

Abstract
Skin is a self-renewing tissue that is required to go through extensive proliferation throughout the lifespan of an organism. Telomere shortening acts as a mitotic clock that prevents aberrant proliferation such as cancer. A consequence of this protection is cellular senescence and ageing. The telomerase enzyme complex maintains telomere length in germline cells and in cancer cells. Telomerase is also active in certain somatic cells such as those in the epidermis but is almost undetectable in the dermis. Increasing evidence indicates that telomerase plays a significant role in maintenance of skin function and proliferation. Mutations in telomerase component genes in the disease dyskeratosis congenita result in numerous epidermal abnormalities. Studies also indicate that telomerase activity in epidermal stem cells might have roles that go beyond telomere elongation. Telomeres in skin cells may be particularly susceptible to accelerated shortening because of both proliferation and DNA-damaging agents such as reactive oxygen species. Skin might present an accessible tissue for manipulation of telomerase activity and telomere length with the potential of ameliorating skin diseases associated with ageing.
Keywords: ageing, keratinocytes, reactive oxygen species, senescence, skin, telomerase, telomeres
Telomeres and telomerase
Telomeres are critical structures at the end of eukaryotic chromosomes made up of numerous copies of G-rich repeats. In mammals, the telomere repeat is TTAGGG reiterated thousands of times (1). Telomeres protect the ends of chromosomes from degradation and from being recognized as double-stranded breaks. Without telomeres, chromosomes will fuse and genetic instability will occur (2). The telomere repeats bind a large number of proteins called the shelterin complex that stabilizes the telomere to fold back onto itself into what has been referred to as a T loop (Fig. 1) (3). Telomere shortening, telomere damage or expression of mutant telomere-binding proteins can disrupt the shelterin complex and lead to activation of a DNA damage response and cellular senescence (4). Telomere shortening occurs during normal DNA replication because of what has been called the end replication problem, which simply means that DNA polymerase cannot completely replicate the 5′ ends of newly synthesized DNA strands (5). It has been proposed that telomere shortening acts as a mitotic clock to prevent unregulated cell proliferation such as occurs in cancer (6). This mechanism of cancer prevention, however, is believed to come at a cost. Accumulating evidence indicates that telomere-mediated replicative senescence can lead to ageing (7).
Figure 1.

Telomere Structure. Human telomeres are regions of DNA that cap the ends of chromosomes and consist of 8–15 kb of repeats of the hexamer TTAGGG that terminates in a 3′ G-rich overhang. The DNA of telomeres is found complexed with a large number of proteins, including the six that comprise the shelterin complex: telomeric-repeat binding factor 1 (TRF1), TRF2, RAP1, TIN2, TPP1 and POT1. These proteins help create a protective structure at chromosome ends, known as the telomeric loop (or t loop). The single-stranded G-rich overhang invades the double-stranded helix of the telomere, protecting the single-stranded DNA (now in the displacement, or D loop) from detection by DNA damage machinery. See text for references.
Telomeres can be maintained by the enzyme complex telomerase (8). Telomerase was originally discovered in the unicellular eukaryote, Tetrahymena, by Elizabeth Blackburn and Carol Greider (9). For their work on telomerase and telomeres, Blackburn, Greider and Jack Szostek recently shared the 2009 Nobel Prize in Medicine. Telomerase consists of a reverse transcriptase component called TERT and an RNA component called TERC (also referred to as TR or TER) that is utilized by TERT to add telomere repeats to the chromosome end (Fig. 2) (10). Other proteins are also involved in the telomerase complex including dyskerin (DKC), which stabilizes small nucleolar RNAs (snoRNAs) such as TERC (8).
Figure 2.
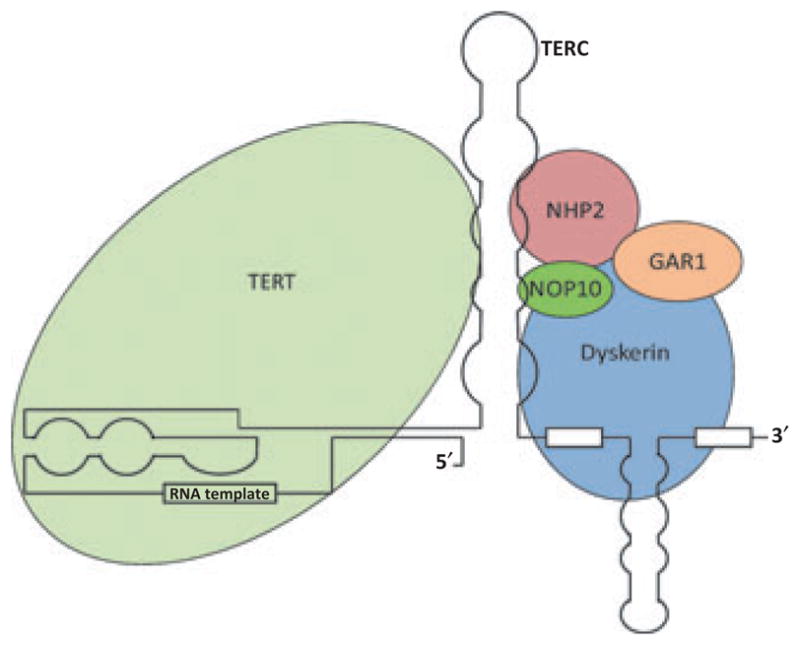
Telomerase Complex. The telomerase complex consists of the reverse transcriptase component (TERT), the RNA component (TERC), the protein Dyskerin, and other associated proteins (NHP2, NOP10, and GAR1). Telomerase adds telomeric repeats (TTAGGG) to the 3′ hydroxyl end of the leading strand of the telomere, with a sequence in the RNA component of the complex serving as the template for nucleotide addition. See text for references.
Expression of telomerase components is tightly regulated in human cells (11). Telomerase is active in germline cells but most differentiated somatic cells do not exhibit much if any telomerase activity (12). However, detectable levels are observed in certain stem cell components, in various hematopoietic lineages, and in cells of the basal epidermis (13–15). Telomerase is highly active in over 90% of all cancers (16). This rate approaches 100% in squamous cell carcinomas and, in fact, the small numbers of cancers that do not have active telomerase are usually not of epithelial origin and maintain telomeres through a modified form of mitotic recombination called alternative lengthening of telomeres (ALT) (17). For example, the ALT mechanism of telomere elongation is more frequent in liposarcomas, fibrosarcomas and sarcomas (18). TERT has been generally viewed as the rate-limiting component of telomerase activity, and much work has been performed to understand how it is regulated during development and carcinogenesis [for reviews see (11,19–22)]. TERT levels can be upregulated in cancer by a variety of mechanisms, including transcriptional upregulation, protein stabilization and gene amplification (23). Low levels of TERT expression are detectable in human epithelial cells, which goes along with the observation that these cells have some telomerase activity (15,24,25). Fibroblasts, on the other hand, have little to no TERT and, concomitantly, have barely detectable telomerase activity (26). The RNA component TERC is expressed at readily detectable levels in most cell types, including skin fibroblasts and epidermal cells (27). Evidence is now accumulating, however, that in addition to upregulation of TERT, upregulation of TERC levels can also increase the amount of telomerase activity in stem cells and other cells, and there have been several reports that many cancers exhibit amplification of the TERC gene and high TERC expression (23,28,29). In normal cells, it would appear that both TERT and TERC levels are fine-tuned to carefully regulate telomerase activity but that this regulation becomes dysfunctional in cancer cells.
Mouse models of telomerase and telomere dysfunction
Mouse models of telomerase deficiency have provided evidence that telomere shortening may be important for skin ageing. Inbred strains of mice are somewhat unusual in that they have fairly active telomerase in most somatic tissues and very long telomeres (30,31). Telomerase knockout mice (either by knocking out TERT or TERC) do not generally exhibit a phenotype until their telomeres are shortened to a critically short length after several generations (32–34). Mice with critically short telomeres exhibit problems with highly proliferative tissue, including epidermal abnormalities such as poor wound healing, ulcerative skin lesions, early hair loss and early hair greying. Wound healing is a particular problem in telomerase knockout mice. Histologic sections revealed a marked delay in wound re-epithelization, epithelial gaps and incomplete coagulum formation, particularly in older telomerase knockout mice, and this was not directly associated with proliferative index (33). Telomerase knockout mice also exhibit a deficiency in epidermal stem cell mobilization after treatment with TPA (12-O-tetradecanoylphorbol), and epidermal cells from telomerase knockout mice have lower clonogenic capacity compared to normal age-matched controls (34). The exact number of generations that it takes for telomerase knockout mice to display a phenotype depends on genetic background, which may reflect different starting telomere lengths or other factors. In mixed genetic background, an early ageing phenotype is not observed until the 5th or 6th generation (33), whereas it occurs in the 3rd or 4th generations in a straight C57BL6 background (34). More recently, it has been demonstrated using a mouse strain (i.e. CAST/EiJ) that starts with telomeres that are more similar in length to human cells (35) that knockout of telomerase can lead to tissue proliferation problems within one or two generations (36,37). Heterozygosity of TERC, but not necessarily TERT, is associated with decreases in telomere length (38), and this is particularly apparent in the CAST/EiJ strain where, after several generations, TERC heterozygosity results in a phenotype that is similar to telomerase knockout mice (36). Thus, telomerase deficiency is associated with skin defects in mice.
Mouse models have also revealed that dysregulation of genes that code for telomere-binding proteins can also lead to epidermal abnormalities. For example, it was demonstrated that epidermal-specific conditional knockout of a gene called TPP1, which codes for a telomere-binding protein (see Fig. 1), led to severe skin problems, including hyperpigmentation and impaired hair follicle morphogenesis and perinatal death (39). Knockout of TPP1 was associated with telomere end-to-end fusions and genetic instability. Similarly, epidermal-specific knockout of another telomere-binding protein, TRF1, resulted in epidermal abnormalities, including skin hyperpigmentation and epithelial dysplasia (40). Mice that conditionally overexpress the telomere-binding protein, TRF2, display increases in skin cancer, premature skin degeneration, skin hyperpigmentation and skin dryness (41). These results indicate that telomere function, and not just telomerase per se, is important for normal skin function and development.
Telomeres and telomerase in human disease
While mouse studies are of interest, mice are not humans and, as mentioned earlier, telomere length is much longer (up to ten times longer) in inbred strains of mice than in humans. In addition, telomerase is generally considered to be more highly active in mouse somatic cells when compared to human somatic cells. To really understand the role of telomere shortening in human ageing and human disease requires a human model system. A human disease called dyskeratosis congenita (DC) has been shown to be linked to mutations in telomerase component genes including TERT, TERC and DKC (42). DC patients often present with dyskeratotic nails, early hair greying and hair loss and dyspigmented and reticulated skin (43). Other problems such as leukoplakia, bone marrow failure, a slightly higher risk of cancer, and more recently pulmonary fibrosis, have also been associated with DC (44). DC patients have very short telomeres, and the observation that epidermal problems are associated with this disease would suggest that telomere maintenance is an important component of epidermal homeostasis (42). DC mutations can come in many forms. The DKC gene is X-linked and recessive mutations often become apparent in males (45). Mutations in TERT or TERC can be autosomal dominant (46,47). While some of these mutations might act in a dominant negative fashion, as both TERT and TERC act as dimers, there are also several reports of mutations in TERT and TERC that result in products that are non-functional or exhibit low levels of expression (48). In these latter cases, haploinsufficiency is believed to be the reason for the disease traits (49). One such family that we have been studying in Iowa exhibits an autosomal dominant pattern of inheritance (46). The mutation in this family is a large deletion of the 3′ end of TERC that causes instability and little to no expression.
Other human diseases, including liver fibrosis, aplastic anaemia, acute myeloid leukaemia and pulmonary fibrosis, have also been linked to mutations in genes that code for telomerase components or telomere-binding proteins (Table 1) (46,49–67). This indicates that defects in telomere maintenance may play an underappreciated role in diseases associated with ageing. That epidermal abnormalities are associated with DC indicates that maintenance of telomere length is important for skin homeostasis and function (see discussion below).
Table 1.
Telomerase mutations linked to human disease
| Disease/syndrome | Clinical presentation | Telomerase component | Mutations and effects | References |
|---|---|---|---|---|
| Dyskeratosis congenita (DC) | Triad of mucosal leukoplakia, nail dystrophy, abnormal skin pigmentation. Also, bone marrow failure, symptoms of premature ageing, aplastic anaemia, higher risk of cancer (esp. AML) | TERT (telomerase reverse transcriptase) (hTERT gene) | Heterozygous mutations found in autosomal dominant DC | 49,52,54 |
| TERC (telomerase RNA component) (hTR gene) | Heterozygous mutations found in autosomal dominant DC | 46 | ||
| Dyskerin (DKC1 gene) | Missense mutations found in X-linked DC | 55 | ||
| TINF2 (TINF2 gene) | Mutations found in sporadic DC or autosomal dominant DC | 56,57 | ||
| NOP10 (NOP10 gene), NHP2 (NHP2 gene) | Mutations found in autosomal recessive DC | 58,59 | ||
| Hoyerall-Hreiderasson syndrome (HH) | Severe variant of DC characterized by progressive pancytopenia, microcephaly, ataxia and growth retardation | Dyskerin | Missense mutations found in this X-linked syndrome | 60,61 |
| Aplastic anaemia (AA) | Hypocellular bone marrow and low blood cell counts | TERT, TERC | Heterozygous mutations lead to haploinsufficiency in telomerase | 62,63 |
| Familial liver disease | Liver fibrosis with inflammation and nodular regenerative hyperplasia | TERT, TERC | Heterozygous loss of function mutations lead to telomere erosion | 51 |
| Idiopathic pulmonary fibrosis (IPF) | Progressive scarring of the lung of unknown aetiology; leads to respiratory failure | TERT, TERC | Heterozygous loss of function mutations lead to haploinsufficiency in telomerase | 64–66 |
| Acute myeloid leukaemia (AML) | Abnormal proliferation and differentiation of hematopoietic progenitor cells | TERT | Missense mutations lead to shortened telomeres | 67 |
Telomerase in human skin
The role of telomerase activity in the human epidermis is not completely clear. As mentioned earlier, telomerase can be detected in keratinocytes of the basal epidermis but not in skin fibroblasts (68). Telomerase activity has also been localized to the bulge component of the hair follicle (69). Whether this activity efficiently maintains the telomeres in human epidermis remains somewhat controversial. There is evidence that telomeres shorten with age in human skin or after skin grafting (70–73). Other studies have demonstrated only minimal shortening of telomeres in the human epidermis with age, less than that observed in fibroblasts, suggesting that telomerase in the epidermis may provide a certain degree of telomere maintenance in vivo (74,75). Certain environmental factors can increase telomerase activity in the epidermis. For example, it has been shown that telomerase activity is increased in the epidermis after it has been exposed to ultraviolet (UV) light or even poison ivy (14). There is also speculation that telomerase can be increased in the epidermis upon inflammation (76). Thus, telomerase may be activated in the epidermis as it is needed for cell proliferation and repair of damage. Shortening of telomeres is believed to provide a barrier for epidermal cell proliferation (i.e. cancer) in vivo. Telomerase is almost always activated in squamous cell carcinomas of the skin, indicating that upregulation of telomerase is important for skin carcinogenesis (77,78). Whether telomere shortening in skin cells is a cause of skin ageing is not entirely clear and will be discussed in more detail later in this review.
It should be noted that telomerase regulation in keratinocytes cultured in vitro might be somewhat different than that which occurs in vivo. When grown in culture, epidermal keratinocytes exhibit low levels of telomerase at early passage but by mid-passage it is turned off to hardly detectable levels (79). Co-culture with irradiated or mitomycin C-treated feeders can prolong the time that telomerase is kept on in culture but, even in these conditions, telomerase activity decreases with passaging (80). Even with some telomerase activity, keratinocytes in culture still exhibit telomere shortening (80). In addition, increased telomerase activation is a prerequisite for keratinocyte immortalization in vitro (81,82). A number of studies have been performed to determine how telomerase is upregulated in skin keratinocytes during immortalization and transformation. For example, it has been shown that expression of the E6 protein from high-risk mucosal human papillomaviruses (HPV), such as HPV-16 and from cutaneous HPV types, such as HPV-5 and HPV-8, can activate telomerase in human skin keratinocytes (83,84). This activation of telomerase, along with the abrogation of the p53 and pRb pathways by HPV E6 and E7, has been demonstrated to be a necessary component of cellular immortalization by HPV (81). HPV E6-mediated activation of telomerase has been shown to occur through transcriptional upregulation of TERT, although upregulation of TERC might also play a role (85,86). The mechanism by which HPV E6 activates TERT is not completely clear but likely involves degradation of a transcriptional repressor (i.e. NF-X1) and /or activation of a transcriptional activator (i.e. c-myc) (87,88). Other studies have shown that expression of c-myc can activate telomerase in human skin keratinocytes (89). Activation of TERT in human keratinocytes is associated with histone acetylation of the chromatin in the TERT promoter (90,91). As with any eukaryotic promoter, regulation of TERT and TERC is complex and further studies are necessary to determine exactly how these genes are regulated during normal development, differentiation, ageing and carcinogenesis.
As mentioned earlier, telomerase activation is a prerequisite for immortalization of skin keratinocytes. Many human cells types, such as fibroblasts, can be immortalized by overexpression of TERT alone (92). Interestingly, several studies have now demonstrated that skin keratinocytes have a greatly increased proliferative capacity after high-level expression of exogenous expression of TERT (93,94). In fact, it has been argued that under the right growth conditions (i.e. with irradiated feeder fibroblasts) that telomerase activation alone is sufficient for immortalization of keratinocytes (93). TERT immortalization of skin keratinocytes is often associated with eventual loss of p16INK4a, even in feeder culture conditions, indicating that p16INK4a still provides a potential barrier to excessive proliferation in the presence of high telomerase activity (81,86,94). Exogenous expression of TERC can also lead to telomerase activation and an extension of lifespan in keratinocytes, suggesting that TERC levels are also rate limiting for telomerase activity in this cell type (86). These observations are likely due to the fact that keratinocytes express low levels of TERT and the combination of low TERT with exogenous TERC results in enough telomerase to maintain telomeres. In fibroblasts, TERC expression alone has little effect because fibroblasts do not express TERT. Thus, skin keratinocytes and skin fibroblasts differ in their ability to activate telomerase and have extended proliferation through expression of TERT and TERC.
A question that remains is whether telomere shortening plays a significant role in the ageing of skin. One study in mice indicated that telomeres do not exhibit extensive shortening in epidermal stem cells as mice age (95). However, other studies have demonstrated with convincing evidence that telomere shortening occurs in epidermal stem cells as normal mice age (34,96). For humans, DC patients suffer from a number of epidermal abnormalities including poor nail growth, early hair loss, hair greying and skin atrophy (97,98). Skin fibroblasts and keratinocytes from DC patients have short telomeres and proliferative and functional defects, including poor in vitro wound healing in early passage keratinocyte cultures (86,99). Skin fibroblasts from other DC families with different mutations have also been shown to have proliferative defects (100,101). While studies characterizing skin keratinocytes from DC patients are limited, these initial findings point to the likely possibility that telomerase dysfunction and/or telomere shortening in skin fibroblasts and keratinocytes are important for the ageing process in skin.
Exogenous expression of telomerase to correct telomere defects
If telomere shortening is involved in skin ageing, then activation of telomerase should ameliorate skin ageing. As mentioned, TERT, and possibly TERC, expression can extend the lifespan of human keratinocytes, and TERT expression can immortalize human skin fibroblasts in vitro. Functional and proliferative defects in DC keratinocytes and fibroblasts can be improved by activation of telomerase (86,99). Minimal telomerase activation, by expression of TERC alone, can maintain telomere length in DC keratinocytes (86). Expression of both TERT and TERC can lead to extremely high levels of telomerase and significant extension of telomere length in human cells (99). Whether having very long telomeres is ‘good’ for a cell or not is unknown. It is possible that this may lead to susceptibility to transformation. In vivo, it has been demonstrated that exogenous expression of TERT using an adenovirus can improve the rate of wound closure in a rabbit model of wound healing (102). Mice that have been engineered to express TERT specifically in the epidermis, have thickened epidermis and exhibit the ability to regenerate hair faster than non-transgenic mice (103). This effect has been specifically localized to an improved ability of epidermal stem cells to mobilize. It should be noted that at least one study indicated that TERT was acting through a mechanism that did not necessarily depend on the telomere elongation function of TERT but rather on other TERT functions (104,105). Certainly, evidence is accumulating that TERT, has functions that go beyond its role in telomerase activity (106). While TERT transgenics have ‘improved’ skin, these mice also have increased rates of epidermal carcinogenesis (107). Interestingly, breeding TERT transgenic mice to mice that are tumor resistant (by virtue of expressing extra p16INK4a and a more active form of p53) resulted in offspring that did not get tumors and, in fact, these mice exhibited a significantly extended lifespan (108). This was true even though increased TERT expression was supposedly only localized to epithelial cells (the transgenic TERT was driven by a K5 promoter). With age, these mice exhibited less dermatitis and better preservation of the thickness of both the epidermis and the subcutaneous fat layer compared to corresponding age-matched controls. While these highly manipulated mice may not be considered normal, the results indicate that telomerase activation may provide a means to slow down ageing if the extra proliferation afforded by lengthened telomeres can be controlled by other mechanisms.
Interplay between telomeres and oxidative stress
Reactive oxygen species (ROS) have long been considered as a reason for the ageing of skin (109). In fact, many cosmetic products claim to have antioxidant properties that prevent skin ageing. In vitro studies have implicated ROS in accelerating telomere shortening (110). Thus, some of the effects of ROS may be mediated through telomere-associated cell senescence. Using fibroblasts, it was demonstrated that higher levels of oxygen (above ambient) and high doses of hydrogen peroxide cause rapid telomere shortening and concomitant cell senescence (110,111). The telomere strand composed of TTAGGG repeats might be exquisitely sensitive to oxidative stress because they are G-rich and guanines can be modified to 8-oxyguanosine by ROS (112). This base alteration could accelerate telomere shortening by causing the loss of binding of telomere proteins. Accelerated telomere loss after peroxide treatment might be mediated by DNA repair processes in the cell. Also, DNA damage in general could accelerate telomere shortening in the skin (113).
Interestingly, there is evidence that cells with longer telomeres might be more sensitive to oxidative stress (114). This is in contrast to other findings that indicate that cells and mice with short telomeres are more sensitive to DNA-damaging agents (115). Another study suggested that telomere loss in keratinocytes could be delayed by treatment with vitamin C (an antioxidant) or by stimulation of endogenous antioxidants through low level treatment with hydrogen peroxide (116). If telomere shortening is mediating the effects of ROS with regard to ageing, decreasing ROS or increasing antioxidants in cells should provide a potential mechanism to decrease telomere shortening and ageing in the skin. There is a possibility that ROS might have very different effects on skin fibroblasts versus skin keratinocytes. As mentioned earlier, keratinocytes treated with low level peroxide exhibit delayed telomere shortening and extension of lifespan, most likely through modulation of antioxidant levels (116), whereas fibroblasts treated with low levels of peroxide exhibit increased telomere shortening and shortened lifespan (111). In addition, it has been demonstrated that keratinocytes are more resistant to DNA damage and growth inhibition caused by oxidizing agents such as UVB and ionizing radiation compared to fibroblasts and tend to undergo more apoptosis and less senescence compared to fibroblasts in these conditions (117). Thus, while keratinocytes with short telomeres and/or damaged DNA might be removed by apoptosis, fibroblasts may become senescent, remaining in the dermal tissue to affect epidermal growth through processes such as paracrine signalling and extracellular matrix deposition. Indeed, recent studies have demonstrated that senescent fibroblasts secrete higher levels of osteopontin, which causes increased proliferation of premalignant keratinocytes (118). On the other hand, when compared to normal fibroblasts, senescent skin fibroblasts turn down expression of IGF-1 and this causes an inappropriate UVB response in skin keratinocytes (119). It will therefore be important to not only characterize differences in the ways that different cell types respond to ROS and how this affects telomere shortening, but also to determine whether the effects of ROS on one cell type (and concomitant telomere shortening and senescence) have effects on other cell types. Studies such as these will help to define how cellular senescence leads to ageing and cancer.
Conclusions and perspectives
Telomeres have an important role in the life of skin cells, including the ageing of skin. Telomerase, the tightly regulated enzyme complex that maintains telomere length in rapidly proliferating cells such as germline and cancer cells, has been implicated in having a key role in the maintenance of skin cell function and proliferation. Mutations in genes of the telomerase complex lead to many diseases that involve epidermal abnormalities, such as in DC. A delicate balance between telomere length and telomerase activity is required to maintain normal skin cells and avoid cancer (Fig. 3). Experimental studies in mice and cell culture have led to many discoveries about telomeres and telomerase. Studies of human skin keratinocytes have shown the importance for regulation of telomere length and telomerase activity in these cells. It is possible that the shortening of telomeres in cells of the dermis (e.g. fibroblasts) may lead to defects in the epidermis; further studies are needed to elucidate this interaction. As an environmental barrier for the body, the skin’s exposure to ROS would suggest a role for ROS in telomere shortening and ageing in skin cells. Future studies will be instrumental in further defining the relationships between ROS, DNA damage, senescence and ageing and the state of telomeres in skin cells. This may lead to treatments or therapies that manipulate telomerase or other factors that alter the length of telomeres, in order to slow down damage and ageing in skin.
Figure 3.
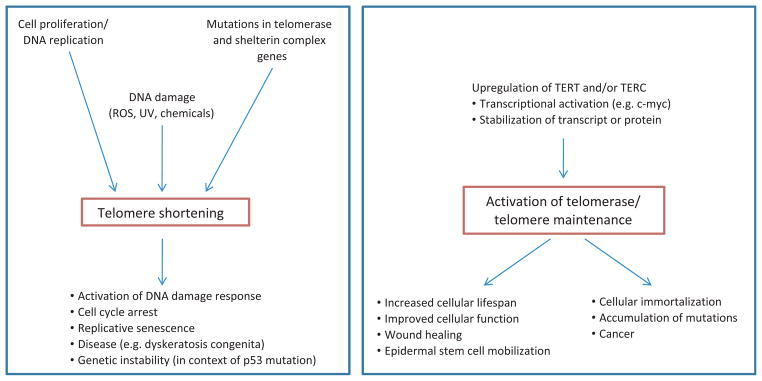
Presented is a summary of factors and activities that lead to either telomere shortening or telomere maintenance, and the possible outcomes in cells, as well as in organisms as a whole. (Left box): Telomere shortening occurs normally during cell division and DNA replication, because of the end replication problem. Mutations in telomerase and shelterin complex genes can lead to accelerated shortening and dysfunctional telomeres. In addition, reactive oxygen species or other DNA-damaging agents cause rapid telomere shortening. The erosion of telomeres to a short critical length activates a DNA damage response, cell cycle arrest and senescence. This causes failure of tissue regeneration and tissue ageing. In the context of p53 mutation, short telomeres can lead to genetic instability, which may play a role in the development of cancer. (Right box): The upregulation of TERT and/or TERC expression through a variety of factors (e.g. transcriptional upregulation, stabilization of the transcript or protein) causes activation of telomerase, which can maintain or even elongate telomeres. In a normal context, this allows stem cell maintenance, cell proliferation and tissue regeneration. Upregulation of TERT has also been shown to be involved in mobilization of epidermal stem cells. Unchecked telomerase activity is associated with cellular immortalization, which is a prerequisite for cancer and can result in accumulation of genetic and epigenetic changes that may increase the likelihood of malignancy. See text for references.
Acknowledgments
The authors thank Francoise Gourronc for critical review of the manuscript. AJK and EMB are supported by R01AG27388 from the National Institute of Aging.
References
- 1.Moyzis RK, Buckingham JM, Cram LS, et al. Proc Natl Acad Sci U S A. 1988;85:6622–6626. doi: 10.1073/pnas.85.18.6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Sullivan RJ, Karlseder J. Nat Rev Mol Cell Biol. 2010;11:171–181. doi: 10.1038/nrm2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Lange T. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 4.d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, et al. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 5.Levy MZ, Allsopp RC, Futcher AB, et al. J Mol Biol. 1992;225:951–960. doi: 10.1016/0022-2836(92)90096-3. [DOI] [PubMed] [Google Scholar]
- 6.Harley CB. Mutat Res. 1991;256:271–282. doi: 10.1016/0921-8734(91)90018-7. [DOI] [PubMed] [Google Scholar]
- 7.Aubert G, Lansdorp PM. Physiol Rev. 2008;88:557–579. doi: 10.1152/physrev.00026.2007. [DOI] [PubMed] [Google Scholar]
- 8.Collins K, Mitchell JR. Oncogene. 2002;21:564–579. doi: 10.1038/sj.onc.1205083. [DOI] [PubMed] [Google Scholar]
- 9.Greider CW, Blackburn EH. Cell. 1985;43:405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- 10.Greider CW, Blackburn EH. Nature. 1989;337:331–337. doi: 10.1038/337331a0. [DOI] [PubMed] [Google Scholar]
- 11.Ducrest AL, Szutorisz H, Lingner J, et al. Oncogene. 2002;21:541–552. doi: 10.1038/sj.onc.1205081. [DOI] [PubMed] [Google Scholar]
- 12.Wright WE, Piatyszek MA, Rainey WE, et al. Dev Genet. 1996;18:173–179. doi: 10.1002/(SICI)1520-6408(1996)18:2<173::AID-DVG10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 13.Engelhardt M, Kumar R, Albanell J, et al. Blood. 1997;90:182–193. [PubMed] [Google Scholar]
- 14.Taylor RS, Ramirez RD, Ogoshi M, et al. J Invest Dermatol. 1996;106:759–765. doi: 10.1111/1523-1747.ep12345811. [DOI] [PubMed] [Google Scholar]
- 15.Bickenbach JR, Vormwald-Dogan V, Bachor C, et al. J Invest Dermatol. 1998;111:1045–1052. doi: 10.1046/j.1523-1747.1998.00420.x. [DOI] [PubMed] [Google Scholar]
- 16.Kim NW, Piatyszek MA, Prowse KR, et al. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 17.Bryan TM, Englezou A, Gupta J, et al. EMBO J. 1995;14:4240–4248. doi: 10.1002/j.1460-2075.1995.tb00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cesare AJ, Reddel RR. Nat Rev Genet. 2010;11:319–330. doi: 10.1038/nrg2763. [DOI] [PubMed] [Google Scholar]
- 19.Liu JP. FASEB J. 1999;13:2091–2104. doi: 10.1096/fasebj.13.15.2091. [DOI] [PubMed] [Google Scholar]
- 20.Smogorzewska A, de Lange T. Annu Rev Biochem. 2004;73:177–208. doi: 10.1146/annurev.biochem.73.071403.160049. [DOI] [PubMed] [Google Scholar]
- 21.Dong CK, Masutomi K, Hahn WC. Crit Rev Oncol Hematol. 2005;54:85–93. doi: 10.1016/j.critrevonc.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 22.Bellon M, Nicot C. J Natl Cancer Inst. 2008;100:98–108. doi: 10.1093/jnci/djm269. [DOI] [PubMed] [Google Scholar]
- 23.Cao Y, Bryan TM, Reddel RR. Cancer Sci. 2008;99:1092–1099. doi: 10.1111/j.1349-7006.2008.00815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yasumoto S, Kunimura C, Kikuchi K, et al. Oncogene. 1996;13:433–439. [PubMed] [Google Scholar]
- 25.Nakano K, Watney E, McDougall JK. Am J Pathol. 1998;153:857–864. doi: 10.1016/S0002-9440(10)65627-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Funk WD, Wang CK, Shelton DN, et al. Exp Cell Res. 2000;258:270–278. doi: 10.1006/excr.2000.4945. [DOI] [PubMed] [Google Scholar]
- 27.Ogoshi M, Le T, Shay JW, et al. J Invest Dermatol. 1998;110:818–823. doi: 10.1046/j.1523-1747.1998.00180.x. [DOI] [PubMed] [Google Scholar]
- 28.Pelosi G, Del Curto B, Trubia M, et al. Clin Cancer Res. 2007;13:1995–2004. doi: 10.1158/1078-0432.CCR-06-2483. [DOI] [PubMed] [Google Scholar]
- 29.Hopman AH, Theelen W, Hommelberg PP, et al. J Pathol. 2006;210:412–419. doi: 10.1002/path.2070. [DOI] [PubMed] [Google Scholar]
- 30.Blasco MA, Funk W, Villeponteau B, et al. Science. 1995;269:1267–1270. doi: 10.1126/science.7544492. [DOI] [PubMed] [Google Scholar]
- 31.Prowse KR, Greider CW. Proc Natl Acad Sci U S A. 1995;92:4818–4822. doi: 10.1073/pnas.92.11.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blasco MA, Lee HW, Hande MP, et al. Cell. 1997;91:25–34. doi: 10.1016/s0092-8674(01)80006-4. [DOI] [PubMed] [Google Scholar]
- 33.Rudolph KL, Chang S, Lee HW, et al. Cell. 1999;96:701–712. doi: 10.1016/s0092-8674(00)80580-2. [DOI] [PubMed] [Google Scholar]
- 34.Flores I, Cayuela ML, Blasco MA. Science. 2005;309:1253–1256. doi: 10.1126/science.1115025. [DOI] [PubMed] [Google Scholar]
- 35.Hemann MT, Greider CW. Nucleic Acids Res. 2000;28:4474–4478. doi: 10.1093/nar/28.22.4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hao LY, Armanios M, Strong MA, et al. Cell. 2005;123:1121–1131. doi: 10.1016/j.cell.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 37.Armanios M, Alder JK, Parry EM, et al. Am J Hum Genet. 2009;85:823–832. doi: 10.1016/j.ajhg.2009.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiang YJ, Hemann MT, Hathcock KS, et al. Mol Cell Biol. 2004;24:7024–7031. doi: 10.1128/MCB.24.16.7024-7031.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tejera AM, Stagno d’Alcontres M, Thanasoula M, et al. Dev Cell. 2010;18:775–789. doi: 10.1016/j.devcel.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martinez P, Thanasoula M, Munoz P, et al. Genes Dev. 2009;23:2060–2075. doi: 10.1101/gad.543509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stout GJ, Blasco MA. Dis Model Mech. 2009;2:139–156. doi: 10.1242/dmm.002121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kirwan M, Dokal I. Clin Genet. 2008;73:103–112. doi: 10.1111/j.1399-0004.2007.00923.x. [DOI] [PubMed] [Google Scholar]
- 43.Marciniak RA, Johnson FB, Guarente L. Trends Genet. 2000;16:193–195. doi: 10.1016/s0168-9525(00)01984-3. [DOI] [PubMed] [Google Scholar]
- 44.Garcia CK, Wright WE, Shay JW. Nucleic Acids Res. 2007;35:7406–7416. doi: 10.1093/nar/gkm644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mitchell JR, Wood E, Collins K. Nature. 1999;402:551–555. doi: 10.1038/990141. [DOI] [PubMed] [Google Scholar]
- 46.Vulliamy T, Marrone A, Goldman F, et al. Nature. 2001;413:432–435. doi: 10.1038/35096585. [DOI] [PubMed] [Google Scholar]
- 47.Vulliamy TJ, Walne A, Baskaradas A, et al. Blood Cells Mol Dis. 2005;34:257–263. doi: 10.1016/j.bcmd.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 48.Xin ZT, Beauchamp AD, Calado RT, et al. Blood. 2006;109:524–532. doi: 10.1182/blood-2006-07-035089. [DOI] [PubMed] [Google Scholar]
- 49.Armanios M, Chen JL, Chang YP, et al. Proc Natl Acad Sci U S A. 2005;44:15960–15964. doi: 10.1073/pnas.0508124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Du HY, Pumbo E, Ivanovich J, et al. Blood. 2009;113:309–316. doi: 10.1182/blood-2008-07-166421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Calado RT, Regal JA, Kleiner DE, et al. PLoS ONE. 2009;4:e7926. doi: 10.1371/journal.pone.0007926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dokal I, Vulliamy T. Blood Rev. 2008;22:141–153. doi: 10.1016/j.blre.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 53.Armanios M. Annu Rev Genomics Hum Genet. 2009;10:45–61. doi: 10.1146/annurev-genom-082908-150046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dokal I, Vulliamy T. Blood Rev. 2003;17:217– 225. doi: 10.1016/s0268-960x(03)00020-1. [DOI] [PubMed] [Google Scholar]
- 55.Heiss NS, Knight SW, Vulliamy TJ, et al. Nat Genet. 1998;19:32–38. doi: 10.1038/ng0598-32. [DOI] [PubMed] [Google Scholar]
- 56.Walne AJ, Vulliamy T, Beswick R, et al. Blood. 2008;112:3594–3600. doi: 10.1182/blood-2008-05-153445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Savage SA, Giri N, Baerlocher GM, et al. Am J Hum Genet. 2008;82:501–509. doi: 10.1016/j.ajhg.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vulliamy T, Beswick R, Kirwan M, et al. Proc Natl Acad Sci U S A. 2008;105:8073–8078. doi: 10.1073/pnas.0800042105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walne AJ, Vulliamy T, Marrone A, et al. Hum Mol Genet. 2007;16:1619–1629. doi: 10.1093/hmg/ddm111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hreidarsson S, Kristjansson K, Johannesson G, et al. Acta Paediatr Scand. 1988;77:773–775. doi: 10.1111/j.1651-2227.1988.tb10751.x. [DOI] [PubMed] [Google Scholar]
- 61.Knight SW, Heiss NS, Vulliamy TJ, et al. Br J Haematol. 1999;107:335–339. doi: 10.1046/j.1365-2141.1999.01690.x. [DOI] [PubMed] [Google Scholar]
- 62.Yamaguchi H, Baerlocher GM, Lansdorp PM, et al. Blood. 2003;102:916–918. doi: 10.1182/blood-2003-01-0335. [DOI] [PubMed] [Google Scholar]
- 63.Yamaguchi H, Calado RT, Ly H, et al. N Engl J Med. 2005;352:1413–1424. doi: 10.1056/NEJMoa042980. [DOI] [PubMed] [Google Scholar]
- 64.Armanios MY, Chen JJ, Cogan JD, et al. N Engl J Med. 2007;356:1317–1326. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 65.Cronkhite JT, Xing C, Raghu G, et al. Am J Respir Crit Care Med. 2008;178:729–737. doi: 10.1164/rccm.200804-550OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tsakiri KD, Cronkhite JT, Kuan PJ, et al. Proc Natl Acad Sci U S A. 2007;104:7552–7557. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Calado RT, Regal JA, Hills M, et al. Proc Natl Acad Sci U S A. 2009;106:1187–1192. doi: 10.1073/pnas.0807057106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Boukamp P. Curr Mol Med. 2005;5:171–177. doi: 10.2174/1566524053586644. [DOI] [PubMed] [Google Scholar]
- 69.Ramirez RD, Wright WE, Shay JW, et al. J Invest Dermatol. 1997;108:113–117. doi: 10.1111/1523-1747.ep12285654. [DOI] [PubMed] [Google Scholar]
- 70.Counter CM, Press W, Compton CC. Lancet. 2003;361:1345–1346. doi: 10.1016/S0140-6736(03)13042-5. [DOI] [PubMed] [Google Scholar]
- 71.Nakamura K, Izumiyama-Shimomura N, Sawabe M, et al. J Invest Dermatol. 2002;119:1014– 1019. doi: 10.1046/j.1523-1747.2002.19523.x. [DOI] [PubMed] [Google Scholar]
- 72.Friedrich U, Griese E, Schwab M, et al. Mech Ageing Dev. 2000;119:89–99. doi: 10.1016/s0047-6374(00)00173-1. [DOI] [PubMed] [Google Scholar]
- 73.Butler MG, Tilburt J, DeVries A, et al. Cancer Genet Cytogenet. 1998;105:138–144. doi: 10.1016/s0165-4608(98)00029-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Krunic D, Moshir S, Greulich-Bode KM, et al. Biochim Biophys Acta. 2009;1792:297–308. doi: 10.1016/j.bbadis.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 75.Sugimoto M, Yamashita R, Ueda M. J Dermatol Sci. 2006;43:43–47. doi: 10.1016/j.jdermsci.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 76.Jurisic D, Kirin I, Rabic D, et al. Med Hypotheses. 2007;68:1093–1095. doi: 10.1016/j.mehy.2006.09.037. [DOI] [PubMed] [Google Scholar]
- 77.Harle-Bachor C, Boukamp P. Proc Natl Acad Sci U S A. 1996;93:6476–6481. doi: 10.1073/pnas.93.13.6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu A, Ichihashi M, Ueda M. Cancer. 1999;86:2038–2044. doi: 10.1002/(sici)1097-0142(19991115)86:10<2038::aid-cncr22>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 79.Miyata Y, Okada K, Fujimoto A, et al. J Dermatol Sci. 2004;34:221–230. doi: 10.1016/j.jdermsci.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 80.Fu B, Quintero J, Baker CC. Cancer Res. 2003;63:7815–7824. [PubMed] [Google Scholar]
- 81.Kiyono T, Foster SA, Koop JI, et al. Nature. 1998;396:84–88. doi: 10.1038/23962. [DOI] [PubMed] [Google Scholar]
- 82.Dickson MA, Hahn WC, Ino Y, et al. Mol Cell Biol. 2000;20:1436–1447. doi: 10.1128/mcb.20.4.1436-1447.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Klingelhutz AJ, Foster SA, McDougall JK. Nature. 1996;380:79–82. doi: 10.1038/380079a0. [DOI] [PubMed] [Google Scholar]
- 84.Bedard KM, Underbrink MP, Howie HL, et al. J Virol. 2008;82:3894–3902. doi: 10.1128/JVI.01818-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Veldman T, Horikawa I, Barrett JC, et al. J Virol. 2001;75:4467–4472. doi: 10.1128/JVI.75.9.4467-4472.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gourronc FA, Robertson M, Herrig AK, et al. Exp Dermatol. 2010;19:279–288. doi: 10.1111/j.1600-0625.2009.00916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gewin L, Myers H, Kiyono T, et al. Genes Dev. 2004;18:2269–2282. doi: 10.1101/gad.1214704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Veldman T, Liu X, Yuan H, et al. Proc Natl Acad Sci U S A. 2003;100:8211–8216. doi: 10.1073/pnas.1435900100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cerezo A, Kalthoff H, Schuermann M, et al. J Cell Sci. 2002;115:1305–1312. doi: 10.1242/jcs.115.6.1305. [DOI] [PubMed] [Google Scholar]
- 90.James MA, Lee JH, Klingelhutz AJ. Int J Cancer. 2006;119:1878–1885. doi: 10.1002/ijc.22064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xu M, Luo W, Elzi DJ, et al. Mol Cell Biol. 2008;28:4819–4828. doi: 10.1128/MCB.01969-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bodnar AG, Ouellette M, Frolkis M, et al. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 93.Ramirez RD, Morales CP, Herbert BS, et al. Genes Dev. 2001;15:398–403. doi: 10.1101/gad.859201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Darbro BW, Lee KM, Nguyen NK, et al. Oncogene. 2006;25:7421–7423. doi: 10.1038/sj.onc.1209729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stern MM, Bickenbach JR. Aging Cell. 2007;6:439–452. doi: 10.1111/j.1474-9726.2007.00318.x. [DOI] [PubMed] [Google Scholar]
- 96.Flores I, Canela A, Vera E, et al. Genes Dev. 2008;22:654–667. doi: 10.1101/gad.451008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Reimann C, Kloeckener-Gruissem B, Niemeyer CM, et al. J Eur Acad Dermatol Venereol. 2007;22:897–898. doi: 10.1111/j.1468-3083.2007.02530.x. [DOI] [PubMed] [Google Scholar]
- 98.Mason PJ, Wilson DB, Bessler M. Curr Mol Med. 2005;5:159–170. doi: 10.2174/1566524053586581. [DOI] [PubMed] [Google Scholar]
- 99.Westin ER, Chavez E, Lee KM, et al. Aging Cell. 2007;6:383–394. doi: 10.1111/j.1474-9726.2007.00288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dokal I, Bungey J, Williamson P, et al. Blood. 1992;80:3090–3096. [PubMed] [Google Scholar]
- 101.Wong JM, Collins K. Genes Dev. 2006;20:2848–2858. doi: 10.1101/gad.1476206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mogford JE, Liu WR, Reid R, et al. Hum Gene Ther. 2006;17:651–660. doi: 10.1089/hum.2006.17.651. [DOI] [PubMed] [Google Scholar]
- 103.Gonzalez-Suarez E, Samper E, Ramirez A, et al. EMBO J. 2001;20:2619–2630. doi: 10.1093/emboj/20.11.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Siegl-Cachedenier I, Flores I, Klatt P, et al. J Cell Biol. 2007;179:277–290. doi: 10.1083/jcb.200704141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sarin KY, Cheung P, Gilison D, et al. Nature. 2005;436:1048–1052. doi: 10.1038/nature03836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee J, Sung YH, Cheong C, et al. Oncogene. 2008;27:3754–3760. doi: 10.1038/sj.onc.1211037. [DOI] [PubMed] [Google Scholar]
- 107.Gonzalez-Suarez E, Geserick C, Flores JM, et al. Oncogene. 2005;24:2256–2270. doi: 10.1038/sj.onc.1208413. [DOI] [PubMed] [Google Scholar]
- 108.Tomas-Loba A, Flores I, Fernandez-Marcos PJ, et al. Cell. 2008;135:609–622. doi: 10.1016/j.cell.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 109.Yaar M, Eller MS, Gilchrest BA. J Investig Dermatol Symp Proc. 2002;7:51–58. doi: 10.1046/j.1523-1747.2002.19636.x. [DOI] [PubMed] [Google Scholar]
- 110.von Zglinicki T. Trends Biochem Sci. 2002;27:339–344. doi: 10.1016/s0968-0004(02)02110-2. [DOI] [PubMed] [Google Scholar]
- 111.von Zglinicki T, Saretzki G, Docke W, et al. Exp Cell Res. 1995;220:186–193. doi: 10.1006/excr.1995.1305. [DOI] [PubMed] [Google Scholar]
- 112.Kawanishi S, Oikawa S. Ann N Y Acad Sci. 2004;1019:278–284. doi: 10.1196/annals.1297.047. [DOI] [PubMed] [Google Scholar]
- 113.Kosmadaki MG, Gilchrest BA. Micron. 2004;35:155–159. doi: 10.1016/j.micron.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 114.Rubio MA, Davalos AR, Campisi J. Exp Cell Res. 2004;298:17–27. doi: 10.1016/j.yexcr.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 115.Gonzalez-Suarez E, Goytisolo FA, Flores JM, et al. Cancer Res. 2003;63:7047–7050. [PubMed] [Google Scholar]
- 116.Yokoo S, Furumoto K, Hiyama E, et al. J Cell Biochem. 2004;93:588–597. doi: 10.1002/jcb.20208. [DOI] [PubMed] [Google Scholar]
- 117.D’Errico M, Lemma T, Calcagnile A, et al. Mutat Res. 2007;614:37–47. doi: 10.1016/j.mrfmmm.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 118.Pazolli E, Luo X, Brehm S, et al. Cancer Res. 2009;69:1230–1239. doi: 10.1158/0008-5472.CAN-08-2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lewis DA, Travers JB, Somani AK, et al. Oncogene. 2010;29:1475–1485. doi: 10.1038/onc.2009.440. [DOI] [PMC free article] [PubMed] [Google Scholar]