrom PJ, Hausser M, Nelson SB, Turrigiano GG. A proportional but slower NMDA potentiation follows AMPA potentiation in LTP. Nat Neurosci. 2004;7:518–524. doi: 10.1038/nn1220. [DOI] [PubMed] [Google Scholar]
- 36.Kenakin T. Biased agonism. F1000 Biol Rep. 2009;1 doi: 10.3410/B1-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- 38.Kobilka BK, Deupi X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci. 2007;28:397–406. doi: 10.1016/j.tips.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 39.Russo A, Soh UJ, Trejo J. Proteases display biased agonism at protease-activated receptors: location matters. Mol Interv. 2009;9:87–96. doi: 10.1124/mi.9.2.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Costa-Mattioli M, Gobert D, Harding H, Herdy B, Azzi M, Bruno M, Bidinosti M, Ben Mamou C, Marcinkiewicz E, Yoshida M, et al. Translational control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature. 2005;436:1166–1173. doi: 10.1038/nature03897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blank T, Nijholt I, Grammatopoulos DK, Randeva HS, Hillhouse EW, Spiess J. Corticotropin-releasing factor receptors couple to multiple G-proteins to activate diverse intracellular signaling pathways in mouse hippocampus: role in neuronal excitability and associative learning. J Neurosci. 2003;23:700–707. doi: 10.1523/JNEUROSCI.23-02-00700.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bourtchouladze R, Patterson SL, Kelly MP, Kreibich A, Kandel ER, Abel T. Chronically increased Gsalpha signaling disrupts associative and spatial learning. Learn Mem. 2006;13:745–752. doi: 10.1101/lm.354106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kelly MP, Stein JM, Vecsey CG, Favilla C, Yang X, Bizily SF, Esposito MF, Wand G, Kanes SJ, Abel T. Developmental etiology for neuroanatomical and cognitive deficits in mice overexpressing Galphas, a G-protein subunit genetically linked to schizophrenia. Mol Psychiatry. 2009;14:398–415. 347. doi: 10.1038/mp.2008.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gingrich MB, Junge CE, Lyuboslavsky P, Traynelis SF. Potentiation of NMDA receptor function by the serine protease thrombin. J Neurosci. 2000;20:4582–4595. doi: 10.1523/JNEUROSCI.20-12-04582.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov. 2010;9:373–386. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Howell DC, Johns RH, Lasky JA, Shan B, Scotton CJ, Laurent GJ, Chambers RC. Absence of proteinase-activated receptor-1 signaling affords protection from bleomycin-induced lung inflammation and fibrosis. Am J Pathol. 2005;166:1353–1365. doi: 10.1016/S0002-9440(10)62354-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- 48.Salah-Uddin H, Thomas DR, Davies CH, Hagan JJ, Wood MD, Watson JM, Challiss RA. Pharmacological assessment of m1 muscarinic acetylcholine receptor-gq/11 protein coupling in membranes prepared from postmortem human brain tissue. J Pharmacol Exp Ther. 2008;325:869–874. doi: 10.1124/jpet.108.137968. [DOI] [PubMed] [Google Scholar]
- 49.Washburn MS, Moises HC. Electrophysiological and morphological properties of rat basolateral amygdaloid neurons in vitro. J Neurosci. 1992;12:4066–4079. doi: 10.1523/JNEUROSCI.12-10-04066.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.