

Abstract
Cholesteryl Ester Storage Disease (CESD) and Wolman disease are autosomal recessive later-onset and severe infantile disorders, respectively, which result from the deficient activity of lysosomal acid lipase (LAL). LAL is encoded by LIPA (10q23.31) and the most common mutation associated with CESD is an exon 8 splice junction mutation (c.894G>A; E8SJM), which expresses only ~3–5% of normally spliced LAL. However, the frequency of c.894G>A is unknown in most populations. To estimate the prevalence of CESD in different populations, the frequencies of the c.894G>A mutation were determined in 10,000 LIPA alleles from healthy African-American, Asian, Caucasian, Hispanic and Ashkenazi Jewish individuals from the greater New York metropolitan area and 6,578 LIPA alleles from African-American, Caucasian, and Hispanic subjects enrolled in the Dallas Heart Study. The combined c.894G>A allele frequencies from the two cohorts ranged from 0.0005 (Asian) to 0.0017 (Caucasian and Hispanic), which translated to carrier frequencies of 1 in 1,000 to ~1 in 300, respectively. No African-American heterozygotes were detected. Additionally, by surveying the available literature, c.894G>A was estimated to account for 60% (95% CI: 51%–69%) of reported mutations among multi-ethnic CESD patients. Using this estimate, the predicted prevalence of CESD in the Caucasian and Hispanic populations is ~0.8 per 100,000 (~1 in 130,000; 95% CI: ~1 in 90,000 to 1 in 170,000).
Conclusion
These data indicate that CESD may be under-diagnosed in the general Caucasian and Hispanic populations, which is important since clinical trials of enzyme replacement therapy for LAL deficiency are currently being developed. Moreover, future studies on CESD prevalence in African and Asian populations may require full-gene LIPA sequencing to determine heterozygote frequencies since c.894G>A is not common in these racial groups.
Keywords: lysosomal acid lipase deficiency, allele frequency, carrier frequency, disease prevalence, splice junction mutation, genotyping
INTRODUCTION
The deficient activity of lysosomal acid lipase (LAL), which hydrolyzes cholesteryl esters and triglycerides, results in either Cholesteryl Ester Storage Disease (CESD) or the more severe Wolman disease (1–2). LAL is encoded by the LIPA gene (10q23.31) (3) and homozygous and compound heterozygous LIPA mutations that result in little or no LAL activity cause Wolman disease, a severe infantile-onset disorder characterized biochemically by the massive storage of cholesteryl esters and triglycerides primarily in the liver, intestines, and adrenals leading to hepatosplenomegaly, adrenal calcification, vomiting, diarrhea, anemia, failure to thrive, and death typically before 1 year of age (4). In contrast, LIPA mutations with residual LAL activity result in CESD, which is characterized by hepatosteatosis, hepatomegaly, splenomegaly, dyslipidemia, accelerated atherosclerosis, and premature demise (1–2, 5–6).
Although over 40 LIPA mutations have been identified in patients with Wolman disease and CESD, the most common LIPA mutation is the c.894G>A exon 8 splice junction mutation (i.e., E8SJM; p.delS275_Q298; rs116928232). This mutation results in a transcript with an in-frame deletion of exon 8 that encodes a mutant enzyme with no residual LAL activity; however, the splicing defect also allows ~3–5% normally spliced LAL with enzyme activity (5, 7–9). Since the c.894G>A mutation results in residual LAL activity, it has only been found among patients with CESD and not Wolman disease (8, 10). In contrast, neighboring exon/intron 8 splice junction mutations, c.892C>T (E8SJM-3) and c.894+1G>A (E8SJM+1), are rare null alleles that have been detected in patients with Wolman disease. Interestingly, c.894G>A heterozygotes recently were found to have elevated total cholesterol compared to normolipidemic Caucasian controls, which may also impart an increased risk of coronary heart disease (CHD) (11). The association of LIPA as a susceptibility gene for CHD has been strengthened further by recent genome-wide association studies (12–14).
The frequency of CESD in different populations is unknown, but it is generally thought that the disease is often unrecognized. For example, it can be misdiagnosed in patients with hepatomegaly following a liver biopsy as non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), or cryptogenic fatty liver disease (15). To estimate the prevalence of CESD, a previous study investigated the frequency of the c.894G>A CESD mutation in the German population and identified an allele frequency of 0.0025 (1 in 202 carrier frequency) (16). Among the reported CESD patients who had undergone LIPA mutation analyses, c.894G>A previously was estimated to account for ~50% of all CESD alleles (16). As such, with this assumption, the identified 0.0025 c.894G>A allele frequency predicted a CESD prevalence of ~2.4 per 100,000 in the German population.
Since no LIPA c.894G>A allele frequencies exist for other racial or ethnic groups, including the African-American, Asian or Hispanic populations, efforts were directed to determine the c.894G>A frequencies in individuals from five major racial and ethnic groups from both the New York metropolitan area and the Dallas Heart Study. The frequency of the c.894G>A mutation in various populations and its prevalence among CESD patients can be used to estimate the prevalence of CESD, which may encourage hepatologists, lipidologists, pathologists, geneticists, and metabolic physicians to identify CESD patients since clinical trials of enzyme replacement therapy for LAL deficiency are underway and may provide safe and effective therapy.
MATERIALS AND METHODS
c.894G>A Prevalence in CESD
The prevalence of the c.894G>A mutation among unrelated multi-ethnic CESD patients was estimated by searching available literature and CESD case reports. The PubMed database (NCBI) was searched using the keywords (CESD OR Cholesteryl Ester Storage Disease) AND (mutation) from 1966 to September 2012. Studies were considered for inclusion if LIPA mutation status was reported for unique patient(s) with clinically confirmed CESD. Only a single affected patient was included in the LIPA mutation prevalence calculation when reports included multiple affected family members with the same mutations. Additionally, the Human Gene Mutation Database (HGMD) Professional (http://www.biobase-international.com/product/hgmd) was interrogated for CESD-causing LIPA mutations.
Study Population
New York Metropolitan Area
Peripheral blood samples from healthy donors who indicated their racial/ethnic background and gave informed consent for the use of their DNA for research were obtained from the New York Blood Center with Institutional Review Board (IRB) approval (17–18). In addition, blood samples were obtained with informed consent from unrelated, healthy 100% Ashkenazi Jewish (AJ) individuals from the greater New York metropolitan area as previously defined (19–20). Allpersonal identifiers were removed, and isolated DNA samples were tested anonymously. Genomic DNA was isolated using the Puregene® DNA Purification kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. For this study, 1,000 individuals each from five different populations (African-American, Asian, Caucasian, Hispanic, and AJ) were subjected to c.894G>A genotyping.
Dallas Heart Study
The Dallas Heart Study is a multi-ethnic population-based probability sample of Dallas County residents (ages 18–85), weighted to include approximately 50% of African-American subjects. Sampling design and recruitment procedures have been previously described in detail (21). The study was transformed from a cross-sectional study to a longitudinal study in 2007 (DHS-2) when all prior DHS participants were invited to the clinic for a repeat evaluation. Race or ethnic identity was self-reported according to a list of categories used in the United States census. A total of 3,401 individuals submitted blood samples and provided written consent for genetic analyses. For this study, 1,720 African-American, 1,089 Caucasian, and 480 Hispanic participants from the DHS-2 cohort were subjected to c.894G>A genotyping. The study was approved by the IRB of the University of Texas Southwestern Medical Center at Dallas.
Genotyping
LIPA c.894G>A genotyping was performed using custom-designed TaqMan® allelic discrimination assays (Applied Biosystems, Carlsbad, CA) as described in the Supplemental Data. Selected mutation carriers were confirmed by bidirectional sequencing with independent M13-tagged LIPA amplification primers (forward: 5′-TGCTTTGAAGGGCAAAATAC-3′; reverse: 5′-TTTCTATTTGGAAAGGGTTTGC-3′) and analyzed using Mutation Surveyor software v3.30 (SoftGenetics).
CESD Prevalence
Prevalence of CESD was calculated using identified c.894G>A allele frequencies and adjusting for the prevalence of c.894G>A among CESD patients, as previously reported (16).
RESULTS
Prevalence of the c.894G>A Mutation in CESD
To estimate the prevalence of the c.894G>A mutation among unrelated multi-ethnic patients with CESD, a literature survey was conducted which revealed that c.894G>A accounted for 60% (95% CI: 51%–69%) of reported multi-ethnic CESD mutations (n=55 patients) (Table 1). These results are consistent with the previous c.894G>A frequency estimate of ~50% among CESD patients (16).
TABLE 1.
LIPA Mutations among 55 Unrelated Patients with Cholesteryl Ester Storage Disease (CESD)
| Mutation | Frequency (n) | Ethnicity | Reference |
|---|---|---|---|
| Missense/Nonsense Mutations | |||
| c.193C>T (p.R65X) | 0.018 (2) | French | Redonnet-Vernhet, et al. 1997 (33) |
| Czech | Lohse, et al. 2000 (34) | ||
| c.254A>G (p.Q85R) | 0.009 (1) | Brazilian | Pagani, et al. 1998 (9) |
| c.260G>T (p.G87V) | 0.009 (1) | Sicilian | Pagani, et al. 1996 (10) |
| c.283T>A (p.W95R) | 0.009 (1) | Czech | Elleder, et al. 1999 (35) |
| c.356A>G (p.N119S) | 0.009 (1) | Australian | Hooper, et al. 2008 (36) |
| c.386A>G (p.H129R) | 0.009 (1) | Swiss | Ries, et al. 1998 (37) |
| c.386A>C (p.H129P) | 0.018 (2) | Austrian | Gasche, et al. 1997 (38) |
| Austrian | Ries, et al. 1998 (37) | ||
| c.419G>A (p.W140X) | 0.009 (1) | Croatian | Fasano, et al. 2012 (39) |
| c.435T>A (p.D145E) | 0.009 (1) | Czech | Elleder, et al. 1999 (35) |
| c.599T>C (p.L200P) | 0.009 (1) | NR | Maslen, et al. 1995 (40) |
| c.605C>T (p.P202L) | 0.009 (1) | Italian | Pagani, et al. 1996 (10) |
| c.652C>T (p.R218X) | 0.009 (1) | Italian | Pisciotta, et al. 2009 (41) |
| c.796G>T (p.G266X) | 0.009 (1) | Polish-German | Aslanidis, et al. 1996 (5) |
| c.863C>T (p.T288I) | 0.018 (2) | Italian | Pagani, et al. 1998 (9) |
| c.866C>G (p.S289C) | 0.009 (1) | African-American | Anderson, et al. 1999 (32) |
| c.881T>C (p.L294S) | 0.009 (1) | Italian | Pagani, et al. 1996 (10) |
| c.883C>T (p.H295Y) | 0.045 (5) | Sicilian | Pagani, et al. 1994 (42) |
| Italian | Redonnet-Vernhet, et al. 1997 (33) | ||
| Italian | Fasano, et al. 2012 (39) | ||
| c.1024G>A (p.G342R) | 0.018 (2) | African-American | Anderson, et al. 1999 (32) |
| Greek | Fasano, et al. 2012 (39) | ||
| c.1070T>C (p.L357P) | 0.009 (1) | Canadian-Norwegian | Seedorf, et al. 1995 (43) |
| Splicing Mutations | |||
| c.676-2A>G | 0.009 (1) | Italian | Pagani, et al. 1996 (10) |
| c.894G>A | 0.600 (65) | Polish-German | Klima, et al. 1993 (7) |
| NR | Maslen, et al. 1995 (40) | ||
| Canadian-Norwegian | Seedorf, et al. 1995 (43) | ||
| German | Ameis, et al. 1995 (44) | ||
| Spanish | Muntoni, et al. 1995 (8) | ||
| Sicilian | Pagani, et al. 1996 (10) | ||
| Polish-German | Aslanidis, et al. 1996 (5) | ||
| Italian, French | Redonnet-Vernhet, et al. 1997 (33) | ||
| Austrian | Gasche, et al. 1997 (38) | ||
| Brazilian | Pagani, et al. 1998 (9) | ||
| Austrian | Ries, et al. 1998 (37) | ||
| Brazilian | Du, et al. 1998 (45) | ||
| Czech | Elleder, et al. 2000 (46) | ||
| NR | Anderson, et al. 1999 (32) | ||
| German | vom Dahl, et al. 1999 (47) | ||
| Czech | Elleder, et al. 2000 (46) | ||
| Irish, Czech | Lohse, et al. 2000 (34) | ||
| German | Rassoul, et al. 2001 (48) | ||
| German | Drebber, et al. 2005 (49) | ||
| NR | Tadiboyina, et al. 2005 (50) | ||
| Australian | Hooper, et al. 2008 (36) | ||
| Italian | Pisciotta, et al. 2009 (41) | ||
| Italian, Greek, Croatian | Fasano, et al. 2012 (39) | ||
| Small Insertions/Deletions | |||
| c.397_398delTC (p.V134Ffs*4) | 0.009 (1) | NR | Anderson, et al. 1999 (32) |
| c.594insT (p.A199Cfs*13) | 0.009 (1) | NR | Tadiboyina, et al. 2005 (50) |
| c.635delC (p.P212Lfs*5) | 0.009 (1) | Czech | Elleder, et al. 2000 (46) |
| c.684delT (p.F228Lfs*13) | 0.009 (1) | Irish | Lohse, et al. 2000 (34) |
| c.967_968delAG (p.S323Lfs*44) | 0.009 (1) | German | Ameis, et al. 1995 (44) |
| c.980delC (p.T327Nfs*4) | 0.009 (1) | NR | Anderson, et al. 1999 (32) |
| c.1028delG (p.G343Vfs*15) | 0.009 (1) | Czech | Lohse, et al. 2000 (34) |
| Gross Deletions | |||
| intron 1 to exon 4 | 0.009 (1) | Swiss | Ries, et al. 1998 (37) |
| Complex Rearrangements | |||
| c.229-33_c.230dup, c.232_245del (p.G77fs*5) | 0.009 (1) | Italian | Pisciotta, et al. 2009 (41) |
| Undetermined Mutations | 0.064 (7) | Polish-German | Klima, et al. 1993 (7) |
| Italian | Pagani, et al. 1996 (10) | ||
| NR | Anderson, et al. 1999 (32) | ||
| Czech | Elleder, et al. 1999 (35) | ||
NR: not reported.
c.894G>A Allele and Carrier Frequencies
The identified c.894G>A allele and carrier frequencies are summarized in Table 2 and a representative sequence chromatogram of a confirmed heterozygote is illustrated in Figure 1. The c.894G>A allele frequencies from the New York metropolitan area cohort ranged from 0.0005 (Asian) to 0.0015 (Caucasian and Hispanic), which translated to carrier frequencies of 1 in 1,000 to 1 in 333, respectively. The c.894G>A allele frequencies from the Dallas Heart Study cohort ranged from 0.0018 (Caucasian) to 0.0021 (Hispanic), which translated to carrier frequencies of 1 in 272 to 1 in 240, respectively. No African-American carriers were detected in either cohort. Additionally, the Caucasian and Hispanic c.894G>A frequency data from the New York and Dallas cohorts were combined, which resulted in similar allele frequencies of 0.0017 and carrier frequencies of 1 in 298 and 1 in 296, respectively. Of note, with a c.894G>A carrier frequency of ~1 in 300 and a detectability of only 0.60, Bayesian analysis indicates that the residual risk of being a CESD carrier after negative c.894G>A carrier screening in the Caucasian and Hispanic populations would be ~1 in 500.
TABLE 2.
LIPA c.894G>A and Predicted Cholesteryl Ester Storage Disease (CESD) Frequencies
| Population | Carriers/Screenees | Allele Frequency (95% CI) | Carrier Frequency | Predicted c.894G>A Homozygotes | Predicted Affecteda (95% CI) |
|---|---|---|---|---|---|
| Caucasian (New York) | 3/1,000 | 0.0015 (0.000–0.003) | 0.0030 (1 in 333) | ~0.2 in 100,000 | ~0.6 in 100,000; 1 in 160,000 (1 in 114,898 to 1 in 212,553) |
| Caucasian (Dallas) | 4/1,089 | 0.0018 (0.000–0.004) | 0.0037 (1 in 272) | ~0.3 in 100,000 | ~0.9 in 100,000; 1 in 106,733 (1 in 76,646 to 1 in 141,790) |
| Caucasian (New York + Dallas) | 7/2,089 | 0.0017 (0.000–0.003) | 0.0034 (1 in 298) | ~0.3 in 100,000 | ~0.8 in 100,000; 1 in 128,246 (1 in 92,095 to 1 in 170,369) |
| Caucasian (Germany)b | 10/2,023 | 0.0025 (0.001–0.004) | 0.0049 (1 in 202) | ~0.6 in 100,000 | ~1.7 in 100,000; 1 in 58,932 (1 in 42,320 to 1 in 78,289) |
| Combined Caucasianc | 17/4,112 | 0.0021 (0.001–0.003) | 0.0041 (1 in 242) | ~0.4 in 100,000 | ~1.2 in 100,000; 1 in 84,250 (1 in 60,501 to 1 in 111,922) |
| Hispanic (New York) | 3/1,000 | 0.0015 (0.000–0.003) | 0.0030 (1 in 333) | ~0.2 in 100,000 | ~0.6 in 100,000; 1 in 160,000 (1 in 114,898 to 1 in 212,553) |
| Hispanic (Dallas) | 2/480 | 0.0021 (0.000–0.005) | 0.0042 (1 in 240) | ~0.4 in 100,000 | ~1.2 in 100,000; 1 in 82,944 (1 in 59,563 to 1 in 110,187) |
| Hispanic (New York + Dallas) | 5/1,480 | 0.0017 (0.000–0.003) | 0.0034 (1 in 296) | ~0.3 in 100,000 | ~0.8 in 100,000; 1 in 126,167 (1 in 90,602 to 1 in 167,607) |
| Ashkenazi Jewish | 2/1,000 | 0.0010 (0.000–0.002) | 0.0020 (1 in 500) | ~0.1 in 100,000 | ~0.3 in 100,000; 1 in 360,000 (1 in 258,520 to 1 in 478,243) |
| Asian | 1/1,000 | 0.0005 (0.000–0.001) | 0.0010 (1 in 1,000) | ~0.1 in 400,000 | ~0.1 in 100,000; 1 in 1,440,000 (1 in 1,034,080 to 1 in 1,912,974) |
| African-American (New York) | 0/1,000 | - | - | - | - |
| African-American (Dallas) | 0/1,720 | - | - | - | - |
Figure 1.
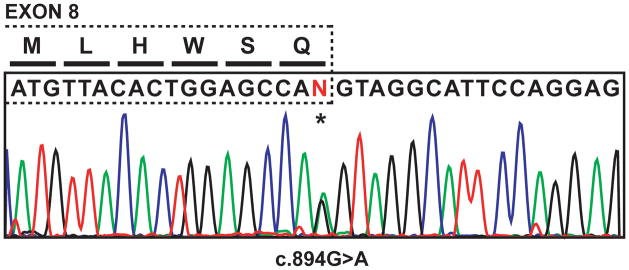
Illustration of LIPA exon 8 and DNA sequence chromatogram from a representative heterozygous c.894G>A carrier. Mutation position is highlighted by an asterisk.
Predicted Prevalence of CESD
The predicted prevalence of affected individuals with CESD in the different populations, assuming Hardy-Weinberg equilibrium, is also summarized in Table 2. CESD prevalence was calculated using the identified c.894G>A allele frequencies and adjusting for the prevalence of c.894G>A among CESD patients. Assuming that the c.894G>A mutation accounts for 60% (95% CI: 51%–69%) of known multi-ethnic CESD mutations, the predicted prevalence of CESD in the Caucasian population of the greater New York metropolitan area is ~0.6 per 100,000 (1 in 160,000; 95% CI: 1 in 114,898 to 1 in 212,553) and ~0.9 per 100,000 among Caucasians from the Dallas Heart Study (1 in 106,733; 95% CI: 1 in 76,646 to 1 in 141,790). When combined, the prevalence in the general Caucasian population was determined to be ~0.8 per 100,000 (1 in 128,246; 95% CI: 1 in 92,095 to 1 in 170,369). For the Hispanic population, CESD prevalence was estimated at ~0.6 in 100,000 and ~1.2 in 100,000 in the New York and Dallas cohorts, respectively, and ~0.8 in 100,000 (1 in 126,167; 95% CI: 1 in 90,602 to 1 in 167,607) in the combined Hispanic population.
DISCUSSION
The lack of reported frequency data for the LIPA c.894G>A mutation prompted our genotyping study using 5,000 DNA samples from a multiracial and multi-ethnic, healthy adult population and 3,289 additional multiracial and multi-ethnic adult samples from the Dallas Heart Study. To the best of our knowledge, this is the first report detailing the c.894G>A allele and carrier frequencies in the African-American, Asian, Caucasian, Hispanic, and AJ populations. Importantly, these data allow for predictions of CESD prevalence in these populations, which indicate that CESD may be under-diagnosed in the general population.
Recently, a phase I/II clinical trial of enzyme replacement therapy was conducted with recombinant human LAL in patients with CESD (22). Intravenous administration of enzyme ranging from 0.35 – 3.0 mg/kg proved safe and well-tolerated and resulted in decreased levels of hepatic transaminases in all patients with evidence of tissue lipid mobilization. These encouraging results suggest the future possibility of a treatment for CESD patients. As such, it is important that hepatologists, lipidologists, and pathologists are made aware of the clinical presentation of this under-recognized disease and its frequency to identify potential patients. In particular, hepatologists and pathologists can order the appropriate histologic liver biopsy stains (e.g., immunostaining with a lysosomal membrane protein to document lysosomal cholesteryl ester and triglyceride storage) (15). Additionally, lipidologists can identify some patients with dyslipidemia for LAL enzyme or LIPA gene screening prior to liver biopsy (23–25). These efforts should detect and biochemically and/or molecularly confirm CESD patients who are candidates for enzyme replacement therapy. Thus, the frequencies of the common c.894G>A mutation in different populations can determine those racial and ethnic groups in which the mutation is present or very rare to absent.
Previously, a single c.894G>A screening study reported a 1 in 202 heterozygote frequency in the German population (16). Importantly, our survey of reported CESD patient genotypes indicated that the LIPA c.894G>A mutation accounted for ~60% (95% CI: 51%–69%) of multi-ethnic CESD alleles. However, the majority of ascertained CESD patients were of European ancestry. Consequently, the reported c.894G>A allele frequency (0.0025) translates to a predicted CESD prevalence of ~1.7 in 100,000 (1 in 58,932; 95% CI: 1 in 42,320 to 1 in 78,289) for the German population.
Genotyping the c.894G>A mutation in individuals of different racial and ethnic groups from the New York metropolitan area and from the Dallas Heart Study indicated that c.894G>A frequencies vary between populations and are highest among populations of European ancestry. Interestingly, the c.894G>A frequencies were equally high among Hispanics. The identified carrier frequencies for the combined Caucasian and Hispanic populations were both ~1 in 300, which translated to a predicted CESD prevalence of ~0.8 per 100,000 (~1 in 130,000) using the c.894G>A prevalence among affected multi-ethnic individuals (60%). Importantly, these data suggest that CESD may be under-diagnosed in the general North American population, particularly among Caucasians and Hispanics, as the predicted frequency of affected individuals markedly exceeds the relative paucity of cases reported in the literature. Additionally, selected populations of central European ancestry may have an even higher prevalence of CESD based on the c.894G>A frequency reported for the German population and the number of CESD case reports involving European patients. For example, when combining the New York and Dallas cohorts with the German data, the combined Caucasian carrier frequency increased to 1 in 242, which translated to a predicted CESD prevalence of ~1.2 in 100,000 (1 in 84,250; 95% CI: 1 in 60,501 to 1 in 111,922).
Other lysosomal storage disorders with prevalence estimates similar to that for CESD in the Caucasian and Hispanic populations include metachromatic leukodystrophy (1 in 92,000), Fabry disease (1 in 117,000), mucopolysaccharidosis types I (1 in 88,000), II (1 in 136,000), III-A (1 in 114,000) and IV (1 in 169,000), and Pompe disease (1 in 146,000) (26–30). Given the estimated population of Caucasians and Hispanics currently living in the United States (31), the predicted prevalence of CESD translates to potentially over 3,000 affected Caucasian and Hispanic individuals in the United States at the time of this writing.
Although c.894G>A was rare in the Asian and African-American populations, it is not currently known if this allele is the major CESD-causing mutation in these populations as it is for populations of European decent. This is evidenced by a previously reported African-American CESD patient who did not carry c.894G>A, unlike the majority of reported CESD cases, but instead was a compound heterozygote for two different LIPA missense mutations (p.S289C and p.G342R) (32). Similarly, the recent NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/) also did not identify any c.894G>A heterozygotes in 4,406 African-American chromosomes. Consequently, future studies on the prevalence of CESD in African and Asian populations may require full-gene LIPA sequencing to determine CESD heterozygote frequencies since c.894G>A is not common in these racial groups.
In addition to multi-ethnic population studies on LIPA allele frequencies, future focused studies are warranted that interrogate the c.894G>A and other LIPA mutations among patients with evidence of hepatomegaly and/or steatosis. Since this was not the primary aim of the reported study, limited data were available to assess the relevant phenotypes of identified c.894G>A carriers. However, available data from four patients from the Dallas Heart Study suggest that c.894G>A heterozygotes may have elevated hepatic triglyceride content indicative of steatosis (data not shown), which is supported by the recent identification of increased total cholesterol levels in c.894G>A carriers compared to non-carriers (11). Future LIPA sequencing studies directed at patients with NAFLD, NASH, and/or cryptogenic fatty liver disease will undoubtedly further refine the role of CESD in fatty liver disease.
Supplementary Material
Acknowledgments
The genotyping testing methodology and reagents used in this study were generously provided by Synageva BioPharma (Lexington, MA). The authors thank Dr. Helen H. Hobbs (Department of Molecular Genetics, University of Texas Southwestern Medical Center, Dallas, TX, USA) for use of critical resources funded by her grants RO1DK090066 and PO1 HL20948.
FINANCIAL SUPPORT
This research was supported in part by the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH), through grants KL2TR000069 (S.A.S.) and UL1TR000067 (I.P.).
LIST OF ABBREVIATIONS
- AJ
Ashkenazi Jewish
- CESD
Cholesteryl Ester Storage Disease
- CHD
coronary heart disease
- CI
confidence interval
- E8SJM
exon 8 splice junction mutation
- IRB
institutional review board
- LAL
lysosomal acid lipase
- LIPA
lipase A
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
Footnotes
CONFLICT OF INTEREST
R.J.D. is a consultant for and owns stock options of Synageva BioPharma, the company developing enzyme replacement therapy for LAL deficiency.
Contributor Information
Stuart A. Scott, Email: stuart.scott@mssm.edu.
Benny Liu, Email: beliu007@hotmail.com.
Irina Nazarenko, Email: irina.nazarenko@mssm.edu.
Suparna Martis, Email: suparna.martis@mssm.edu.
Julia Kozlitina, Email: julia.kozlitina@UTSouthwestern.edu.
Yao Yang, Email: yao.yang@mssm.edu.
Charina Ramirez, Email: charina.ramirez@childrens.com.
Yumi Kasai, Email: yumi.kasai@mssm.edu.
Tommy Hyatt, Email: tommy.hyatt@UTSouthwestern.edu.
Inga Peter, Email: inga.peter@mssm.edu.
Robert J. Desnick, Email: robert.desnick@mssm.edu.
References
- 1.Assmann G, Seedorf U. Acid Lipase Deficiency: Wolman Disease and Cholesteryl Ester Storage Disease. In: Valle D, Beaudet AL, Vogelstein B, Kinzler K, Antonarakis S, Ballabio A, editors. OMMBID: The Online Metabolic & Molecular Bases of Inherited Disease. 2001. [Google Scholar]
- 2.Grabowski GA, Charnas L, Du H. Lysosomal Acid Lipase Deficiencies: The Wolman Disease/Cholesteryl Ester Storage Disease Spectrum. In: Valle D, Beaudet AL, Vogelstein B, Kinzler K, Antonarakis S, Ballabio A, editors. OMMBID: The Online Metabolic & Molecular Bases of Inherited Disease. 2010. [Google Scholar]
- 3.Aslanidis C, Klima H, Lackner KJ, Schmitz G. Genomic organization of the human lysosomal acid lipase gene (LIPA) Genomics. 1994;20:329–331. doi: 10.1006/geno.1994.1180. [DOI] [PubMed] [Google Scholar]
- 4.Wolman M. Wolman disease and its treatment. Clin Pediatr (Phila) 1995;34:207–212. doi: 10.1177/000992289503400406. [DOI] [PubMed] [Google Scholar]
- 5.Aslanidis C, Ries S, Fehringer P, Buchler C, Klima H, Schmitz G. Genetic and biochemical evidence that CESD and Wolman disease are distinguished by residual lysosomal acid lipase activity. Genomics. 1996;33:85–93. doi: 10.1006/geno.1996.0162. [DOI] [PubMed] [Google Scholar]
- 6.Hoeg JM, Demosky SJ, Jr, Pescovitz OH, Brewer HB., Jr Cholesteryl ester storage disease and Wolman disease: phenotypic variants of lysosomal acid cholesteryl ester hydrolase deficiency. Am J Hum Genet. 1984;36:1190–1203. [PMC free article] [PubMed] [Google Scholar]
- 7.Klima H, Ullrich K, Aslanidis C, Fehringer P, Lackner KJ, Schmitz G. A splice junction mutation causes deletion of a 72-base exon from the mRNA for lysosomal acid lipase in a patient with cholesteryl ester storage disease. J Clin Invest. 1993;92:2713–2718. doi: 10.1172/JCI116888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muntoni S, Wiebusch H, Funke H, Ros E, Seedorf U, Assmann G. Homozygosity for a splice junction mutation in exon 8 of the gene encoding lysosomal acid lipase in a Spanish kindred with cholesterol ester storage disease (CESD) Hum Genet. 1995;95:491–494. doi: 10.1007/BF00223858. [DOI] [PubMed] [Google Scholar]
- 9.Pagani F, Pariyarath R, Garcia R, Stuani C, Burlina AB, Ruotolo G, Rabusin M, et al. New lysosomal acid lipase gene mutants explain the phenotype of Wolman disease and cholesteryl ester storage disease. J Lipid Res. 1998;39:1382–1388. [PubMed] [Google Scholar]
- 10.Pagani F, Garcia R, Pariyarath R, Stuani C, Gridelli B, Paone G, Baralle FE. Expression of lysosomal acid lipase mutants detected in three patients with cholesteryl ester storage disease. Hum Mol Genet. 1996;5:1611–1617. doi: 10.1093/hmg/5.10.1611. [DOI] [PubMed] [Google Scholar]
- 11.Muntoni S, Wiebusch H, Jansen-Rust M, Rust S, Schulte H, Berger K, Pisciotta L, et al. Heterozygosity for lysosomal acid lipase E8SJM mutation and serum lipid concentrations. Nutr Metab Cardiovasc Dis. 2012 doi: 10.1016/j.numecd.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 12.IBC 50K CAD Consortium. Large-scale gene-centric analysis identifies novel variants for coronary artery disease. PLoS Genet. 2011;7:e1002260. doi: 10.1371/journal.pgen.1002260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coronary Artery Disease (C4D) Genetics Consortium. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet. 2011;43:339–344. doi: 10.1038/ng.782. [DOI] [PubMed] [Google Scholar]
- 14.Wild PS, Zeller T, Schillert A, Szymczak S, Sinning CR, Deiseroth A, Schnabel RB, et al. A genome-wide association study identifies LIPA as a susceptibility gene for coronary artery disease. Circ Cardiovasc Genet. 2011;4:403–412. doi: 10.1161/CIRCGENETICS.110.958728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hulkova H, Elleder M. Distinctive histopathological features that support a diagnosis of cholesterol ester storage disease in liver biopsy specimens. Histopathology. 2012;60:1107–1113. doi: 10.1111/j.1365-2559.2011.04164.x. [DOI] [PubMed] [Google Scholar]
- 16.Muntoni S, Wiebusch H, Jansen-Rust M, Rust S, Seedorf U, Schulte H, Berger K, et al. Prevalence of cholesteryl ester storage disease. Arterioscler Thromb Vasc Biol. 2007;27:1866–1868. doi: 10.1161/ATVBAHA.107.146639. [DOI] [PubMed] [Google Scholar]
- 17.Scott SA, Khasawneh R, Peter I, Kornreich R, Desnick RJ. Combined CYP2C9, VKORC1 and CYP4F2 frequencies among racial and ethnic groups. Pharmacogenomics. 2010;11:781–791. doi: 10.2217/pgs.10.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martis S, Peter I, Hulot JS, Kornreich R, Desnick RJ, Scott SA. Multi-ethnic distribution of clinically relevant CYP2C genotypes and haplotypes. Pharmacogenomics J. 2012 doi: 10.1038/tpj.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scott SA, Edelmann L, Kornreich R, Desnick RJ. Warfarin pharmacogenetics: CYP2C9 and VKORC1 genotypes predict different sensitivity and resistance frequencies in the Ashkenazi and Sephardi Jewish populations. Am J Hum Genet. 2008;82:495–500. doi: 10.1016/j.ajhg.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scott SA, Edelmann L, Liu L, Luo M, Desnick RJ, Kornreich R. Experience with carrier screening and prenatal diagnosis for 16 Ashkenazi Jewish genetic diseases. Hum Mutat. 2010;31:1240–1250. doi: 10.1002/humu.21327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Victor RG, Haley RW, Willett DL, Peshock RM, Vaeth PC, Leonard D, Basit M, et al. The Dallas Heart Study: a population-based probability sample for the multidisciplinary study of ethnic differences in cardiovascular health. Am J Cardiol. 2004;93:1473–1480. doi: 10.1016/j.amjcard.2004.02.058. [DOI] [PubMed] [Google Scholar]
- 22.Jones S, Enns G, Balwani M, Breen C, Sharma R, Deegan P, Malinova V, et al. Recombinant human lysosomal acid lipase (LAL) demonstrates pharmacodynamic activity in cholesteryl ester storage disease (CESD), the late onset form of LAL deficiency Society for the Study of Inborn Errors of Metabolism Annual Symposium; 2012; 2012. Accepted abstract. [Google Scholar]
- 23.Vajro P, Lenta S, Socha P, Dhawan A, McKiernan P, Baumann U, Durmaz O, et al. Diagnosis of nonalcoholic fatty liver disease in children and adolescents: position paper of the ESPGHAN Hepatology Committee. J Pediatr Gastroenterol Nutr. 2012;54:700–713. doi: 10.1097/MPG.0b013e318252a13f. [DOI] [PubMed] [Google Scholar]
- 24.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. 2012;55:2005–2023. doi: 10.1002/hep.25762. [DOI] [PubMed] [Google Scholar]
- 25.Hamilton J, Jones I, Srivastava R, Galloway P. A new method for the measurement of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin Chim Acta. 2012;413:1207–1210. doi: 10.1016/j.cca.2012.03.019. [DOI] [PubMed] [Google Scholar]
- 26.Lowry RB, Applegarth DA, Toone JR, MacDonald E, Thunem NY. An update on the frequency of mucopolysaccharide syndromes in British Columbia. Hum Genet. 1990;85:389–390. doi: 10.1007/BF00206770. [DOI] [PubMed] [Google Scholar]
- 27.Nelson J, Crowhurst J, Carey B, Greed L. Incidence of the mucopolysaccharidoses in Western Australia. Am J Med Genet A. 2003;123A:310–313. doi: 10.1002/ajmg.a.20314. [DOI] [PubMed] [Google Scholar]
- 28.Von Figura K, Gieselmann V, Jacken J. Metachromatic leukodystrophy. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. pp. 3695–3724. [Google Scholar]
- 29.Desnick RJ, Ioannou YA, Eng CM. Alpha-galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Kinzler KE, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Diseases. New York: McGraw-Hill; 2001. pp. 3733–3774. [Google Scholar]
- 30.Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254. doi: 10.1001/jama.281.3.249. [DOI] [PubMed] [Google Scholar]
- 31.United States Census Bureau. [Accessed October 2, 2012];Population and Housing Unit Estimates. http://www.census.gov/popest/index.html.
- 32.Anderson RA, Bryson GM, Parks JS. Lysosomal acid lipase mutations that determine phenotype in Wolman and cholesterol ester storage disease. Mol Genet Metab. 1999;68:333–345. doi: 10.1006/mgme.1999.2904. [DOI] [PubMed] [Google Scholar]
- 33.Redonnet-Vernhet I, Chatelut M, Basile JP, Salvayre R, Levade T. Cholesteryl ester storage disease: relationship between molecular defects and in situ activity of lysosomal acid lipase. Biochem Mol Med. 1997;62:42–49. doi: 10.1006/bmme.1997.2626. [DOI] [PubMed] [Google Scholar]
- 34.Lohse P, Maas S, Elleder M, Kirk JM, Besley GT, Seidel D. Compound heterozygosity for a Wolman mutation is frequent among patients with cholesteryl ester storage disease. J Lipid Res. 2000;41:23–31. [PubMed] [Google Scholar]
- 35.Elleder M, Poupetova H, Ledvinova J, Hyanek J, Zeman J, Sykora J, Stozicky F, et al. Lysosomal acid lipase deficiency. Overview of Czech patients. Cas Lek Cesk. 1999;138:719–724. [PubMed] [Google Scholar]
- 36.Hooper AJ, Tran HA, Formby MR, Burnett JR. A novel missense LIPA gene mutation, N98S, in a patient with cholesteryl ester storage disease. Clin Chim Acta. 2008;398:152–154. doi: 10.1016/j.cca.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 37.Ries S, Buchler C, Schindler G, Aslanidis C, Ameis D, Gasche C, Jung N, et al. Different missense mutations in histidine-108 of lysosomal acid lipase cause cholesteryl ester storage disease in unrelated compound heterozygous and hemizygous individuals. Hum Mutat. 1998;12:44–51. doi: 10.1002/(SICI)1098-1004(1998)12:1<44::AID-HUMU7>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 38.Gasche C, Aslanidis C, Kain R, Exner M, Helbich T, Dejaco C, Schmitz G, et al. A novel variant of lysosomal acid lipase in cholesteryl ester storage disease associated with mild phenotype and improvement on lovastatin. J Hepatol. 1997;27:744–750. doi: 10.1016/s0168-8278(97)80092-x. [DOI] [PubMed] [Google Scholar]
- 39.Fasano T, Pisciotta L, Bocchi L, Guardamagna O, Assandro P, Rabacchi C, Zanoni P, et al. Lysosomal lipase deficiency: molecular characterization of eleven patients with Wolman or cholesteryl ester storage disease. Mol Genet Metab. 2012;105:450–456. doi: 10.1016/j.ymgme.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 40.Maslen CL, Babcock D, Illingworth DR. Occurrence of a mutation associated with Wolman disease in a family with cholesteryl ester storage disease. J Inherit Metab Dis. 1995;18:620–623. doi: 10.1007/BF02436008. [DOI] [PubMed] [Google Scholar]
- 41.Pisciotta L, Fresa R, Bellocchio A, Pino E, Guido V, Cantafora A, Di Rocco M, et al. Cholesteryl Ester Storage Disease (CESD) due to novel mutations in the LIPA gene. Mol Genet Metab. 2009;97:143–148. doi: 10.1016/j.ymgme.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 42.Pagani F, Zagato L, Merati G, Paone G, Gridelli B, Maier JA. A histidine to tyrosine replacement in lysosomal acid lipase causes cholesteryl ester storage disease. Hum Mol Genet. 1994;3:1605–1609. doi: 10.1093/hmg/3.9.1605. [DOI] [PubMed] [Google Scholar]
- 43.Seedorf U, Wiebusch H, Muntoni S, Christensen NC, Skovby F, Nickel V, Roskos M, et al. A novel variant of lysosomal acid lipase (Leu336-->Pro) associated with acid lipase deficiency and cholesterol ester storage disease. Arterioscler Thromb Vasc Biol. 1995;15:773–778. doi: 10.1161/01.atv.15.6.773. [DOI] [PubMed] [Google Scholar]
- 44.Ameis D, Brockmann G, Knoblich R, Merkel M, Ostlund RE, Jr, Yang JW, Coates PM, et al. A 5′ splice-region mutation and a dinucleotide deletion in the lysosomal acid lipase gene in two patients with cholesteryl ester storage disease. J Lipid Res. 1995;36:241–250. [PubMed] [Google Scholar]
- 45.Du H, Sheriff S, Bezerra J, Leonova T, Grabowski GA. Molecular and enzymatic analyses of lysosomal acid lipase in cholesteryl ester storage disease. Mol Genet Metab. 1998;64:126–134. doi: 10.1006/mgme.1998.2707. [DOI] [PubMed] [Google Scholar]
- 46.Elleder M, Chlumska A, Hyanek J, Poupetova H, Ledvinova J, Maas S, Lohse P. Subclinical course of cholesteryl ester storage disease in an adult with hypercholesterolemia, accelerated atherosclerosis, and liver cancer. J Hepatol. 2000;32:528–534. doi: 10.1016/s0168-8278(00)80407-9. [DOI] [PubMed] [Google Scholar]
- 47.vom Dahl S, Harzer K, Rolfs A, Albrecht B, Niederau C, Vogt C, van Weely S, et al. Hepatosplenomegalic lipidosis: what unless Gaucher? Adult cholesteryl ester storage disease (CESD) with anemia, mesenteric lipodystrophy, increased plasma chitotriosidase activity and a homozygous lysosomal acid lipase -1 exon 8 splice junction mutation. J Hepatol. 1999;31:741–746. doi: 10.1016/s0168-8278(99)80356-0. [DOI] [PubMed] [Google Scholar]
- 48.Rassoul F, Richter V, Lohse P, Naumann A, Purschwitz K, Keller E. Long-term administration of the HMG-CoA reductase inhibitor lovastatin in two patients with cholesteryl ester storage disease. Int J Clin Pharmacol Ther. 2001;39:199–204. doi: 10.5414/cpp39199. [DOI] [PubMed] [Google Scholar]
- 49.Drebber U, Andersen M, Kasper HU, Lohse P, Stolte M, Dienes HP. Severe chronic diarrhea and weight loss in cholesteryl ester storage disease: a case report. World J Gastroenterol. 2005;11:2364–2366. doi: 10.3748/wjg.v11.i15.2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tadiboyina VT, Liu DM, Miskie BA, Wang J, Hegele RA. Treatment of dyslipidemia with lovastatin and ezetimibe in an adolescent with cholesterol ester storage disease. Lipids Health Dis. 2005;4:26. doi: 10.1186/1476-511X-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.