

Abstract
We previously reported the discovery of BRD0476 (1), a small molecule generated by diversity-oriented synthesis that suppresses cytokine-induced β-cell apoptosis. Herein, we report the synthesis and biological evaluation of 1 and analogs with improved aqueous solubility. By replacing naphthyl with quinoline moieties, we prepared active analogs with up to a 1400-fold increase in solubility from 1. In addition, we demonstrated that compound 1 and analogs inhibit STAT1 signal transduction induced by IFN-γ.
INTRODUCTION
Type-1 diabetes (T1D) is a chronic metabolic disorder that 78,000 children worldwide develop each year.1 This disease is characterized by autoimmune destruction of pancreatic β-cells, resulting in decreased insulin production. The secretion of pro-inflammatory cytokines by macrophages in the pancreatic islets of Langerhans is believed to trigger intracellular signaling cascades leading to β-cell apoptosis.2 Small molecules that restore β-cell viability in the presence of cytokines may have potential to augment insulin replacement therapy. However, relatively few small molecules are known to possess protective effects from cytokines.3–6
In a campaign to discover new therapies and probes for T1D, our group developed a phenotypic assay using the rat INS-1E β-cell line to screen for small-molecule suppressors of apoptosis in the presence of the cytokines tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interferon-γ (IFN-γ).7 High-throughput screening identified several hits that increased β-cell viability. Subsequent medicinal-chemistry optimization of a screening hit belonging to a library derived from diversity-oriented synthesis (DOS)8–10 led to the discovery of BRD0476 (1, ML18711) (Figure 1), a small molecule with sub-micromolar activity (EC50 = 0.78 μM).12 1 represents a novel chemotype with a highly functionalized and stereochemically rich medium-sized (8-membered) lactam ring.13–14
Figure 1.
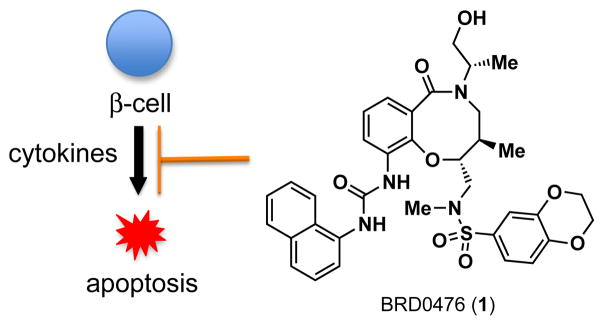
DOS-generated BRD0476 (1) suppresses cytokine-induced β-cell apoptosis.
Compound 1 exhibits poor aqueous solubility, which would adversely affect the bioavailability of 1 in animals and limit its use in vivo. In this study, we describe our efforts toward the synthesis and biological evaluation of analogs of 1 with improved solubility. In addition, we show that the biological activity of 1 and analogs largely results from inhibiting cell-signaling pathways activated by IFN-γ.
RESULTS AND DISCUSSION
To rapidly access compound 1 and urea analogs, we developed an efficient, solution-phase synthesis of aniline 4 in four steps from 2, a DOS-generated intermediate previously prepared using an aldol-based “build/couple/pair” strategy (Scheme 1).12–13 Treatment of 2 with TBSOTf resulted in a protecting group switch from a N-Boc-carbamate to a N-silyl-carbamate intermediate, which was desilylated (TBAF) to afford a secondary crude amine. Direct cleavage of the Boc-carbamate to a free amine under acidic conditions was not feasible due to competing deprotection of the p-methoxybenzyl (PMB) ether. Capping the amine intermediate with 1,4-benzodioxan-6-sulfonyl chloride provided sulfonamide 3 in 74% isolated yield for three steps. Hydrogenation (H2, Pd/C) of the nitro moiety generated 4 in gram quantities.
Scheme 1. Synthesis of advanced aniline intermediate 4.a.

aReagents and conditions: (a) TBSOTf, 2,6-lutidine, CH2Cl2; (b) TBAF, AcOH, THF; (c) 1,4-benzodioxan-6-sulfonyl chloride, 2,6-lutidine, CH2Cl2, 74% yield (3 steps); (d) H2, Pd/C, EtOH, 91% yield.
Two strategies were pursued to install the urea moiety for compound 1 and analogs. One strategy involved addition of isocyanates directly to aniline 4. However, these reactions were sluggish and required microwave heating under basic conditions to achieve moderate yields at best. Furthermore, quinolyl isocyanates were not commericially available for the preparation of quinoline urea analogs, and their syntheses proved problematic using standard protocols for isocyanate formation. Instead, the urea moiety could be installed using a two-step protocol by subjecting 4 to diphosgene (ClCO2CCl3) to form isocyanate intermediate 5,15 followed by addition of various aryl and alkyl amines (Scheme 2). Isocyanate 5 did not require purification for further reactions and could be stored at −20 °C for relatively short periods of time (at least one week). Moreover, addition of amines to 5 was facile and proceeded at room temperature under neutral conditions to afford PMB-protected urea derivatives 6 in moderate to excellent yields (36–99%). Deprotection of the PMB ether with DDQ or TFA provided 1 and urea analogs 7 (see Table 1 for complete structures).
Scheme 2. General synthesis of compound 1 and urea analogs.a.

aReagents and conditions: (a) diphosgene (ClCO2CCl3), proton sponge, CH2Cl2, 96% yield; (b) R2NH, PhMe, 36–99% yield; (c) DDQ, pH 7.4 buffer, CH2Cl2, 63% and 50% yield (for 1 and 7b, respectively); (d) TFA, CH2Cl2, 44–87% yield (for 7a,c–i).
Table 1.
SAR and solubility for compound 1 and analogs.
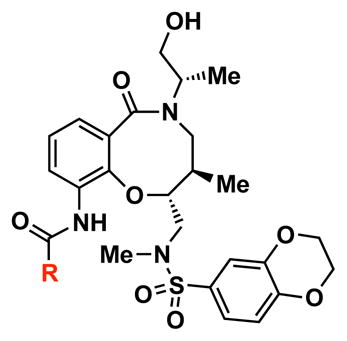
| ||||
|---|---|---|---|---|
| cmpd | R | IC50 (μM)a | maximum activity (%)b | solubility (μM)c |
| 1 |
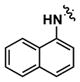
|
1.66 | 71 | 0.1 |
| 7a |
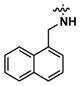
|
>20 | n.d.d | 0.1 |
| 7b |
|
4.74 | 15 | 11.2 |
| 7c |
|
1.05 | 21 | 0.7 |
| 7d |
|
4.10 | 31 | 84.9 |
| 7e |
|
>20 | n.d. | 30.3 |
| 7f |
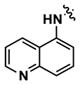
|
1.62 | 41 | 138.5 |
| 7g |
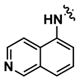
|
4.65 | 59 | 60.5 |
| 7h |
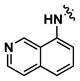
|
n.d. | n.d. | 101.3 |
| 7i |
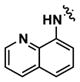
|
3.26 | 69 | 1.8 |
IC50 values were determined using three replicates in the caspase-3/7 assay in INS-1E β-cells.
Maximum activity is reported as % rescue from cytokine-induced caspase-3/7 activation.
Solubility was measured in a phosphate buffered saline solution (pH 7.4).
n.d., not determined.
In our earlier report,12 we determined the potency and maximum activity of compound 1 and analogs using the CellTiter-Glo viability assay with INS-1E β-cells.16 In the present study, caspase-3/7 activity was measured as a more direct readout of apoptosis. 1 demonstrated a small protective effect with standard concentrations of cytokines in the caspase assay (condition A, Figure 2). Lowering the concentrations of IL-1β and TNF-α by 20- and 10-fold, respectively, as well as increasing IFN-γ 4-fold resulted in higher rescue efficacy of 1, despite lowering caspase-3/7 activity for this mixture (condition B). Further increasing the concentration of IFN-γ to 500 ng/mL resulted in equal levels of cytokine-induced caspase-3/7 activation, but elicited the strongest protective effects from 1, reducing caspase activity by 74% (condition C). Using these cytokine concentrations, 1 generated a dose-response with an IC50 value of 1.66 μM and a maximum activity of 71% (Table 1). We used this assay for the biological evaluation of additional analogs.
Figure 2.
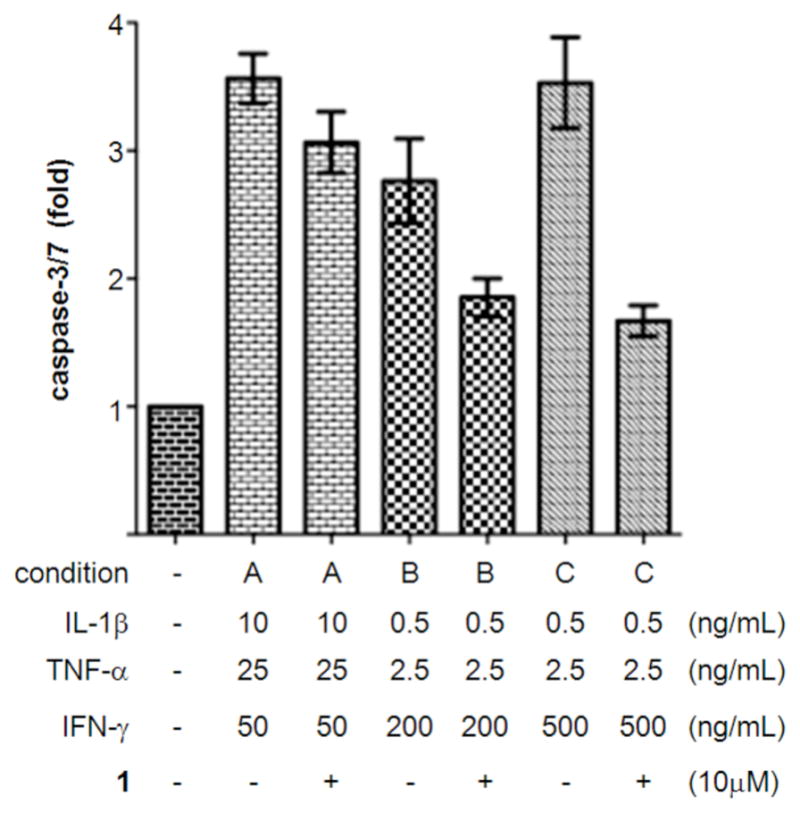
Optimization of cytokine concentrations for the caspase-3/7 assay in INS-1E β-cells. Effects of cytokines and compounds were measured after 48 h. Data represent the mean ± standard error for three independent experiments with six replicates.
Using a thermodynamic solubility assay, the aqueous solubility of compound 1 was determined to be 0.1 μM in phosphate buffered saline (pH 7.4) (Table 1). This low solubility may be attributed to the lipophilic naphthyl moiety. In previous SAR studies, this group was found to be a crucial structural requirement in suppressing cytokine-induced β-cell apoptosis.12 An approach to increase aqueous solubility while retaining the naphthyl moiety involved disrupting molecular planarity between the naphthyl and urea groups to lower crystal packing energy of solutes.17 1-Naphthylmethyl urea 7a was designed to disrupt planarity by interrupting delocalization of the naphthyl- and urea-conjugated pi system with a methylene spacer. However, 7a did not show enhanced solubility and was inactive.
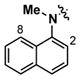
N-Methyl-N-1-naphthyl urea 7b was also designed to increase aqueous solubility by disrupting molecular planarity. The addition of the methyl group to the urea nitrogen may prevent access to planar conformations, which should be hindered by a steric interaction between the N-methyl and C(2)- or C(8)-hydrogen of the quinoline moiety. In fact, 7b exhibited a 110-fold increase in solubility (11.2 μM), which correlated to a decreased melting point for 7b (144–146 °C) compared to 1 (170–172 °C). However, 7b was less efficacious in inhibiting apoptosis (maximum activity = 15%)
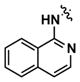
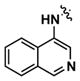
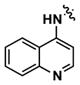
Alternatively, quinoline analogs 7c–i should retain the steric properties of the naphthyl urea moiety crucial for biological activity, while increasing aqueous solubility. Introduction of nitrogen atoms would decrease the lipophilicity of the naphthyl component and provide an additional hydrogen bond acceptor that should enhance solubility.18–19 Indeed, quinoline analogs 7c–i demonstrated an increase in solubility, in which 5-quinolyl urea 7f showed a remarkable 1400-fold increase in solubility compared to 1. 7f was equally potent (1.62 μM) to 1 in the caspase assay but had a lower maximum activity (41%). Likewise, the exceptionally soluble 4-isoquinolyl (7d) and 5-isoquinolyl (7g) ureas exhibited similar biological activity. 1-Isoquinolyl (7c) and 8-quinolyl (7i) ureas were less soluble (0.7 and 1.8 μM, respectively) than other quinoline analogs, which may be due to hydrogen bonding of the urea hydrogen (N-H) and quinoline nitrogen. However, 7i possessed the best activity among the quinoline analogs, inhibiting apoptosis similarly to 1 with a favorable maximum activity of 69%.
As indicated in the optimization of cytokine concentrations for the caspase-3/7 assay, compound 1 and active analogs may directly perturb cell-signaling pathways induced by IFN-γ. This cytokine activates the JAK/STAT signaling pathway,20–21 and STAT1 has previously been shown to regulate apoptosis in INS-1E β-cells.22–23 Binding of IFN-γ to cell surface receptors leads to activation of JAK1/JAK2, which subsequently phosphorylate cytoplasmic STAT1 at Tyr701. Upon phosphorylation, STAT1 dimerizes and translocates to the nucleus to regulate gene transcription.
When INS-1E β-cells were treated with IFN-γ, rapid phosphorylation of STAT1 at 30 min was observed by Western blotting (Figure 3). In addition to Tyr701, phosphorylation was also observed at Ser727.20–21 Co-treatment with IFN-γ and compound 1 led to a complete reduction of STAT1 phosphorylation, while inactive analog 7a did not result in inhibition. Further, total STAT1 levels were slightly increased by IFN-γ alone or in combination with 1 and analogs. Taken together, these results suggest that STAT1 phosphorylation may be an excellent biomarker for compound activity. To this end, the 8-quinolyl urea analog 7i with improved aqueous solubility also significantly decreased levels of pSTAT1 in an analogous manner to 1.
Figure 3.
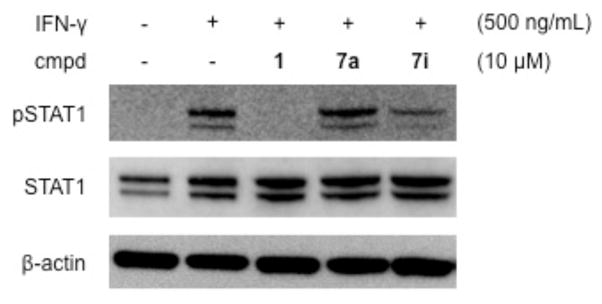
Regulation of STAT1 phosphorylation by IFN-γ, 1, and analogs in INS-1E β-cells. Effects of cytokines and compounds were analyzed by Western blot after 30 min.
To examine the effect of inhibiting STAT1 phosphorylation on subsequent nuclear translocation, cells were treated with IFN-γ in the presence or absence of compounds, fixed, and immunofluorescence was performed to monitor subcellular localization of STAT1 (Figure 4).24 STAT1 was located in both the cytoplasm and nucleus of untreated cells, while treatment with IFN-γ induced a substantial accumulation of STAT1 in the nucleus. Remarkably, compounds 1 and 7i nearly abolished this nuclear localization of STAT1. As a negative control, compound 7a did not inhibit nuclear translocation of STAT1 in the presence of IFN-γ.
Figure 4.
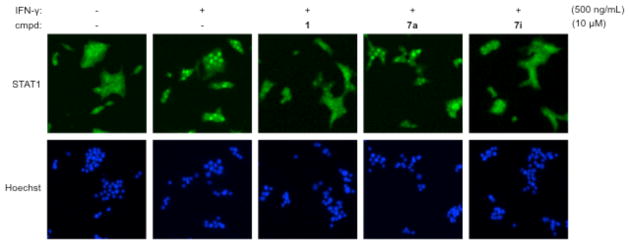
Regulation of STAT1 nuclear translocation by IFN-γ, 1, and analogs in INS-1E β-cells. Effects of cytokines and compounds were assessed after 30 min by immunofluorescence imaging.
CONCLUSION
In summary, we developed an efficient synthesis of compound 1 and analogs, which suppress cytokine-induced β-cell apoptosis. Quinoline-containing analogs showed improved aqueous solubility from 1, of which 8-quinolyl urea 7i retained optimal biological activity. In addition, we have demonstrated that these small molecules inhibit the JAK-STAT signaling pathway by inhibiting IFN-γ-induced STAT1 phosphorylation and nuclear translocation. Studies to improve the pharmacokinetic properties of 1 and 7i, as well as develop a quantitative immunofluorescence assay for STAT1 nuclear translocation, are ongoing and will be described in subsequent reports.
EXPERIMENTAL SECTION
Chemistry
The purity of all compounds was >95% as determined by UPLC-MS. The synthesis of 3, 4, and 7i is described below. See Supporting Information for detailed syntheses and spectral characterization of all compounds.
N-(((2R,3R)-5-((S)-1-((4-methoxybenzyl)oxy)propan-2-yl)-3-methyl-10-nitro-6-oxo-3,4,5,6-tetrahydro-2H-benzo[b][1,5]-oxazocin-2-yl)methyl)-N-methyl-2,3-dihydrobenzo[b][1,4]dioxine-6-sulfonamide (3)
TBSOTf (1.98 mL, 8.61 mmol) was added dropwise to a solution of N-Boc-protected amine 212 (1.6 g, 2.87 mmol) and 2,6-lutidine (1.35 mL, 11.59 mmol) in CH2Cl2 (29 mL) at rt. The reaction mixture was stirred at rt for 1h, diluted with sat. NH4Cl, and extracted into CH2Cl2. The organic layers were dried over MgSO4, filtered, and concentrated in vacuo to provide a N-silyl carbamate intermediate. LCMS (ESI+) m/z: 616.39 (M+H). This crude intermediate was dissolved in THF (57 mL) and cooled to 0 °C. To the resulting solution was added AcOH (0.181 mL, 3.16 mmol) followed by dropwise addition of TBAF (1.0 M in THF, 3.16 mL, 3.16 mmol). The reaction mixture was further stirred for 30 min at 0 °C, diluted with sat. NH4Cl, and extracted into EtOAc. The organic layers were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo to provide a secondary amine. LCMS (ESI+) m/z: 458.41 (M+H). This crude intermediate and 2,6-lutidine (1.67 mL, 14.35 mmol) were dissolved in CH2Cl2 (29 mL), and 1,4-benzodioxan-6-sulfonyl chloride (808 mg, 3.44 mmol) was added to the resulting solution at rt. The reaction mixture was further stirred for 20 h at rt, diluted with sat. NH4Cl, and extracted into CH2Cl2. The organic layers were dried over MgSO4, filtered, and concentrated in vacuo. Purification on silica gel (gradient of 10–50% EtOAc in hexanes) provided 1.39 g (2.12 mmol, 74% yield for 3 steps) of 3 as a white foam. 1H NMR (300 MHz, CDCl3) δ 7.86 (dd, J = 8.1, 1.2 Hz, 1H), 7.70 (dd, J = 7.5, 1.5 Hz, 1H), 7.30-7.21 (ovrlp m, 5H), 6.95 (d, J = 8.4 Hz, 1H), 6.84 (d, J = 8.7 Hz, 2H), 4.55 (d, J = 11.7 Hz, 1H), 4.47 (d, J = 11.7 Hz, 1H), 4.39 (m, 1H), 4.29 (m, 4H), 3.90-3.77 (ovrlp m, 3H), 3.77 (ovrlp s, 3H), 3.66 (dd, J = 9.9, 4.5 Hz, 1H), 3.38-3.20 (ovrlp m, 2H), 3.11 (d, J = 12.6 Hz, 1H), 2.81 (s, 3H) 2.70 (m, 1H), 1.38 (d, J = 6.9 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H); LCMS (ESI+) m/z: 656.38 (M+H).
N-(((2R,3R)-10-amino-5-((S)-1-((4-methoxybenzyl)oxy)propan-2-yl)-3-methyl-6-oxo-3,4,5,6-tetrahydro-2H-benzo[b][1,5]oxazocin-2-yl)methyl)-N-methyl-2,3-dihydrobenzo[b][1,4]dioxine-6-sulfonamide (4)
Under hydrogen (H2) atmosphere, a solution of 3 (1.39 g, 2.12 mmol) and palladium (10% on activated carbon, 226 mg, 0.212 mmol) in EtOH (42 mL) was stirred at 35°C for 3.5 h. The reaction mixture was cooled, filtered, and washed through a pad of celite, and concentrated in vacuo. Purification on silica gel (gradient of 10–60% EtOAc in hexanes) provided 1.21 g (1.93 mmol, 91% yield) of 4 as a white foam. 1H NMR (300 MHz, CDCl3) δ 7.34-7.26 (ovrlp m, 4H), 7.02 (d, J = 8.4 Hz, 1H), 6.97 (d, J = 7.5 Hz, 1H), 6.85 (d, J = 8.7 Hz, 2H), 6.79 (d, J = 7.8 Hz, 2H), 4.67 (m, 1H), 4.54 (d, J = 11.4 Hz, 1H), 4.47-4.43 (ovrlp m, 3H), 4.36-4.33 (ovrlp m, 4H), 3.81-3.74 (ovrlp m, 2H), 3.79 (ovrlp s, 3H), 3.67 (dd, J = 10.2, 4.5 Hz, 1H), 3.49 (dd, J = 15.6, 10.5 Hz, 1H), 3.30 (d, J = 13.8 Hz, 1H), 3.06 (d, J = 15.0 Hz, 1H), 2.94-2.86 (ovrlp m, 1H), 2.90 (ovrlp s, 3H), 2.01 (m, 1H), 1.32 (d, J = 6.9 Hz, 3H), 0.83 (d, J = 6.9 Hz, 3H); HRMS (ESI+) m/z calculated for C32H39N3O8SNa (M+Na): 648.2356, found 648.2352.
N-(((2R,3R)-5-((S)-1-hydroxypropan-2-yl)-3-methyl-6-oxo-10-(3-(quinolin-8-yl)ureido)-3,4,5,6-tetrahydro-2H-benzo[b][1,5]oxazocin-2-yl)methyl)-N-methyl-2,3-dihydrobenzo[b][1,4]dioxine-6-sulfonamide (7i)
To a solution of disphosgene (11 μL, 0.095 mmol) in CH2Cl2 (0.5 mL) at 0°C was added dropwise a solution of aniline 4 (100 mg, 0.160 mmol) and proton sponge (67 mg, 0.314 mmol) in CH2Cl2 (0.5 mL). The reaction mixture was warmed to rt, stirred for 15 min, and concentrated in vacuo. The residue was redissolved in CH2Cl2, washed with 1M HCl and 1M NaOH, dried over MgSO4, filtered, and concentrated in vacuo to provide 120 mg (0.184 mmol, 96% yield) of isocyanate 5. LCMS (ESI+) m/z: 652.25 (M+H). Crude 5 (80 mg, 0.123 mmol) and 8-aminoquinoline (89 mg, 0.615 mmol) were dissolved in toluene (2.1 mL) and stirred at rt for 2.5 h. The reaction mixture was concentrated in vacuo and directly purified on silica gel (gradient of 0–5% MeOH in CH2Cl2) to provide 88 mg (0.111 mmol, 90% yield) of quinolyl urea 6i. LCMS (ESI+) m/z: 796.37 (M+H). To a solution of 6i (53 mg, 0.067 mmol) in CH2Cl2 (2.3 mL) was added TFA (1.1 mL) dropwise at rt. The reaction mixture was further stirred at rt for 15 min, concentrated in vacuo, and directly purified on silica gel (gradient of 0–5% MeOH in CH2Cl2) to provide the 39 mg of 7i (0.058 mmol, 87% yield). 1H NMR (300 MHz, CDCl3) δ 9.63 (s, 1H), 8.86 (d, J = 2.7 Hz, 1H), 8.49 (d, J = 7.2 Hz, 1H), 8.27 (d, J = 8.1 Hz, 1H), 8.18 (s, 1H), 8.07 (dd, J = 6.6 Hz, 3.0 Hz, 1H) 7.53 (ovrlp m, 3H), 7.32 (d, J = 1.8 Hz, 1H), 7.26 (ovrlp dd, J = 8.1 Hz, 1.5 Hz, 1H), 7.16 (ovrlp m, 2H), 6.85 (d, J = 8.4 Hz, 1H), 5.65 (br s, 1H), 4.22 (ovrlp m, 4H), 4.10 (m, 1H), 3.78 (dd, J = 11.1 Hz, 2.1 Hz, 1H), 3.79-3.47 (ovrlp m, 4H), 3.31 (d, J = 13.5 Hz, 1H), 3.02 (ovrlp d, J = 15.6 Hz, 1H), 2.95 (ovrlp s, 3H), 2.26 (m, 1H), 1.41 (d, J = 6.9 Hz, 3H), 0.83 (d, J = 6.3 Hz, 3H); HRMS (ESI+) m/z calculated for C34H38N5O8S (M+H): 676.2441, found 676.2441.
Supplementary Material
Acknowledgments
Financial support from the NIH-NIDDK (Type 1 Diabetes Pathfinder Award, to B.K.W.) is gratefully acknowledged. K.P. was sponsored in part by a NIH/NIGMS MARC U*STAR T34 08663 National Research Award and the Howard Hughes Medical Institute (HHMI) Undergraduate Science Education Program at UMBC. We thank Tamara Gilbert for experimental assistance as well as Dr. Danny Chou, Dr. Jeremy Duvall, and Prof. Stuart Schreiber (Broad Institute) for helpful discussions.
ABBREVIATIONS
- JAK
Janus kinase
- STAT
signal transducer and activation of transcription
- T1D
type-1 diabetes
Footnotes
The authors declare no competing financial interest.
Supporting Information. New compound characterization and procedures for compound synthesis, solubility determination, and biological assays. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.International Diabetes Federation. IDF Diabetes Atlas. 5. Brussels, Belgium: International Diabetes Federation; 2011. http://www.idf.org/diabetesatlas. [Google Scholar]
- 2.Grunnet LG, Aikin R, Tonnesen MF, Paraskevas S, Blaabjerg L, Størling J, Rosenberg L, Billestrup N, Maysinger D, Mandrup-Poulsen T. Pro-inflammatory cytokines activate the intrinsic apoptotic pathway in β-cells. Diabetes. 2009;58:1807–1815. doi: 10.2337/db08-0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matsuda T, Ferreri K, Todorov I, Kuroda Y, Smith CV, Kandeel F, Mullen Y. Silymarin protects pancreatic β-cells against cytokine-mediated toxicity: implication of c-Jun NH2-terminal kinase and janus kinase/signal transducer and activator of transcription pathways. Endocrinology. 2005;146:175–185. doi: 10.1210/en.2004-0850. [DOI] [PubMed] [Google Scholar]
- 4.Kim EK, Kwon KB, Song MY, Han MJ, Lee JH, Lee YR, Ryu DG, Park BH, Park JW. Flavonoids protect against cytokine-induced pancreatic β-cell damage through suppression of nuclear factor kappaB activation. Pancreas. 2007;35:e1–9. doi: 10.1097/mpa.0b013e31811ed0d2. [DOI] [PubMed] [Google Scholar]
- 5.Larsen L, Tonnesen M, Ronn SG, Storling J, Jorgensen S, Mascagni P, Dinarello CA, Billestrup N, Mandrup-Poulsen T. Inhibition of histone deacetylases prevents cytokine-induced toxicity in β-cells. Diabetologia. 2007;50:779–789. doi: 10.1007/s00125-006-0562-3. [DOI] [PubMed] [Google Scholar]
- 6.Chou DH-C, Holson EB, Wagner FF, Tang AJ, Maglathlin RL, Lewis TA, Schreiber SL, Wagner BK. Chem Biol. 2012;19:669–673. doi: 10.1016/j.chembiol.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chou DH-C, Bodycombe NE, Carrinski HA, Lewis TA, Clemons PA, Schreiber SL, Wagner BK. Small-molecule suppressors of cytokine-induced β-cell apoptosis. ACS Chem Biol. 2010;5:729–734. doi: 10.1021/cb100129d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schreiber SL. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science. 2000;287:1964–1969. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- 9.Burke MD, Schreiber SL. A planning strategy for diversity-oriented synthesis. Angew Chem Int Ed. 2004;43:46–58. doi: 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]
- 10.Nielsen TE, Schreiber SL. Towards the optimal screening collection: a synthesis strategy. Angew Chem Int Ed. 2008;47:48–56. doi: 10.1002/anie.200703073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.This compound is registered with the NIH Molecular Libraries Program (probe ML187) and is available upon request.
- 12.Chou DH-C, Duvall JR, Gerard B, Liu H, Pandya BA, Suh B-C, Forbeck EM, Faloon P, Wagner BK, Marcaurelle LA. Synthesis of a novel suppressor of β-cell apoptosis via diversity-oriented synthesis. ACS Med Chem Lett. 2011;2:698–702. doi: 10.1021/ml200120m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marcaurelle LA, Comer E, Dandapani S, Duvall JR, Gerard B, Kesavan S, Lee MD, Liu H, Lowe JT, Marie JC, Mulrooney CA, Pandya BA, Rowley A, Ryba TD, Suh BC, Wei J, Young DW, Akella LB, Ross NT, Zhang YL, Fass DM, Reis SA, Zhao WN, Haggarty SJ, Palmer M, Foley MA. An aldol-based build/couple/pair strategy for the synthesis of medium-large-sized rings: discovery of macrocyclic histone deacetylase inhibitors. J Am Chem Soc. 2010;132:16962–16976. doi: 10.1021/ja105119r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerard B, Duvall JR, Lowe JT, Murillo T, Wei J, Akella LB, Marcaurelle LA. Synthesis of a stereochemically diverse library of medium-sized lactam and sultams via S(N)Ar cycloetherification. ACS Comb Sci. 2011;13:365–374. doi: 10.1021/co2000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sigurdsson ST, Seeger B, Kutzke U, Eckstein F. A mild and simple method for the preparation of isocyanates from aliphatic amines using trichloromethyl chloroformate. Synthesis of an isocyanate containing an activated double disulfide. J Org Chem. 1996;61:3883–3884. doi: 10.1021/jo9521920. [DOI] [PubMed] [Google Scholar]
- 16.Merglen A, Theander S, Rubi B, Chaffard G, Wollheim CB, Maechler P. Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology. 2003;145:667–668. doi: 10.1210/en.2003-1099. [DOI] [PubMed] [Google Scholar]
- 17.Ishikawa M, Hashimoto Y. Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J Med Chem. 2011;54:1539–1554. doi: 10.1021/jm101356p. [DOI] [PubMed] [Google Scholar]
- 18.Guzman-Perez A, Wester RT, Allen MC, Brown JA, Buchholz AR, Cook ER, Day WW, Hamanaka ES, Kennedy SP, Knight DR, Kowalczyk PJ, Marala RB, Mularski CJ, Novomisle WA, Ruggeri RB, Tracey WR, Hill RJ. Discovery of zoniporide: a potent and selective sodium-hydrogen exchanger type 1 (NHE-1) inhibitor with high aqueous solubility. Bioorg Med Chem Lett. 2001;11:803–807. doi: 10.1016/s0960-894x(01)00059-2. [DOI] [PubMed] [Google Scholar]
- 19.Pérez-Melero C, Maya ABS, del Rey B, Peláez R, Caballero E, Medarde M. A new family of quinoline and quinoxaline analogous of combrestatins. Bioorg Med Chem Lett. 2004;14:3771–3774. doi: 10.1016/j.bmcl.2004.04.098. [DOI] [PubMed] [Google Scholar]
- 20.Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol. 2003;3:900–911. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- 21.Schindler CS, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282:20059–20063. doi: 10.1074/jbc.R700016200. [DOI] [PubMed] [Google Scholar]
- 22.Moore F, Naamane N, Colli ML, Bouckenooghe T, Ortis F, Gurzov EN, Igoillo-Esteve M, Mathieu C, Bontempi G, Thykjaer T, Ørntoft TF, Eizirik DL. STAT1 is a master regulator of pancreatic β-cell apoptosis and islet inflammation. J Biol Chem. 2011;286:929–941. doi: 10.1074/jbc.M110.162131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klampfer L, Huang J, Sasazuki T, Shirasawa S, Augenlicht L. Inhibition of interferon γ signaling by the short chain fatty acid butyrate. Mol Cancer Res. 2003;1:855–862. [PubMed] [Google Scholar]
- 24.Klampfer L, Huang J, Swaby LA, Augenlicht L. Requirement of histone deacetylase activity for signaling by STAT1. J Biol Chem. 2004;279:30358–30368. doi: 10.1074/jbc.M401359200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.