

Abstract
Mammalian thioredoxin reductase catalyzes the reduction of the redox-active disulfide bond of thioredoxin and is similar in structure and mechanism to glutathione reductase except for a C-terminal 16 amino acid extension containing a rare vicinal selenylsulfide bond. This vicinal selenylsulfide bond is essentially a substrate for the enzyme’s N-terminal redox center. Here we report the synthesis of peptide substrates for the truncated enzyme missing the C-terminal redox center. We developed a procedure for the synthesis of peptides containing cyclic vicinal disulfide/selenylsulfide bonds as well as their corresponding acyclic heterodimers. Vicinal disulfide bonds form 8-membered ring structures and are difficult to synthesize due to their propensity to dimerize during oxidation. Our procedure makes use of two key improvements for on-resin disulfide bond formation presented previously by Galande and coworkers [Galande AK, Weissleder R, Tung C-H. An effective method of on-resin disulfide bond formation in peptides. J. Comb. Chem. 2005; 7: 174–177.]. First, the addition of an amine base to the deprotection solution allows for complete removal of the StBu group, allowing it to be replaced with a 5-Npys group. The second enhancement is the direct use of a Cys(Mob) or Sec(Mob) derivative as the nucleophilic partner instead of utilizing a naked sulfhydryl or selenol. These improvements result in the formation of a vicinal disulfide (or selenylsulfide) bond in high purity and yield. A direct comparison with the Galande procedure is presented.
We also present a novel strategy for the formation of an acyclic, interchain selenylsulfide linked peptide (linking H-PTVTGC-OH and H-UG-OH). Cysteine analogs of the cyclic and acyclic peptides were also synthesized. The results show that the ring structure contributes a factor of 52 to the rate, but the presence of selenium in the peptide is more important to catalysis than the presence of the ring.
INTRODUCTION
Thioredoxin reductase (TR), thioredoxin (Trx), and NADPH make up the thioredoxin system. This system, along with the glutaredoxin system, functions to maintain cellular redox homeostasis [1]. Mammalian TR has two particularly interesting features. First it contains selenium in the form of selenocysteine (Sec, U) – the 21st amino acid [2], and second this selenocysteine residue is part of a vicinal selenylsulfide bridge during the catalytic cycle. Other eukaryotic homologs have replaced the Sec residue with a conventional cysteine (Cys) residue so that these enzymes contain a rare vicinal disulfide bridge as part of their catalytic cycles [3–4].
In the Brookhaven Protein Data Bank (PDB), a disulfide bond between neighboring Cys residues, henceforth referred to as a “vicinal disulfide bond” (i, i + 1), is a rare occurrence. In a study by Pongor and coworkers, there were only 32 occurrences of this type of disulfide bond from 28,000 deposited structures [5]. The reason for the infrequency of this type of disulfide bond is most likely due to the strain imparted on the intervening peptide bond between adjacent half-cystinyl residues [6]. This strain is due to the existence of an 8-membered ring motif which contains a central amide bond possessing partial double bond character. The ring strain imparts flexional torsion onto the region of a peptide or protein where it is found in nature [7–8]. For example, NMR studies have indicated that the most flexible part of human and bass hepcidin, as well as the Janus-faced atracotoxin (J-ACTX) is the region of the protein where this ring is found [9–11].
This 8-membered ring motif almost always exists as part of a type VIII β-turn, helping to stabilize this type of high energy turn structure [5]. It is also found in various enzymes such as methanol dehydrogenase, mercuric ion reductase, as well as TR [12–14]. In these systems, the Cys-Cys dyad cycles between reduced and oxidized states as part of a redox cycle. In addition to a catalytic role for this motif, it has been proposed that this flexible ring is part of a conformational switch, controlling active and inactive states of the protein. This is apparently the case in the nicotinic acetylcholine receptor where it has been proposed that switching between cis and trans forms of the ring regulates the on/off state of the receptor [7,15]. A regulatory role for this motif has also been proposed for cytochrome C oxidase from Utricularia [16].
Hepcidin, J-ACTX, and a fragment of the acetylcholine receptor (used as a model for the whole protein), are small proteins (18 – 37 residues) that have been synthesized by solid-phase peptide synthesis (SPPS) for biophysical experiments [9, 11, 17]. In these cases, very low yields of the final target product are reported. This is most likely due to the general inefficiency of forming the various disulfide bonds within these proteins. Vicinal disulfide bridges present a difficult challenge for regioselective disulfide bond formation because one might expect polymerization and oligomerization to be preferential to the formation of the strained 8-membered ring.
We have taken a “chemical approach” to studying the enzyme mechanism of TR by breaking it into two pieces [18–19]. This approach is possible because of TR’s structural and mechanistic similarity to glutathione reductase. As shown in Figure 1, TR contains an additional thiol-disulfide exchange step resulting from the presence of a 16 amino acid C-terminal extension containing either a vicinal disulfide bond or vicinal selenylsulfide bond. This additional thiol-disulfide exchange step is in the form of the reduction and opening of the 8-membered ring motif. We have constructed a truncated version of the enzyme lacking the amino acid sequence possessing the ring motif so that we could isolate this ring-opening step from the rest of the catalytic cycle by using peptide disulfides/selenylsulfides as substrates.
Figure 1.

(A) The proposed flow of electrons from NADPH to the disulfide bond of Trx in high Mr TRs, such as the Sec-containing mouse TR and the Cys-containing TR from Drosophila melanogaster. The X (Nuc′) represents either a sulfur atom from Cys or a selenium atom from Sec, which is the presumptive nucleophile for the reduction of Trx. The neighboring Cys residue (labeled as Res′) would then resolve the mixed selenylsulfide or mixed disulfide bond between TR and Trx. The “prime” designation is used to show residues that are on the opposite subunit as TR is a homodimer. (B) Truncated TR/peptide system in which an oxidized peptide (corresponding to the missing C-terminal redox center of the enzyme), acts a substrate for the truncated enzyme.
As part of our interest in this enzyme, we wished to determine what the contributions of ring strain and entropy of this 8-membered ring were to the catalytic cycle. In order to accomplish this, we needed a procedure for making peptides containing vicinal disulfide bonds. We found that other protocols for making disulfide bonds were not applicable to this unique system. Here we report a highly efficient procedure for making such vicinal disulfide/selenylsulfide bonds using a variation of chemistry we have recently reported [20], as well as open chain analogs (e.g. interchain selenylsulfide linked peptide PTVTGC/UG), so that the peptide bond between Cys and Sec/Cys in the open chain form is “hydrolyzed” in comparison with the cyclic form. These peptides were then used in our enzymatic assay of TR to compare the rate of disulfide/selenylsulfide reduction of each peptide substrate. The synthesis of each type of peptide substrate requires new strategies for disulfide and selenylsulfide bond formation that will be of interest to peptide chemists. The results provide a unique insight into the catalytic mechanism of TR that was heretofore unachievable.
MATERIALS AND METHODS
Materials
Fmoc-protected amino acids and HBTU were purchased from Synbiosci Corporation (Livermore, CA). Resins for solid phase peptide synthesis (SPPS) were purchased from Novabiochem (San Diego, CA) and Applied Biosystems (Foster City, CA). Boc-Cys(5-Npys)-OH was purchased from Bachem (King of Prussia, PA). Fmoc-Sec(Mob)-OH was manufactured using a procedure previously reported [21–22]. Solvents for peptide synthesis and all other required reagents were purchased from Fisher Scientific (Pittsburgh, PA). DNA primers were purchased from IDT (Coralville, IA).
Peptide Synthesis
All peptides for this study were synthesized on a Symphony™ multiple peptide synthesizer (Protein Technologies Inc., Tuscon, AZ). via Fmoc protocol, utilizing either 2-chlorotrityl chloride resin, PAL-PEG resin, or Fmoc-Gly-PEG-PS resin. Double coupling using standard HBTU activation was employed for peptide elongation. A typical on-resin coupling procedure is as follows: 20% piperidine/DMF (2 × 10 min); DMF washes (6 × 30 sec); 5 eq. Fmoc amino acid and HBTU in 0.4 M NMM/DMF (2 × 30 min); DMF washes (3 × 30 sec). Cleavage of peptides from their respective resins was accomplished through treatment of the dried resin with 96:2:2 TFA/TIS/H2O for 2 h. Following filtration of the resin, the cleavage supernatant was evaporated to one fifth its original volume in a stream of nitrogen, followed by precipitation into cold anhydrous diethyl ether.
High Pressure Liquid Chromatography (HPLC)
All analytical HPLC chromatography was carried out on a Shimadzu analytical HPLC system which utilized LC-10AD pumps, a SPD-10A UV-Vis detector, and a SCL-10A controller. A Shimadzu Shim-pack™ VP-ODS C18 analytical column (4.6 μm pore size, 150 × 4.6 mm) was used in these separations. Beginning with 100% Buffer A, a 1.4 mL/min gradient elution increase of 1% Buffer B/min for 50 min was used for all peptide chromatograms (Buffer A = 0.1% TFA/H2O; Buffer B = 0.1% TFA/ACN). Peptide elution profiles were detected at 214 nm and 254 nm. Preparative HPLC purification was carried out on a Shimadzu preparatory HPLC system utilizing LC-8A pumps, a SPD-10A UV-Vis detector, and a SCL-10A controller. A Waters SymmetryPrep™ C18 preparatory column (7 μm pore size, 1900 × 150 mm) was used in these separations. Beginning with 100% Buffer A, a 17 mL/min gradient elution increase of 1% Buffer B/min for 50 min was used for all preparative chromatograms.
Mass Spectrometry
MALDI-TOF mass spectrometry data was collected on a Voyager DE-Pro instrument under positive ionization and in reflectron mode. All samples were run using a matrix of 10 mg/mL α-cyano-4-hydroxycinnamic acid (CHCA) vacuum dried from a solution of 1:1 H2O/ACN buffered to 0.1% TFA.
Formation of oxidized vicinal Cys-Sec and Cys-Cys dyad peptides
40 μmol of peptides Boc-PTVTGC(StBu)X(Mob)G were synthesized via the previously described standard Fmoc protocol on preloaded Fmoc-Gly-PEG-PS resin (0.19 mmol/g; carboxylate C-terminus) or PAL-PEG-Fmoc resin (0.20 mmol/g; carboxamide C-terminus), in which X corresponded to either Sec or Cys. α-Fmoc protection was converted to Boc via treatment of the resin with 20% piperidine/DMF (2 × 10 min) followed by incubation with Boc2O (10 eq.) in 5 mL 0.1 M NMM 1:1 DMF/DCM for 40 min. Removal of the StBu protecting group from Cys was accomplished by treating the resin-bound peptides with 20% βME in DMF (buffered to 0.1 M in NMM) (2 × 2h). The resin was washed thoroughly with DMF and DCM, and the deprotected Cys residue was then functionalized and activated through treatment with 2,2′-dithiobis(5-nitropyridine) (DTNP) in DCM (2 × 5 eq; 30 min each) to form the corresponding 5-Npys conjugate. The resin was then thoroughly washed with DCM and DMF in order to remove any residual DTNP, and on-resin cyclization was initiated via treatment with 1% TFA/DCM (3 × 30 min). This treatment was repeated until the absence of any yellow coloration in the supernatant. Following acidolytic cleavage and precipitation of the peptide, preparatory HPLC purification was carried out as previously described.
Formation of “open chain” Cys-Cys disulfide
Interchain peptide linked PTVTGC/CG was synthesized via solution fragment coupling of H-PTVTGC(SH)-OH with H-C(5-Npys)G-OH. The peptide H-PTVTGC(SH)-OH was synthesized via standard Fmoc protocol from 2-chlorotrityl chloride resin (1.40 mmol/g), in which a ~5-fold excess of resin was treated with 40 μmol of Fmoc-Cys(Trt)-OH in 0.1 M NMM/DCM for 40 min. Following this initial loading step, the remaining attachment sites on the resin were capped via treatment with 1:0.5:8.5 MeOH/NMM/DCM for an additional 40 min. The remaining amino acid residues were attached using the previously described standard Fmoc protocol. Following peptide elongation, α-Fmoc removal, and acidolytic cleavage, preparatory HPLC purification of this fragment was carried out as previously described.
The H-C(5-Npys)G-OH fragment was synthesized using standard Fmoc protocol from 2-chlorotrityl chloride resin (1.40 mmol/g). The Fmoc-Gly-OH fragment (40 μmol) was incubated with a ~5-fold excess of resin in 0.1 M NMM/DCM for 40 min, followed by MeOH capping of any unreacted attachment sites as described above. Boc-Cys(5-Npys) was then coupled using the previously-described protocol. Removal of the dipeptide fragment from the resin was effected by treatment of the resin with neat TFA for 20 min. Following evaporation of the TFA solvent, the crude isolate was dissolved in 4 mL H2O and injected directly into preparative HPLC for purification.
Solution fragment coupling was carried out by incubating a 2-fold excess of the H-C(5-Npys)G-OH dipeptide over H-PTVTGC(SH)-OH coupling partner and dissolved in a minimal volume (~4 mL) of pH 5.5 aqueous Et3N buffer. All couplings were complete in 10 min as judged by analytical HPLC. The product was then purified by preparative HPLC.
Formation of “open chain” Cys-Sec selenysulfide and Cys-Cys disulfide
Interchain selenylsulfide linked peptide PTVTGC/UG was synthesized via solution fragment coupling of H-PTVTGC(5-Npys)-OH with H-U(Mob)G-OH in neat TFA. The peptide fragment H-PTVTGC(5-Npys)-OH was synthesized from a resin-bound Fmoc-PTVTGC(StBu) precursor via standard Fmoc protocol from 2-chlorotrityl chloride resin as described above. Following peptide elongation, the α-Fmoc protecting group was converted to a Boc group via treatment of the resin with 20% piperidine/DMF (2 × 10 min) followed by incubation with Boc2O (10 eq.) in 5 mL 0.1 M NMM 1:1 DMF/DCM for 40 min. After removing the StBu protecting group by treatment with βME as described above, the resulting free sulfhydryl was converted to a 5-Npys derivative by treatment of the resin with 10 eq. DTNP in 5 mL DCM for 1 h. Following thorough washing of the resin with DCM, the peptide was cleaved from the resin with neat TFA and immediately purified by preparatory HPLC as previously above.
H-U(Mob)G-OH was synthesized using standard Fmoc protocol from 2-chlorotrityl chloride resin similarly to the C(5-Npys)G fragment described above, using Fmoc-U(Mob) as the second residue instead of Boc-C(5-Npys). Following α-Fmoc removal, the dipeptide fragment was released from the resin using 1% TFA/DCM and concentrated. This fragment was utilized immediately in the following coupling procedure without further purification.
Fragment coupling was carried out by incubating an approximate 6-fold excess of the H-U(Mob)G-OH dipeptide over H-PTVTGC(5-Npys)-OH coupling partner in a minimal volume (~2 mL) of TFA for 1 h. Following evaporation of the TFA supernatant in a stream of nitrogen, the residue was precipitated in cold anhydrous diethyl ether. The crude isolate was then dissolved in 4 mL H2O and purified by preparative HPLC.
Cloning of truncated mouse TR
A truncated form of TR (missing the C-terminal 8 amino acids- TRtrunc), was produced by amplifying the coding region (via PCR) of TR corresponding to the desired truncated product. For this experiment, plasmid pTR3, which contained the full-length sequence of the mouse thioredoxin reductase-3 gene (accession #AF171053), was used as the template DNA. The PCR was performed on a Perkin-Elmer GenAmp PCR System 2400 (Life Sciences Inc., Boston, MA) and contained 100 ng template DNA, 50 pmol of upstream and downstream primers, 200 μM dNTP’s, 2U of Vent DNA polymerase, which is supplied with Thermo pol buffer, in a final volume of 100 μL. The PCR cycle consisted of initial denaturation at 96°C for 90 sec, followed by 25 cycles of 96°C for 45 sec, 50°C for 30 sec, and 72°C for 190 sec, and then chilled to 4°C. The upstream primer was of sequence 5′–AACAGACCATGGGAGGGCAGCAGAGCTTTG-3′ and the downstream primer was of sequence 5′–ACAGCCGCTCTTCAGCATTACTCCAGGCCGGAGCGCTTGGAGAT-3′. The reaction product was verified by analytical agarose electrophoresis and purified via the QIAquick PCR Purification Kit from Qiagen (Valencia, Ca) according to the product manual. The sequence was verified by the University of Vermont DNA Sequencing Facility using an ABI 3100-Avant Genetic Analyzer. The PCR product was then inserted into plasmid pTYB3 using restriction enzymes Nco I and Sap I to produce a plasmid capable of producing the recombinant enzyme in E. coli cells. Production and purification of the recombinant enzyme has been previously described [18].
Assay of Cyclic and Acyclic Peptides as Substrates for TRtrunc
Peptide turnover rates were monitored on a Varian-Cary 50 spectrophotometer by a decrease in absorbance at 340 nm. This loss in absorbance indicates consumption of NADPH. The assay contained 50 mM potassium phosphate, pH 7.0, 1 mM EDTA, 150 μM NADPH, 0.0–7.5 mM interchain selenylsulfide linked peptide PTVTGC/UG, 0.0–20.0 mM for cyclic peptides PTVTGCXG(ox) (X = Cys or Sec), or 0.0–20.0 mM for interchain disulfide linked peptide PTVTGC/CG. The reaction was monitored for 2 min and the data was fit to a straight line to calculate the change in absorbance (Δ340 nm/min). The activity is reported as the number of moles of NADPH oxidized per min per mole of enzyme using an extinction coefficient of 6.22 mM−1•cm−1 for NADPH.
RESULTS AND DISCUSSION
In order to do the type of enzymatic assay for this study, we needed two different types of peptides: intramolecular cyclic disulfides/selenylsulfides and intermolecular acyclic disulfides/selenylsulfides. Although Kamber [23] has previously reported synthesizing a vicinal disulfide peptide containing a pair of Cys(Acm) derivatives using I2 as the oxidant, we have found that I2 is not a good reagent for peptides containing Sec because of a high degree of deselenization that occurs during oxidation. In order to achieve the highest purity and yield upon cleavage from the solid-support, we envisioned an on-resin oxidation/deprotection procedure.
We have previously reported that we could simultaneously deprotect and oxidize a short vicinal selenylsulfide bond peptide of sequence Cys(5-Npys)-Sec(Mob)-Gly-PAL resin on the solid support [20]. However, similar success was not achieved when we attempted to extend this same procedure to a longer 8-mer peptide containing neighboring Cys and Sec residues. As our earlier report had been inspired by work done by Galande and coworkers for making disulfide bonds on-resin, we decided to undertake a careful step-by-step analysis of their protocol [24].
In this procedure, two (non-vicinal) Cys side chains in the peptide disulfide precursor are orthogonally protected as their StBu and Mmt conjugates. The synthetic approach begins with treating the peptide attached to the solid support with a 20% βME/DMF solution to reductively remove the StBu group. The free sulfhydryl is then treated with excess DTNP in DCM to convert the Cys residue into a Cys(5-Npys) derivative. Treatment of the resulting resin with a 1% TFA/DCM solution serves the dual purpose of Mmt removal from the attacking sulfhydryl group as well as to activate the mixed disulfide bond of the electrophilic Cys(5-Npys) group by protonating the pyridyl nitrogen atom, resulting in directed disulfide formation.
Our initial attempts at using the previously established procedure of Galande for synthesizing peptide PTVTGCCG(ox) (peptide I in Table 1) resulted in the formation of a complex product mixture (Figure 2A). Careful MALDI analysis of the analytical HPLC peaks in the chromatogram showed minimal cyclic, monomeric-oxidized product, with most of the chromatogram populated by oligomers, StBu adducts, and other uncharacterized components, implying an inefficient removal of the StBu protecting group at the beginning of the reaction sequence. Further, we found that the StBu group was recalcitrant to removal even after extended incubation with excess βME (Supplementary Data). It was hypothesized that by carrying out the βME deprotection step in 0.1 M NMM/DMF, the process would be accelerated due to the higher proportion of βME thiolate at the increased pH. Incubation of a small amount of resin with cleavage cocktail after 90 min of treatment using these augmented conditions showed complete removal of the StBu group from Cys as demonstrated by MALDI-MS analysis (Supplementary Data).
Table 1.
Structure of the C-terminal 8-mer peptides used as substrates for the truncated mouse TR in this study. Shown in the table are the cyclized vicinal (disulfide/selenylsulfide) and interchain (acyclic) forms of the peptide.
| Peptides | %Yield | |
|---|---|---|
| Cyclic Peptides |
(I) |
24 |
|
(II) |
14 | |
|
(III) |
84 | |
|
(IV) |
53 | |
| Acyclic Peptide |
(V) |
87 |
|
(VI) |
79 |
Figure 2.

(A) Procedure described by Galande and coworkers [24]. HPLC chromatogram of the crude isolate of peptide I – PTVTGCCG(ox) following cleavage from the resin. The synthesis of this peptide did not include addition of an amine base in the StBu deprotection step. Virtually no desired product is formed in this reaction. (B) Modified Galande procedure in which an amine base is added to the StBu deprotection step. HPLC chromatogram of the crude isolate of peptide I following cleavage from the resin. The cyclized product comprises 44% of the crude yield, while 41% corresponds to a 5-Npys-conjugated impurity and 12% corresponds to dimerization products. (C) HPLC chromatogram of the crude isolate of peptide I following cleavage from the resin using our modified procedure which includes addition of amine base in the StBu deprotection step and use of a Cys(Mob) nucleophile (rather than a Cys(SH) sulfhydryl). The cyclized product comprises 81% of the crude yield, while 6% corresponds to dimerization products. The solid line is A214 and the dashed line is A254.
A comparison of Figure 2B with Figure 2A illustrates how crucial the addition of amine base in the StBu deprotection step of the precursor peptide is to the final outcome of the synthesis. The addition of base to the deprotection step results in a significantly simplified chromatogram and greatly increased yield of the desired product. While this modified procedure did improve the synthesis, the desired oxidized product was only present as 44% of the crude mixture. The remaining peaks were identified as oligomeric side products and a large amount of impurity identified in the chromatogram as Peak A (representing 41% of the total area). The HPLC chromatogram clearly showed this peak having an enhanced absorbance at 254 nm, and once isolated exhibited a deep yellow color. Both of these facts are consistent with the existence of a 5-Npys-conjugated impurity.
We hypothesized (based on our earlier work) that these undesired products were due to the presence of a free sulfhydryl generated by deprotection of Cys(Mmt) upon addition of the 1% TFA solution. We believed we could use a protected Cys(Mob) derivative directly as the nucleophile to achieve cyclization with concomitant deprotection. While thioethers have been used previously as nucleophiles [25–27] (especially in the formation of sulfonium cations), their utility as a nucleophile is often overlooked. We then combined the strategy of using a Cys(Mob) nucleophile and the addition of exogenous base to assist in removing the StBu group. This resulted in the formation of a vicinal disulfide bond in very high purity for peptide I, as shown in Figure 2C. The chromatogram shows the peak corresponding to the desired cyclized product comprises 81% of the total area with only 6% dimer present and none of the 5-Npys conjugated impurity.
In peptide chemistry the use of S-Mob nucleophiles, in combination with Cys(5-Npys) electrophiles, is an especially useful combination. The Mob protecting group “tunes” the sulfur atom to the appropriate nucleofugacity for disulfide bond formation, while providing adequate protection during Fmoc SPPS. In the formation of a vicinal disulfide bond, the Mob group aligns the nucleophile in correct position for its attack on the neighboring mixed disulfide, leading to selective formation of the vicinal disulfide, while preventing unwanted side reactions.
These results illustrate two deficiencies in the Galande protocol. It is well known that the StBu group is difficult to remove using mercaptans [28–29], and without the addition of a base our results show that it is unlikely to be removed, consequently little disulfide bond formation will be observed. Also, on-resin disulfide bond formation makes use of a phenomenon known as pseudodilution. Pseudodilution has the effect of promoting intramolecular disulfide bond formation and suppressing intermolecular disulfide bond formation (i.e. dimer and oligomer formation). However in the case of a vicinal disulfide bond, the effect of pseudodilution is diminished due to the energetic unfavorability of ring formation, even at low resin loadings of 0.18 mmol/g utilized here.
This modified procedure was also extended to the synthesis of peptide PTVTGCUG(ox) (peptide II). Here the nucleophile is the selenium atom of a Sec(Mob) group, and though the selenium atom is a stronger nucleophile than sulfur, the requirement for addition of an organic base to the deprotection solution is also present as shown by the results in Figures 3A and B. If NMM is omitted from the deprotection solution, the result is incomplete removal of the StBu group from the adjacent Cys residue promoting significant dimer formation and other side reactions. The neighboring nucleophile will prefer to attack the highly electrophilic, mixed disulfide bond of a Cys(5-Npys) group (on a nearby chain) in comparison to the neighboring mixed disulfide formed in a Cys(StBu) derivative. This effect is most likely enhanced by the difficulty in making a vicinal disulfide bond (Figure 4).
Figure 3.

(A) HPLC chromatogram of the crude isolate of peptide II – PTVTGCUG(ox) following cleavage from the resin using the procedure of Galande for on-resin selenylsulfide bond formation. Note the resulting complicated HPLC chromatogram. (B) HPLC chromatogram of the crude isolate of peptide II following cleavage from the resin using our modified procedure to form resin-bound selenylsulfide peptide product. This synthesis included addition of an amine base in the StBu deprotection step. Cyclized product comprises 71% of the crude yield, while only 12% corresponds to dimerization products. The retention times of cyclized product are slightly different due to a shallower solvent gradient in the HPLC method for (A). The solid line is A214 and the dashed line is A254
Figure 4.

Generalized scheme for formation of a vicinal disulfide/selenylsulfide bond. The key feature of the reaction is that the attacking nucleophile must be either a Cys(Mob) or Sec(Mob) derivative. The Mob group helps to align the nucleophile in the correct orientation for attack on the neighboring mixed disulfide (modeling data not shown). When the neighboring Cys residue is protected in the form of a StBu mixed disulfide, very little vicinal disulfide bond formation occurs (this is true even in the case of a Sec(Mob) nucleophile). Only when each Cys(StBu) mixed disulfide bond has been replaced with a Cys(5-Npys) mixed disulfide bond, will a homogenous product result.
It was suspected that the low yields reported in Table 1 for peptides I and II were due to diketopiperazine formation following Fmoc deprotection of the second residue in the sequence because the identity of the C-terminal residue was glycine-OH [30]. Since this phenomenon is specific for ester-based solid supports, it was decided to carry out a synthesis of a series of carboxamide-terminated peptides using an amide-based resin (PAL-PEG) of similar loading capacity. Peptide amide isolates derived from this resin protocol exhibited greatly increased yields (84% and 53% for peptides III and IV – Table 1).
We then began to develop strategies for the synthesis of interchain peptides of form PTVTGC/XG(ox) peptides. While Cys/Cys interchain peptides have been synthesized previously, there is no such report for an interchain Cys/Sec peptide, most likely due to the difficulty of handling a Sec residue. Our initial attempts at constructing this sequence envisioned a solid-phase fragment condensation in which a solution of H-PTVTGC(SH)-OH was slurried with an excess of the resin-bound Boc-X(5-Npys)G, initiating a “peptide capture” scenario. This strategy resulted in poor yields for both the Cys/Cys and Cys/Sec interchain peptides.
We reasoned that a more straightforward approach be undertaken involving a solution-phase fragment coupling of a nucleophilic and electrophilic pair. For the Cys/Cys interchain disulfide, coupling partners H-PTVTGC(SH)-OH and H-C(5-Npys)G-OH were incubated in aqueous buffer at pH 5.5 to yield the desired open-chain peptide (peptide V) in significantly higher yields than our earlier strategy (Figure 5). At this pH, the reaction was specific only for disulfide formation. It was important to utilize at least a two-fold excess of the electrophilic H-C(5-Npys)G-OH coupling partner in the reaction in order to prevent subsequent dimerization (disulfide) of the H-PTVTGC(SH)-OH fragment. Injection of the entire reaction contents into the preparative HPLC allowed for purification of the product as well as collection of unreacted H-C(5-Npys)G-OH coupling partner. Care was taken to minimize exposure of the peptide to light in order to avoid decomposition of the compound.
Figure 5.

Successful aqueous solution fragment coupling between H-PTVTGC(SH)-OH and H-Cys(5-Npys)G-OH at pH 5.5. An excess of the electrophilic H-Cys(5-Npys)G-OH fragment ensured no H-PTVTGC(SH)-OH dimer formation via a disproportionation reaction with the open-chain product.
During the course of our “peptide capture” strategy, we had difficulty converting Sec(Mob) to Sec(5-Npys) on-resin, we thus decided to reversethe roles of nucleophile and electrophile in the fragment coupling of the Cys/Sec interchain peptide (Figure 6). However, instead of deprotecting the H-U(Mob)G-OH to yield the free selenol (H-U(SeH)G-OH) as the nucleophilic coupling partner, we reasoned that the a peptide containing a Cys(5-Npys) derivative could serve as deprotection substrate for a Sec(Mob) containing peptide, resulting in selenylsulfide bond formation with concomitant Mob removal (Table 2). Using this strategy of using the selenoether as the nucleophile, we were successfully able to couple the two fragments together to form the interchain selenylsulfide linked peptide in high yield (peptide VI). For successful coupling and minimization of side reactions, the reaction time was minimized to 1 h and then immediately injected onto a preparative HPLC.
Figure 6.

Unusual fragment coupling between H-PTVTGC(5-Npys)-OH and H-Sec(Mob)G-OH in neat TFA to yield the interchain Cys-Sec acyclic peptide variant. Coupling partners in this case have exchanged electrophilic and nucleophilic character, with the larger fragment possessing the 5-Npys-activated sulfhydryl. The Sec residue on the dipeptide was left protected as the Mob derivative.
Table 2.
Deprotection substrates for Sec(Mob) residues. The small molecule disulfide DTNP as demonstrated in our previous study [20] or peptides containing Cys(5-Npys) derivatives. The latter results in simultaneous deprotection with concomitant selenylsulfide bond formation.
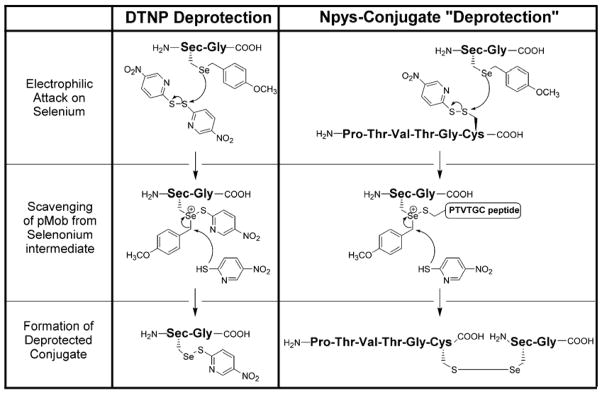
|
We believe that the unique methodology described above is very useful for the creation of selenylsulfide linked peptide fragments. If one considers classic thiol-disulfide exchange chemistry, then the “obvious” strategy would be to deprotect the Sec residue and use the free selenol as the nucleophilic partner, which was our original intent. However, there are several complications inherent in working with Sec-containing peptides that makes such a strategy difficult. First, selenols are extremely sensitive to oxidation and carrying out these manipulations under anaerobic conditions are inherently problematic. Secondly, the purification of selenol-containing peptides is difficult because it has been established that these peptide systems are prone to adhering to RP-HPLC columns, greatly reducing their yield (P.E. Dawson, personal communication). The strategies presented here minimize these problems.
Having developed unique strategies for the synthesis of peptides I, II, V, and VI, we were then able to use them as substrates for TRtrunc. The results of our kinetic assay using these peptide substrates (Table 3) show that the cyclic selenium-containing peptide II is a better substrate than the corresponding acyclic analog (peptide VI) by a factor of 52. In comparison, the cyclic Cys-containing peptide I is only a slightly better substrate than peptide V, its acyclic analog (1.5 fold). Given the uniqueness of the 8-membered ring structure in nature, one might find it surprising that the acyclic selenium-containing peptide VI is a much better substrate than the cyclic Cys-containing peptide I (167 fold). The results indicate that while the ring contributes significantly to catalysis, the presence of a selenium atom in the peptide substrate contributes more to the rate.
Table 3.
Summary of Peptide Turnover Rates using TRtrunc.
| Peptide | Rate (min−1•mM−1)a | |
|---|---|---|
| Se-peptides | (II) PTVTGCUG(ox) – cyclic | 260.0 (± 16) |
| (VI) PTVTGC/UG – acyclic | 5.0 (± 0.3) | |
| S-peptides | (I) PTVTGCCG(ox) – cyclic | 0.03 (± 0.002) |
| (V) PTVTGC/CG – acyclic | 0.02 (± 0.001) | |
| S-substrate | DTNB – acyclic | 3500 (± 230) |
Second order rate constant.
Why might this be so? Figure 1B illustrates how the oxidized peptide acts as a substrate for the truncated form of the enzyme [18–19]. Here we have “disconnected” the C-terminal peptide containing the 8-membered selenylsulfide from the enzyme, allowing us to isolate this one step in the enzymatic cycle. Once the enzyme reduces Trx to form the 8-membered ring, it must be opened by thiol-disulfide exchange. The attacking thiolate can either attack the sulfur atom of the ring or the selenium atom. We have previously proposed that, during the ring opening step of the enzymatic cycle, the Sec residue occupies the leaving group position (see Figure 7) [19]. The reason that the presence of selenium in the peptide is more important than the ring structure to rate can be explained if leaving group pKa is the predominant factor in the ring opening. This point is illustrated clearly by the use of 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) as a substrate for TRtrunc. DTNB is a highly reactive, acyclic disulfide that is turned over at a comparable rate to peptide II – PTVTGCUG(ox) (kcat of 2905 min−1 for DTNB vs. 2058 min−1 for the peptide). However, DTNB binds more tightly to the enzyme resulting in a ten-fold lower Km (0.8 mM vs. 7.9 mM), producing a ten-fold difference in the second order rate constant (Table 3). Yet DTNB is a better substrate than the cyclic, sulfur-containing peptide I by a factor of 105. The leaving group pKa of the thiol of TNB- is 4.75 [31], comparable to that of a selenol (ca. 5.2). while that of a typical thiol is 8.5 [32]. Thus both DTNB and PTVTGCUG(ox) (peptide II) are good substrates for the enzyme because they contain either a highly reactive, polarized disulfide bond with a low pKa leaving group (DTNB), or a reactive selenylsulfide bond with a low pKa leaving group (selenol of the peptide). A combination of ring strain and entropic effects (contributing a factor of 52 to the rate) in the cyclic peptide undoubtedly enhances the reactivity of the less polarized selenylsulfide bond, making its reactivity comparable to that of DTNB. Based upon leaving group pKa, these results support attack at the sulfur atom, placing selenium in the leaving group position in the ring opening step (Figure 7). TRs that contain sulfur in place of selenium [3] must have evolved a mechanism to protonate the leaving group in this step.
Figure 7.

Opening of the 8-membered ring in TR by thiol-disulfide exchange. The attacking thiolate (from CysIC IC = interchange) can attack either the sulfur atom or the selenium atom of the ring. Since leaving group pKa is the predominating factor for this reaction (Table 3), the results support pathway 1 (placing Se in the leaving group position) as the favored pathway. CysCT forms a charge transfer complex with the bound flavin of the enzyme.
CONCLUSIONS
Peptides containing vicinal disulfide bridges are difficult to synthesize due to the strain present in the small ring, thus regioselective pairing strategies tend to result in the formation of dimers and other oligomers. We have presented here a simple and efficient procedure for the formation of vicinal disulfide/selenylsulfide bridges that is based upon two key modifications of a procedure developed by Galande and coworkers for the synthesis of disulfide bonds on-resin, namely the use of a base to facilitate the removal of the StBu group in order to convert the Cys(StBu) group to a Cys(5-Npys) group, and the use of Cys(Mob) and Sec(Mob) nucleophiles. We believe that the modifications we have reported here for on-resin disulfide bond formation will be widely applicable to the method of Galande [24].
We have also developed a unique strategy for the synthesis of interchain selenylsulfide bonds linking Cys and Sec residues that utilizes a Sec(Mob) nucleophile and a peptide containing a Cys(5-Npys) group as a deprotection substrate (Table 2), resulting in simultaneous formation of a selenylsulfide bond with concomitant removal of the Mob group.
The new strategies described here have allowed us to use the synthesized peptides in an enzymatic assay with TRtrunc. While the ring is mildly important to catalysis for the mammalian enzyme, the presence of a selenium atom in the peptide substrate is the predominating factor governing turnover rate of the peptide.
Supplementary Material
Acknowledgments
We wish to thank Ms. Katharine Harris for her previous contributions in our laboratory on disulfide bond forming reactions.
Footnotes
These studies were supported by National Institutes of Health Grants GM070742 to RJH.
References
- 1.Holmgren A, Johansson C, Berndt C, Lonn ME, Hudemann C, Lillig CH. Thiol redox control via thioredoxin and glutaredoxin systems. Biochem Soc Trans. 2005;33:1375–1377. doi: 10.1042/BST0331375. [DOI] [PubMed] [Google Scholar]
- 2.Bock A, Forchhammer K, Heider J, Leinfelder W, Sawers G, Veprek B, Zinoni F. Selenocysteine: the 21st amino acid. Mol Microbiol. 1991;5:515–520. doi: 10.1111/j.1365-2958.1991.tb00722.x. [DOI] [PubMed] [Google Scholar]
- 3.Kanzok SM, Fechner A, Bauer H, Ulschmid JK, Muller HM, Botella-Munoz J, Schneuwly S, Schirmer R, Becker K. Substitution of the thioredoxin system for glutathione reductase in Drosophila melanogaster. Science. 2001;291:643–646. doi: 10.1126/science.291.5504.643. [DOI] [PubMed] [Google Scholar]
- 4.Lacey BM, Hondal RJ. Characterization of mitochondrial thioredoxin reductase from C. elegans. Biochem Biophys Res Commun. 2006;346:629–36. doi: 10.1016/j.bbrc.2006.05.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carugo O, Cemazar M, Zahariev S, Hudaky I, Gaspari Z, Perczel A, Pongor S. Vicinal disulfide turns. Protein Eng. 2003;16:637–639. doi: 10.1093/protein/gzg088. [DOI] [PubMed] [Google Scholar]
- 6.Ruggles EL, Hondal RJ. Synthesis and properties of disulfide-bond containing eight membered rings. Tet Lett. 2006;47:4281–4284. doi: 10.1016/j.tetlet.2006.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Creighton CJ, Reynolds CH, Lee DH, Leo GC, Reitz AB. Conformational analysis of the eight-membered ring of the oxidized cysteinyl-cysteine unit implicated in nicotinic acetylcholine receptor ligand recognition. J Am Chem Soc. 2001;123:12664–12669. doi: 10.1021/ja016505m. [DOI] [PubMed] [Google Scholar]
- 8.Avizonis DZ, Farr-Jones S, Kosen PA, Basus VJ. Conformations and dynamics of the essential cysteinyl-cysteine ring derived from the acetylcholine receptor. J Am Chem Soc. 1996;118:13031–13039. [Google Scholar]
- 9.Hunter HN, Fulton DB, Ganz T, Vogel HJ. The solution structure of human hepcidin, a peptide hormone with antimicrobial activity that is involved in iron uptake and hereditary hemochromatosis. J Biol Chem. 2002;277:37597–37603. doi: 10.1074/jbc.M205305200. [DOI] [PubMed] [Google Scholar]
- 10.Lauth X, Babon JJ, Stannard JA, Singh S, Nizet V, Carlberg JM, Ostland VE, Pennington MW, Norton RS, Westerman ME. Bass hepcidin synthesis, solution structure, antimicrobial activities and synergism, and in vivo hepatic response to bacterial infections. J Biol Chem. 2005;280:9272–9282. doi: 10.1074/jbc.M411154200. [DOI] [PubMed] [Google Scholar]
- 11.Wang X-H, Connor M, Smith R, Maciejewski MW, Howden MEH, Nicholson GM, Macdonald CJ, King GF. Discovery and characterization of a family of insecticidal neurotoxins with a rare vicinal disulfide bridge. Nat Struct Biol. 2000;7:505–513. doi: 10.1038/75921. [DOI] [PubMed] [Google Scholar]
- 12.Blake CC, Ghosh M, Harlos K, Avezoux A, Anthony C. The active site of methanol dehydrogenase contains a disulphide bridge between adjacent cysteine residues. Nat Struct Biol. 1994;1:102–105. doi: 10.1038/nsb0294-102. [DOI] [PubMed] [Google Scholar]
- 13.Moore MJ, Miller SM, Walsh CT. C-terminal cysteines of Tn501 mercuric ion reductase. Biochemistry. 1992;31:1677–1685. doi: 10.1021/bi00121a015. [DOI] [PubMed] [Google Scholar]
- 14.Zhong L, Arner ES, Holmgren A. Structure and mechanism of mammalian thioredoxin reductase: the active site is a redox-active selenolthiol/selenenylsulfide formed from the conserved cysteine-selenocysteine sequence. Proc Natl Acad Sci US A. 2000;97:5854–5859. doi: 10.1073/pnas.100114897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhat AA, Taylor EW. Structural evaluation of distant homology. A 3-D model of the ligand binding domain of the nicotinic acetylcholine receptor based on acetylcholinesterase: Consistency with experimental data. J Mol Model. 1996;2:46–50. [Google Scholar]
- 16.Laakkonen L, Jobson RW, Albert VA. A new model for the evolution of carnivory in the bladderwort plant (utricularia): adaptive changes in cytochrome C oxidase (COX) provide respiratory power. Plant Biol (Stuttg) 2006;8:758–764. doi: 10.1055/s-2006-924459. [DOI] [PubMed] [Google Scholar]
- 17.Zeng H, Moise L, Grant MA, Hawrot E. The solution structure of the complex formed between alpha-bungarotoxin and an 18-mer cognate peptide derived from the alpha 1 subunit of the nicotinic acetylcholine receptor from Torpedo californica. J Biol Chem. 2001;276:22930–22940. doi: 10.1074/jbc.M102300200. [DOI] [PubMed] [Google Scholar]
- 18.Eckenroth B, Harris K, Turanov AA, Gladyshev VN, Raines RT, Hondal RJ. Semisynthesis and characterization of mammalian thioredoxin reductase. Biochemistry. 2006;45:5158–5170. doi: 10.1021/bi0517887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eckenroth BE, Rould MA, Hondal RJ, Everse SJ. Structural and biochemical studies reveal differences in the catalytic mechanisms of mammalian and Drosophila melanogaster thioredoxin reductases. Biochemistry. 2007;46:4694–4705. doi: 10.1021/bi602394p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harris KM, Flemer S, Jr, Hondal RJ. Studies on deprotection of cysteine and selenocysteine side-chain protecting groups. J Pept Sci. 2007;13:81–93. doi: 10.1002/psc.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hondal RJ, Nilsson BL, Raines RT. Selenocysteine in native chemical ligation and expressed protein ligation. J Am Chem Soc. 2001;123:5140–5141. doi: 10.1021/ja005885t. [DOI] [PubMed] [Google Scholar]
- 22.Hondal RJ, Raines RT. Semisynthesis of proteins containing selenocysteine. Methods Enzymol. 2002;347:70–83. doi: 10.1016/s0076-6879(02)47009-7. [DOI] [PubMed] [Google Scholar]
- 23.Kamber B. Cystine peptide from (S-acetamidomethyl-cysteine) peptide through oxidation with iodine: The synthesis of cyclo-L-cystine. Helv Chim Acta. 1971;54:927–930. doi: 10.1002/hlca.19710540319. [DOI] [PubMed] [Google Scholar]
- 24.Galande AK, Weissleder R, Tung C-H. An effective method of on-resin disulfide bond formation in peptides. J Comb Chem. 2005;7:174–177. doi: 10.1021/cc049839r. [DOI] [PubMed] [Google Scholar]
- 25.Cattalini L, Martelli M, Kirschner G. Nucleophilicity of thioethers toward neutral platinum (II) complexes. Inorganic Chem. 1968;7:1488–1492. [Google Scholar]
- 26.Grant SA, Chaudhary K, Stewar L, Peters A, Delisle S, Decken A. Synthesis of a “twisted” transition-state analogue of biotin. Tet Lett. 2004;45:1777–1780. [Google Scholar]
- 27.Yamamoto K, Kobayashi S, Shouji E, Tsuchida E. Oxidative coupling of methyl phenyl sulfide via sulfonium formation using an oxovanadium complex. J Org Chem. 1996;61:1312–1913. [Google Scholar]
- 28.Denis B, Triflieff E. Synthesis of palmitoyl-thioester T-cell epitopes of myelin proteolipid protein (PLP). Comparison of two thiol protecting groups (StBu and Mmt) for on-resin acylation. J Pept Sci. 2000;6:372–377. doi: 10.1002/1099-1387(200008)6:8<372::AID-PSC264>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 29.Moroder L, Gemeiner M, Goehring W, Jaeger E, Thamm P, Wunsch E. New synthesis of somatostatin according to the S-tert-butylthiocysteine procedure. Biopolymers. 1981;20:17–37. doi: 10.1002/bip.1981.360200103. [DOI] [PubMed] [Google Scholar]
- 30.Chan WC, White PD. Fmoc Solid Phase Peptide Synthesis: A Practical Approach. Oxford University Press; New York, NY: 2000. pp. 26–27. [Google Scholar]
- 31.Danehy JP, Elia VJ, Lavelle CJ. The alkaline decomposition of organic disulfides. IV. A limitation on the use of Ellman’s Reagent, 2,2′Dinitro-5,5′-dithiodibenzoic acid. J Org Chem. 1971;36:1003–1005. [Google Scholar]
- 32.Huber RE, Criddle RS. Comparison of the chemical properties of selenocysteine and selenocystine with their sulfur analogs. Arch Biochem Biophys. 1967;122:164–173. doi: 10.1016/0003-9861(67)90136-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.