

Abstract
Background
Intestinal inflammation has been linked with multi-organ failure in burn and other traumatic injuries. We hypothesized that markers of intestinal inflammation are detectible non-invasively.
Methods
Fecal samples were collected from 7 severely burned patients and 15 control patients for the measurement of inflammatory cytokines using a multiplex assay kit (Bio-Rad). In addition, fecal levels of myeloperoxidase (MPO) and elastase were measured using standard procedures.
Results
Compared to a control group, levels of inflammatory cytokines were significantly increased in the burn group. IL-6 was increased 2.16 ± 0.61 pg/mg, (mean ± SEM) to 3.81 ± 0.49; p<0.05, as were IL-8 (3.32 ± 0.76 pg/mg to 20.51 ± 6.65 p<0.05), IL-12 (4.457 ± 0.98pg/mg to 7.371 ± 0.95 p<0.05), IL-13 (3.86 ± 0.32 pg/mg to 11.83 ± 1.47 p<0.05), MCP-1 (2.78 ± 2.61 pg/mg to 6.5 ± 3.97 p<0.05), MPO (13.41 ± 1.40 units/mg protein to 24.52 ± 4.31 p<0.05) and elastase (2.46 ± 0.38 pg/ml to 5.08 ± 0.72 p<0.05).
Conclusions
Our results suggest that markers of intestinal inflammation are measurable by non-invasive means and are increased following burn injury compared to controls. Of note, increased IL-8 correlated with increased MPO and elastase activity, suggesting a role for neutrophil activation in burn-mediated intestinal inflammation. Thus, these inflammatory cytokine profiles may be valuable biomarkers of intestinal inflammation following burn injury.
Keywords: Thermal Injury, Intestinal Immunity, Inflammation, Fecal Cytokines, Trauma
Introduction
Sepsis and multi-organ dysfunction syndrome (MODS) remain an overwhelming cause of mortality following burn injury. It is well known that burn injury can lead to a marked organ and immune dysfunction that is associated with a distinct increase in the circulatory levels of pro-inflammatory cytokines and chemokines. Although the injury itself is a major insult, several lines of evidence suggest that in the post burn period, the gastrointestinal tract is actually playing a central role in driving organ and immune dysfunction and that it may be the primary source of the mediators of inflammation found in systemic circulation. 1–8 Previous findings from our laboratory have shown that in an animal model of burn injury, an elevation in cytokines, chemokines and neutrophil infiltration exists in the gut. 9–12 Furthermore, we found that an elevation in these inflammatory mediators correlates well with actual tissue damage to the gut itself. 9–12
The human colon is the major reservoir of bacteria within the body. Under healthy conditions, it maintains an effective barrier against luminal bacteria; however, it is known that this barrier is disrupted following burn injury. 1, 3, 12–14 This compromised barrier may play a key part in the process that leads to post-burn organ and immune dysfunction but the exact role is unknown. There is evidence that it is not the actual bacteria themselves that translocate and lead to this immune dysfunction; instead it is the cytokine milieu induced by our immune system’s interaction with these bacteria that seems to be the culprit. Thus the situation that is created by the burn injury is one where we have a permeable gut loaded with intestinal bacteria and this sets the stage for an abnormal immune interaction leading to the production of mediators which can fuel organ and immune dysfunction.
Inflammation of the gut is historically difficult to assess by non-invasive means. Over the last three decades there has been developing interest in fecal markers and their role in the inflammatory bowel diseases. 15, 16 Specifically calprotectin and lactoferrin have been used to successfully predict mucosal disease, healing, response to therapy and relapse. There are new data to suggest a role for fecal markers in the prediction of necrotizing enterocolitis in neonates. 15, 16
Because of the significant evidence for gut dysfunction after burn injury, we hypothesized an observable change in inflammatory mediators in the feces of burn victims, and hope that a non-invasive method of monitoring this process will serve as a window into what is actually happening in the gut, increasing our understanding of the pathophysiology regarding these injuries.
Patients and Methods
Sample Group
This study was approved by our Institutional Review Board for human subjects and informed consent was obtained from all participants. Samples were collected from burn patients admitted to the burn unit at Loyola University Health Sciences Campus, Maywood from December 2010 to November 2011. In the care of severely injured burn patients, we routinely place a Fecal Management System (FMS). These systems facilitate routine care of the patient by allowing the egress of feces to occur into a collection device rather than onto the patient. From December 2010 to November 2011 samples of this effluent were collected from 7 individuals who suffered burn injury over various periods during their admission.
Patients selected for observation met the following criteria: they were adult males and females greater than age 18 with superficial or full-thickness burn injury >10% TBSA, and without pre-existing clinical infection, pre-existing clinical or historical evidence of gastrointestinal illness such as ulcerative colitis or Crohn’s disease, pre-existing clinical or historical evidence of gastrointestinal infection such as clostridium difficile (c.diff) or CMV colitis, not taking antibiotics (other than surgical prophylaxis), without the presence of perforated viscera or peritonitis, any form of stoma, and transplant recipient, AIDS, known immunodeficiency, or taking immune suppressing medications, or disseminated cancer.
Control Group
To examine the feces of burn-injured patients we needed comparable samples from otherwise healthy patients. Patients with ‘physiologically insignificant’ burns, i.e. superficial or partial-thickness burns less than 10% of total body surface area (TBSA) that are located in sensitive areas such as the hands and face are also routinely hospitalized for care. These patients do not require the use of a FMS but instead use the toilet as needed. We used this patient population as a control for comparison to those with significant burn injury. During the aforementioned time period samples from 15 of these patients were collected at a minimum of two time points during the course of their hospitalization. These patients were also subject to the above inclusion and exclusion criteria. This study was approved by the Loyola University Medical Center Institutional Review Board and informed consent was obtained from all study subjects.
Sample Handling/Processing
Patient samples were collected roughly once every 7 days of hospitalization via sterile sample cups and immediately frozen at −80°C. Samples of 50mg were sonicated in phosphate- buffered saline with protease inhibitor (Roche Diagnostics) for two rounds of 15 seconds each and then centrifuged at 10,000 rpm for 15 minutes at 4°C. Supernatant was collected for the measurement of inflammatory mediators using a Bio-Plex Pro Human Cytokine 17-plex Assay (Bio-Rad Hercules, CA) according manufacturer specification.
Myeloperoxidase activity in fecal samples was measured by incubating fecal homogenates (10 μl) in a 96-well plate with 290 μl of 50 mM phosphate buffer, 3 μl of substrate solution containing 20 mg/ml o-dianisidine hydrochloride, and 3 μl of 20 mM H2O2. The reaction was stopped by adding 3 μl of 30% sodium azide. Plates were read at 460nm using Synergy 2 Multi-Mode Microplate Reader.
For the measurement of Elastase, fecal lysates (25 μl) were incubated in a 96-well plate at room temperature for 60 min with 1 mM methoxy-succinyl-alanyl-alanyl-prolyl-valyl-p-nitroanilide (Sigma Chemical), 0.1 M HEPES, and 0.5 M NaCl (pH 7.5) in a total volume of 150 μl. Absorbance was measured at 405 nm. These methods had been used previously by our lab. 9, 11, 17 The protein content of each aliquot of stool sample was measured by standard Biorad (Hercules, CA) assay to which all cytokines and chemokines were normalized and the values are expressed per mg protein.
Results
Table 1 shows a comparison of specific patient characteristics. A total of 7 patients met the inclusion and exclusion criteria. Samples from these patients were collected at various time points with at least one sample in each post-injury week. Samples from 3 separate time points during hospitalization were taken from 4 patients, 1–2 samples were taken from the remaining 3. This results in a burn group of 7 patients in total but yielding 14–17 fecal samples. The mean age of these patients is 58 ± 13 ranging from 31 to 81. Burn types consisted of flame and chemical, with flame being the predominant type (86%). The mean total body surface area was 36 ± 14 and ranged from 17 to 57%. One patient suffered concomitant orthopedic trauma in addition to burn injury. Four patients developed subsequent septic complications as a result of injury, and 6 (86%) died as a result of injury or withdrawal of physiologic support after family decision to withdraw care. Our control group consisted of 15 patients with physiologically insignificant burns yielding a total of 23 fecal samples.
Table 1.
Characteristics of burn injured patients
| Patients (n) | 7 |
|
| |
| Age(y) | 58 ± 13 |
|
| |
| Burn Type | |
| Flame | 6(86) |
| Chemical | 1(14) |
|
| |
| Burn Surface Area (%) | 36 ±14 |
|
| |
| Concomitant Traumatic Injury | 1(14) |
|
| |
| Concomitant Ethanol Intoxication | 3(43) |
|
| |
| Inhalation Injury | 5(71) |
|
| |
| Septic complication | 4(57) |
|
| |
| Patient Mortality | 6(86) |
Data represented as n (%) or mean (± standard deviation).
As described in the methods section we used a Bio-Plex Pro Human Cytokine 17-plex Assay which detects: IL-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12, IL-13, IL-17, G-CSF, GM-CSF, TNF-α, INF-γ, MIP-1, MCP-1β. Of those, only the following cytokines were observed to be measurable at a detectable level in the stool samples: IL-6, IL-7, IL-8, IL-12, IL-13, G-CSF, TNF-α, and INF-γ. Among these, levels of IL-6, IL-12, IL-13 were significantly increased in the burn group as compared with the control. As demonstrated in Figure 1, IL-6 was increased 2.16 ± 0.61 pg/mg, (mean ± SEM) to 3.81 ± 0.49; p<0.05, as was IL-12 (4.457 ± 0.98 pg/mg to 7.371 ± 0.95 p<0.05), and IL-13. (3.86 ± 0.32 pg/mg to 11.83 ± 1.47 p<0.05)
Figure 1.
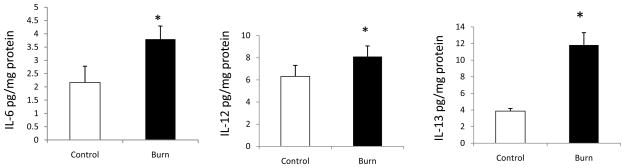
Fecal levels of cytokines following burn injury. Feces collected from patients at various time points after burn injury was used to determine the levels of cytokines using multiplex kit. IL-6 (Panel A), IL-12 (Panel B), and IL-13 (Panel C) are increased in burn groups compared with control groups. Data are shown as means ± SEM from control and burn patients respectively. *P<0.05 compared with control group.
TNF-α and G-CSF were increased from 512±246pg/mg to 933±274 and from 32.52 ± 9.62 to 155.34 ± 58.36 pg/mg in the burn group compared to the control group but this difference was not statistically significant. INF-γ was also increased in the burn compared to control group, 70.85 ± 12.99 pg/mg protein versus 46.67 ± 12.92 but again this difference was not statistically significant.
Figure 2 shows chemokine levels following burn injury. Similar to cytokines, the levels IL-8 and MCP-1 were significantly increased in the feces from burn patients as compared with controls. IL-8 increased from 3.32 ± 0.76 pg/mg to 20.51 ± 6.65 p<0.05 and MCP-1 2.78±0.55pg/mg protein to 6.5±0.99, p<0.05.
Figure 2.

Fecal levels of chemokines IL-8 (Panel A) and MCP-1 (Panel B) following burn injury. Inflammatory chemokines IL-8 and MCP-1 were also increased in the burn group as compared with control groups. Data are shown as means ± SEM from control and burn patients respectively. *P<0.05 compared with control group.
Figure 3 demonstrates the neutrophil markers MPO and elastase. Both MPO and elastase were found to be significantly elevated in samples harvested from burn patients compared to control. The levels of MPO and elastase were elevated from 13.41 ± 1.40 units/mg protein to 24.52 ± 4.31 p<0.05 and 2.46 ± 0.38 pg/ml to 5.08 ± 0.72 p<0.05 respectively.
Figure 3.

Fecal levels of neutrophil markers myeloperoxidase (MPO; Panel A) and Elastase (Panel B) were increased in burn samples in comparison to control samples. Data are shown as means ± SEM from control and burn patients respectively. *P<0.05 compared with control group.
Discussion
The role of the gastrointestinal tract in burn morbidity and mortality has long been recognized. Over the last several decades many advances have been made in burn care such that morbidity and mortality have fallen precipitously. Sepsis remains an overwhelming cause of death and morbidity in burn and other traumatic injury and the role of the gut in inflammation and immunomodulation after burn has been well described. Our results show an increase in IL-6, -8, -12, -13, MCP-1, elastase and MPO in the fecal samples of burned patients as compared to those without physiologically significant burn injury.
We now know that cutaneous burn injury causes gut mucosal atrophy, alters mucosal integrity, and leads to a breakdown in mucosal defense mechanisms. 18–24 This breakdown has been blamed for the translocation of indigenous gut bacteria and the occurrence of sepsis and MOF in burn patients. 1–8 Burn injury also is associated with mesenteric vasoconstriction which results in gut mucosal damage and an increase in bacterial translocation. 25 The gut origin hypothesis of MODS or essentially what is termed ‘bacterial translocation’ initially was based on the concept that gut barrier failure and intestinally derived bacteria and/or endotoxin translocating to the bloodstream and systemic tissues triggered a septic state and promoted the development of MODS. However, conflicting data from human studies indicated that translocating bacteria themselves may not be responsible for the development of gut-induced MODS. 5 A predominant explanation to resolve this notion is the possibility that gut-derived factors contributing to systemic inflammation and organ injury were reaching the systemic circulation via the mesenteric lymphatics rather than the portal venous system. This gut–lymph hypothesis is supported by previous experimental studies indicating that many gut-derived factors, including bacteria, exit the intestine via the intestinal lymphatics rather than the portal blood and that gut origin bacteremia rarely was observed in animal models unless they were highly and rapidly lethal. 13
An important conceptual consequence of the gut–lymph hypothesis is that the lung rather than the liver would be the first major vascular bed to be exposed to mesenteric lymph because mesenteric lymph reaches the systemic circulation via the thoracic duct, which empties into the subclavian vein and hence the pulmonary circulation. This concept that gut-derived toxic and inflammatory factors are reaching the systemic circulation via the intestinal lymphatics with the lung being the first organ exposed to these lymph factors is consistent with extensive evidence documenting a strong link between gut ischemia/injury and the subsequent development of acute lung injury. 13,26,27
Non-invasive assessment of intestinal inflammation was first investigated by those in the field of inflammatory bowel disease who saw tremendous clinical utility in a simple, reliable, reproducible, and non-invasive test with the ability to differentiate IBD from other gastrointestinal conditions, such as IBS, and to monitor disease activity. 15, 16 Fecal markers are a non-invasive way of objectively measuring intestinal inflammation, with the potential to play a primary or adjunctive role in the assessment of disease activity and as such have been evaluated in IBD. To our knowledge, whether intestinal inflammation could be observed non-invasively in burned patients prior to this study has not been determined. A simple, non-invasive way of determining the extent of intestinal inflammation in the burn-injured patient could represent an important adjunct to the existing complement of clinical observational methods available. Based on prior research, we know that intestinal inflammation after burn injury involves various cell types such as dentritic cells, epithelial cells, macrophages, T-cells and neutrophils. While the source of increased fecal cytokines in burned patients is not specifically identified, changes in one or more of the above cell types may contribute to the observed increase.
Our data offer substantial and statistically significant evidence that the inflammatory cytokine profile of fecal effluent differs in burn patients from that of controls. Additionally, the cytokine and chemokine profiles of the burn samples indicate the presence of increased intestinal inflammation. Specifically increased are neutrophil activity and the products of neutrophilic granules MPO and elastase. Neutrophilia and its implication on intestinal permeability is a well-recognized finding in the intestine of burned-injured animals. 11, 12, 14, 26,12,14,28 We are encouraged that these findings indicate comparable evidence of acute inflammation in human burn patients. One potential concern is the possibility of the bowel management system itself causing local inflammation in our sample, which may confound our results. However in clinical practice the BMS is routinely used, and the majority are without complication. Short of erosion, bleeding and necrosis, any degree of inflammatory change due specifically to the indwelling rectal tube should be negligible and not influence the local inflammatory response. An additional concern could be that the burn sample patients received diets that were largely tube feeds as well as stool softening regimens, as opposed to the general diets of the control population. Whether such differential dietary intake impacts the fecal cytokine levels in burn patients compared to controls is beyond the scope of this study and will remain to be established.
We set out to determine whether a non-invasive method could be used to monitor intestinal inflammation in the setting of burn injury. Though we were able to observe changes in cytokine profiles, these can only offer a speculative idea as to what is occurring without direct histologic or radiographic evidence of intestinal injury. As such, the potential for this method represents an adjunct marker that could be used to evaluate a clinical picture overall, rather than a specific diagnostic test. An additional goal was and is to apply our method in such a way that the severity of disturbance in cytokine profile could be correlated with severity of injury. Unfortunately these data did not provide enough support to answer that question. We also hoped to characterize a hypothetical correlation between time course of injury (in this case based upon when each fecal sample was collected) and level of observed inflammatory change. We were unable to find such a correlation. Unquestionably we feel that this failure comes from the small sample size of our burn group. We are confident that as we increase the complement of sample patients, we will have sufficient numbers to properly address these questions.
An additional issue that arises is the significance of the high rate of mortality seen in the burn population and whether the findings of probable intestinal inflammation are in some way related. Notably, 4 of 7 of our burn patients with intestinal inflammation as assumed by the presence of higher levels of inflammatory cytokines in the feces developed infectious complications within 1 month after the samples were collected. This in turn raises the question of whether intestinal inflammation in the setting of burn injury predisposes to, or is perhaps reflective of an impending immune perturbation. A larger retrospective study might then be aimed at correlating (if there is correlation to be found) intestinal inflammation as measured by fecal cytokines with downstream complications.
Conclusion
Sepsis and multi-organ dysfunction are an overwhelming cause of morbidity and mortality in burn-injured patients. Intestinal inflammation is a well-recognized mediator of systemic inflammation and bacterial translocation in burn and traumatic injury. Non-invasive assessment of intestinal inflammation has been used successfully in the IBD population but to date no one had investigated the utility of this method in the burn-injured patient. We hypothesized that non-invasive assessment of the cytokine profile of fecal effluent samples in burn-injured patients would show evidence of inflammation and serve as an important clinical adjunct to existing methods. Our data show that indeed fecal samples of burned patients show increased markers of inflammation. While more studies are needed to confirm this observation with large burn patient population, the findings suggest that fecal levels of inflammatory markers may be considered for determining the gut inflammation after burn injury.
Acknowledgments
The authors would like to thank the physicians and other nurses specially Marcia Halerz, BSN, MBA and Melissa Mounts, BSN at Loyola University Burn Center and the Burn Shock Trauma Institute for their ongoing contributions, without which this study could not have been performed.
Footnotes
This study is supported by NIH grants R01AA015731, F30AA020167 (JLR), Dr. Ralph and Marian C. Falk Medical Research Trust (MS and JLR).
Reference List
- 1.Baron P, Traber LD, Traber DL, Nguyen T, Hollyoak M, Heggers JP, Herndon DN. Gut failure and translocation following burn and sepsis. J Surg Res. 1994;57:197–204. doi: 10.1006/jsre.1994.1131. [DOI] [PubMed] [Google Scholar]
- 2.Baue AE, Durham R, Faist E. Systemic inflammatory response syndrome (SIRS), multiple organ dysfunction syndrome (MODS), multiple organ failure (MOF): are we winning the battle? Shock. 1998;10:79–89. doi: 10.1097/00024382-199808000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Choudhry MA, Fazal N, Namak SY, Haque F, Ravindranath T, Sayeed MM. PGE2 suppresses intestinal T cell function in thermal injury: a cause of enhanced bacterial translocation. Shock. 2001;16:183–8. doi: 10.1097/00024382-200116030-00003. [DOI] [PubMed] [Google Scholar]
- 4.Choudhry MA, Rana SN, Kavanaugh MJ, Kovacs EJ, Gamelli RL, Sayeed MM. Impaired intestinal immunity and barrier function: a cause for enhanced bacterial translocation in alcohol intoxication and burn injury. Alcohol. 2004;33:199–208. doi: 10.1016/j.alcohol.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 5.Deitch EA. Role of the gut lymphatic system in multiple organ failure. Curr Opin Crit Care. 2001;7:92–8. doi: 10.1097/00075198-200104000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Gosain A, Gamelli RL. Role of the gastrointestinal tract in burn sepsis. J Burn Care Rehabil. 2005;26:85–91. doi: 10.1097/01.bcr.0000150212.21651.79. [DOI] [PubMed] [Google Scholar]
- 7.Mainous MR, Ertel W, Chaudry IH, Deitch EA. The gut: a cytokine-generating organ in systemic inflammation? Shock. 1995;4:193–9. [PubMed] [Google Scholar]
- 8.Mannick JA, Rodrick ML, Lederer JA. The immunologic response to injury. J Am Coll Surg. 2001;193:237–44. doi: 10.1016/s1072-7515(01)01011-0. [DOI] [PubMed] [Google Scholar]
- 9.Li X, Rana SN, Schwacha MG, Chaudry IH, Choudhry MA. A novel role for IL-18 in corticosterone-mediated intestinal damage in a two-hit rodent model of alcohol intoxication and injury. J Leukoc Biol. 2006;80:367–75. doi: 10.1189/jlb.1205745. [DOI] [PubMed] [Google Scholar]
- 10.Li X, Schwacha MG, Chaudry IH, Choudhry MA. Heme oxygenase-1 protects against neutrophil-mediated intestinal damage by down-regulation of neutrophil p47phox and p67phox activity and O2- production in a two-hit model of alcohol intoxication and burn injury. J Immunol. 2008;180:6933–40. doi: 10.4049/jimmunol.180.10.6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X, Schwacha MG, Chaudry IH, Choudhry MA. Acute Alcohol intoxication potentiates neutrophil-mediated intestine tissue damage following burn injury. Shock. 2008;29:377–83. doi: 10.1097/shk.0b013e31815abe80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sir O, Fazal N, Choudhry MA, Gamelli RL, Sayeed MM. Neutrophil depletion prevents intestinal mucosal permeability alterations in burn-injured rats. Am J Physiol Regul Integr Comp Physiol. 2000;278:R1224–R1231. doi: 10.1152/ajpregu.2000.278.5.R1224. [DOI] [PubMed] [Google Scholar]
- 13.Magnotti LJ, Deitch EA. Burns, bacterial translocation, gut barrier function, and failure. J Burn Care Rehabil. 2005;26:383–91. doi: 10.1097/01.bcr.0000176878.79267.e8. [DOI] [PubMed] [Google Scholar]
- 14.Sir O, Fazal N, Choudhry MA, Goris RJ, Gamelli RL, Sayeed MM. Role of neutrophils in burn-induced microvascular injury in the intestine. Shock. 2000;14:113–7. doi: 10.1097/00024382-200014020-00006. [DOI] [PubMed] [Google Scholar]
- 15.Judd TA, Day AS, Lemberg DA, Turner D, Leach ST. Update of fecal markers of inflammation in inflammatory bowel disease. J Gastroenterol Hepatol. 2011;26:1493–9. doi: 10.1111/j.1440-1746.2011.06846.x. [DOI] [PubMed] [Google Scholar]
- 16.Lewis JD. The utility of biomarkers in the diagnosis and therapy of inflammatory bowel disease. Gastroenterology. 2011;140:1817–26. doi: 10.1053/j.gastro.2010.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akhtar S, Li X, Chaudry IH, Choudhry MA. Neutrophil chemokines and their role in IL-18-mediated increase in neutrophil O2- production and intestinal edema following alcohol intoxication and burn injury. Am J Physiol Gastrointest Liver Physiol. 2009;297:G340–G347. doi: 10.1152/ajpgi.00044.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fine J, Frank ED, Ravin HA, Rutenberg SH, Schweinburg FB. The bacterial factor in traumatic shock. N Engl J Med. 1959;260:214–20. doi: 10.1056/NEJM195901292600505. [DOI] [PubMed] [Google Scholar]
- 19.Horton JW. Bacterial translocation after burn injury: the contribution of ischemia and permeability changes. Shock. 1994;1:286–90. [PubMed] [Google Scholar]
- 20.Moore FA, Moore EE, Poggetti R, McAnena OJ, Peterson VM, Abernathy CM, Parsons PE. Gut bacterial translocation via the portal vein: a clinical perspective with major torso trauma. J Trauma. 1991;31:629–36. doi: 10.1097/00005373-199105000-00006. [DOI] [PubMed] [Google Scholar]
- 21.Moore FA. The role of the gastrointestinal tract in postinjury multiple organ failure. Am J Surg. 1999;178:449–53. doi: 10.1016/s0002-9610(99)00231-7. [DOI] [PubMed] [Google Scholar]
- 22.Ravin HA, Fine J. Biological implications of intestinal endotoxins. Fed Proc. 1962;21:65–8. [PubMed] [Google Scholar]
- 23.Schweinburg FB, Seligman AM, Fine J. Transmural migration of intestinal bacteria; a study based on the use of radioactive Escherichia coli. N Engl J Med. 1950;242:747–51. doi: 10.1056/NEJM195005112421903. [DOI] [PubMed] [Google Scholar]
- 24.Sori AJ, Rush BF, Jr, Lysz TW, Smith S, Machiedo GW. The gut as source of sepsis after hemorrhagic shock. Am J Surg. 1988;155:187–92. doi: 10.1016/s0002-9610(88)80691-3. [DOI] [PubMed] [Google Scholar]
- 25.Tokyay R, Zeigler ST, Traber DL, Stothert JC, Jr, Loick HM, Heggers JP, Herndon DN. Postburn gastrointestinal vasoconstriction increases bacterial and endotoxin translocation. J Appl Physiol. 1993;74:1521–7. doi: 10.1152/jappl.1993.74.4.1521. [DOI] [PubMed] [Google Scholar]
- 26.Sayeed MM. Exuberant Ca(2+) Signaling in Neutrophils: A Cause for Concern. News Physiol Sci. 2000;15:130–6. [PubMed] [Google Scholar]