

Abstract
Background
Patients with long QT syndrome (LQTS) who harbor multiple mutations (i.e. ≥ 2 mutations in ≥ 1 LQTS-susceptibility gene) may experience increased risk for life-threatening cardiac events.
Objectives
The present study was designed to compare the clinical course of LQTS patients with multiple mutations to those with a single mutation.
Methods
The risk for life-threatening cardiac events (comprising aborted cardiac arrest, implantable defibrillator shock, or sudden cardiac death) from birth through age 40 years, by the presence of multiple vs. single mutations, was assessed among 403 patients from the LQTS Registry.
Results
Patients with multiple mutations (n = 57) exhibited a longer QTc at enrollment compared with those with a single mutation (mean ± SD: 506 ± 72 vs. 480 ± 56 msec, respectively; p = 0.003) and had a higher rate of life threatening cardiac events during follow-up (23% vs. 11%, respectively; p < 0.001). Consistently, multivariate analysis demonstrated that patients with multiple mutations had a 2.3-fold (p = 0.015) increased risk for life threatening cardiac events as compared to patients with a single mutation. The presence of multiple mutations in a single LQTS gene was associated with a 3.2-fold increased risk for life threatening cardiac events (p = 0.010) whereas the risk associated with multiple mutation status involving > 1 LQTS gene was not significantly different from the risk associated with a single mutation (HR 1.7, p = 0.26).
Conclusions
LQTS patients with multiple mutations have a greater risk for life-threatening cardiac events as compared to patients with a single mutation.
Keywords: Aborted cardiac arrest, Long QT syndrome, Mutation, Risk factor, Sudden cardiac death
Introduction
Genetic testing adds important information to the diagnosis, risk stratification, and management of long QT syndrome (LQTS). To date, more than 600 mutations have been identified in 13 LQTS genes. The LQT1, LQT2 and LQT3 genotypes comprise more than 95% of genotype positive LQTS patients and approximately 75% of all patients with LQTS.1–2 LQTS exhibits incomplete penetrance (i.e. the likelihood that a disease causing mutation will have a phenotypic expression in a mutation-positive subject) and variable expressivity (i.e. the level of phenotypic expression). Thus, within the same LQTS family, some LQTS genotype-positive subjects may display a markedly prolonged QTc and experience syncope, aborted cardiac arrest, or death, whereas others have a normal QTc; and may be asymptomatic.
Among the potential factors that may explain the incomplete penetrance and variable expressivity associated with LQTS is the presence of multiple mutations. Multiple mutations (i.e ≥ 2 LQTS-associated mutations in the same individual) could involve either i) the same allele of one LQTS-associated gene, ii) both alleles of the same LQTS-associated gene, or iii) different LQTS genes. Excluding patients with the autosomal recessive form of LQTS associated with deafness (Jervell and Lange-Nielsen syndrome), it has been suggested that the prevalence of multiple mutation status world wide is between 8% and 11% among unrelated patients with LQTS and that patients with multiple mutation-LQTS have a greater risk for cardiac events than those with a single LQTS mutation.3–6
However, there are limited data on the risk for life-threatening cardiac events (including defibrillator shock, aborted cardiac arrest [ACA] and sudden cardiac death [SCD]) among patients with multiple mutations,3, 5 In particular, there are no data comparing the risk of life threatening cardiac events among important subgroups of patients with multiple mutations.
Therefore, the present study aimed to 1) evaluate and compare the risk for life-threatening cardiac events among patients with multiple mutations to those with a single LQTS mutation; and 2) evaluate the effect of multiple mutation status involving the same gene vs. multiple mutation status involving multiple genes on the patient’s clinical course.
Methods
Study population and data collection
The study population of 403 subjects was derived from 196 proband-identified families drawn from the US portion of the International LQTS Registry.7 Patients were divided into two groups based on the presence of either i) multiple, LQTS-associated mutations or ii) a single LQTS-associated mutation. The group of patients with only one LQTS mutation (control group) were drawn from a group of patients consisting of 1) family members of patients with multiple mutations who were tested for all known family mutations, or 2) patients who were tested for a minimum of the following 5 LQTS-associated genes: LQTS 1, 2, 3, 5, and 6 (12.4% of them were tested for all 12 LQTS genes). Patients with the Jervell and Lange-Nielsen syndrome (those with LQTS and sensory hearing loss) were excluded from the study. For each patient, data on personal and family history, cardiac events, and therapy were recorded systematically at each visit or medical contact. Clinical data were recorded on prospectively designed forms and included patient and family history and demographic, electrocardiogram (ECG), therapeutic, and cardiac event information. Measured parameters on the first recorded ECG included QT and R-R intervals in milliseconds, with QT corrected for heart rate by Bazett’s formula. All subjects or their guardian provided informed consent for participation in the Registry and genetic studies. The reported analyses used the LQTS analytic database version 18.
Genotype characterization
The LQTS gene mutations were identified with the use of standard genetic tests performed in academic molecular-genetic laboratories (including the Functional Genomics Center, University of Rochester Medical Center, Rochester, NY; Baylor College of Medicine, Houston, TX; the Windland Smith Rice Sudden Death Genomics Laboratory, Mayo Clinic, Rochester, MN; and Boston Children’s Hospital, Boston, MA) and by commercial genetic laboratories (including GeneDx, Gaithersburg, MD; and Transgenomic (formerly PGxHealth [FAMILION], New Haven, CT). Multiple mutation-LQTS status was defined as the presence of 2 or more mutations in one or more LQTS-associated genes (LQTS 1–12). Patients with multiple mutations were further dichotomized into two subgroups: those with multiple mutations involving the same LQTS gene and those with mutations in different LQTS genes. Regarding the former, cis/trans status of the mutations involving the same gene were not distinguished systematically so both mutations could involve the same allele or both alleles could each have a separate mutation.
End point
The primary end point of the study was the occurrence of a first life-threatening cardiac event, comprising ICD shock for VT/VF, aborted cardiac arrest ([ACA] requiring external defibrillation as part of the resuscitation) or LQTS-related sudden cardiac death ([SCD] abrupt in onset without evident cause, if witnessed, or death that was not explained by any other cause if it occurred in a non-witnessed setting) from birth through age 40 years. The consistency of the results was also assessed for a secondary end point including only ACA or SCD. Follow up after age 40 years was not included to minimize the influence of coronary disease on cardiac events.
Statistical Analysis
Characteristics of patients with either single or multiple mutations were compared with student t-test or Chi square and Fisher exact tests, as appropriate. The probability of a first life-threatening cardiac event by mutation subgroup was graphically displayed using the Kaplan-Meier method, with comparison of cumulative probability of events by the log-rank test. A Mantel-Byar graph8 was used to compare the cumulative probability of life threatening cardiac event between patients without a history of syncope and following the occurrence of syncope.
The Cox proportional-hazards survivorship model was used to evaluate the independent contribution of clinical and genetic factors to the first occurrence of the primary endpoint from birth through age 40 years. Pre-specified covariates in the multivariate model included: gender, baseline QTc ≥ 500msec, time dependent β-blocker therapy, and mutation type subgroup. Patients who did not have an ECG for QTc measurement (n = 15) were identified in the Cox models as “QTc missing”, and all Cox models were adjusted for this QTc missing parameter. The Cox regression models were stratified by decade of birth year. We have also adjusted for the effect of potential lack of independence between subjects using the robust sandwich estimator for family membership.9–10 In a secondary analysis, the effect of each clinical covariate (adding also prior syncope as a time dependent variable) was assessed in the subgroup of patients with multiple mutations by interaction-term analysis (i.e. by including a mutation type-by-risk factor interaction-term in the multivariate models), with interactions tested one at a time. The statistical software used for the analyses was SAS version 9.20 (SAS Institute Inc, Cary, NC). A 2-sided 0.05 significance level was used for hypothesis testing.
Results
Study population
The present study includes 403 patients from 196 families. There were 346 patients with a single LQTS mutation (control group) from 188 families, 57 patients with multiple mutations from 23 families (54 patients had 2 mutations and 3 patients had 3 mutations). Of the 57 patients with multiple LQTS mutations, 24 had ≥ 2 mutations in the same gene and 33 had ≥ 1 mutation involving more than one gene. The spectrum of mutations as categorized by single/multiple mutations and LQTS gene and their respective number of mutation-positive subjects are presented in Table 1s (Supplementary Appendix).
The clinical characteristics of patients by mutation type are presented in Table 1. There were no significant differences between the two subgroups of patients with regard to gender, baseline heart rate, the proportion of proband patients, and the proportion of patients implanted with an ICD. However, patients with multiple mutations exhibited a longer QTc at enrollment, were treated with β-blockers more frequently during follow-up, and had a higher frequency of life threatening cardiac events (dominated by ICD shocks) as compared to patients with a single mutation.
Table 1.
Clinical characteristics
| Single mutation | Multiple mutations | P-value | |
|---|---|---|---|
| N = | 346 | 57 | |
| Male, n (%) | 144 (41.6%) | 23 (40.4%) | 0.857 |
| QTc, msec | 480 ± 56 | 506 ± 72 | 0.003 |
| QTc > 500 msec, n (%) | 94 (27.2%) | 22 (38.6%) | 0.077 |
| QT, msec | 436 ± 86 | 446 ± 92 | 0.431 |
| RR, msec | 838 ± 238 | 787 ± 206 | 0.143 |
| Proband, n (%) | 125 (36.1%) | 20 (35.1%) | 0.880 |
| Age at 1st Syncope, years | 22.5 ± 13.9 | 19.5 ± 14.9 | 0.129 |
| Age at 1st ACA/LQTS death, years | 28.3 ± 13.1 | 27.3 ± 12.5 | 0.593 |
| Syncope, n (%) | 140 (40.5%) | 28 (49.1%) | 0.219 |
| ICD shock, n (%) | 10 (2.9%) | 5 (8.8%) | 0.030 |
| ACA, n (%) | 18 (5.2%) | 3 (5.3%) | 0.985 |
| LQTS death, n (%) | 1 (0.3%) | 2 (3.5%) | – |
| ACA, ICD shock or LQTS death, n (%) | 26 (7.5%) | 9 (15.8%) | 0.040 |
| Beta Blockers used, n (%) | 205 (59.3%) | 42 (73.7%) | 0.038 |
| ICD, n (%) | 64 (18.5%) | 9 (15.8%) | 0.623 |
| Pacemaker, n (%) | 21 (6.1%) | 3 (5.3%) | 0.812 |
| LCSD, n (%) | 4 (1.2%) | 0 (0.0%) | – |
Data are presented as number of patients (%) or mean ± SD
ACA = aborted cardiac arrest, ICD = implantable cardioverter defibrillator, JLNS = Jervell Lange-Nielsen Syndrome, LCSD = left cervical sympathetic denervation.
Risk of life threatening cardiac events by mutation type subgroup
The cumulative probability of the first occurrence of a life-threatening cardiac event (including ICD shock, ACA, or SCD) was significantly higher among patients with multiple LQTS mutations as compared to patients with a single LQTS-associated mutation. Thus, at age 40 years the rate of life threatening cardiac events was 23 percent among multiple mutation patients, and 11 percent among single mutation patients (p < 0.001 for the overall difference during follow-up; Figure 1). In multivariate analysis, we observed consistent results: patients with multiple mutations had a 2.3 fold (p = 0.015) increased risk for life threatening cardiac events as compared to patients with a single mutation. Notably, the presence of multiple mutations involving the same LQTS gene was associated with a greater than 3-fold increased risk for life threatening cardiac events (p = 0.010) whereas the risk associated with multiple mutations in different LQTS genes was not significantly different as compared with single mutations. (HR 1.7, p = 0.26). Results were consistent in a secondary analysis in which the end point of ACA or SCD (i.e. excluding appropriate ICD shocks from the composite end point) was assessed.
Figure 1.
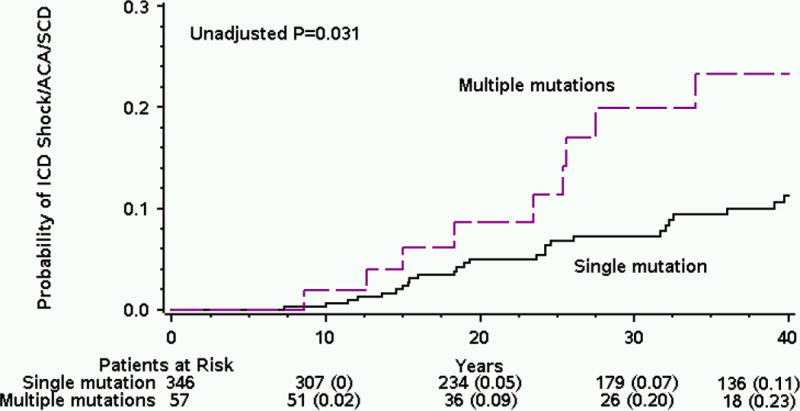
Cumulative probability of life threatening cardiac events by mutation subgroup.
ACA = aborted cardiac arrest; ICD = implantable cardioverter defibrillator; SCD = sudden cardiac death.
Clinical predictors of life threatening cardiac events among patients with multiple mutations
In order to evaluate independent clinical predictors for life threatening cardiac events among patients with multiple mutations, we carried out a secondary multivariate analysis adjusting for clinical variables only. This analysis showed that the development of time-dependent syncope and a prolonged QTc > 500 msec were the only independent predictors of life threatening cardiac events in this high risk subgroup of patients. Thus, prior syncope (used in analysis as a time dependent variable) was associated with a prominent 15-fold (p < 0.001) increased risk for life threatening cardiac events and QTc > 500 msec was associated with a 4-fold (p = 0.038) increased risk. Consistently, Mantel-Byar event free survival analysis showed that the occurrence of syncope in patients with multiple mutations was associated with a significant increase in the risk of a subsequent life threatening event (Fig. 3). This risk factor was associated with a 48% cumulative probability of life threatening cardiac event among patients with multiple mutations from birth through age 40 years.
Nine patients with multiple mutations experienced life threatening cardiac events. Eight out of 9 patients were female, 7 out of 8 patients with available ECG data had a very prolonged QTc (> 500 msec). Notably, 4 of the 9 patients (44%) experienced the life-threatening event during beta-blocker therapy, and 8/9 (89%) experienced syncope before the occurrence of life threatening cardiac event (mostly during the adolescence or early post-adolescence period). Six patients had multiple mutations in the same gene and 3 had multiple mutations in different genes.
Discussion
The present study is the first to assess the risk of life-threatening cardiac events among patients with multiple LQTS mutations and to evaluate outcome among specific subsets of this group as compared to patients with a single-mutation. Our findings provide several important implications useful in risk assessment and management of patients with LQTS. We have shown that: 1) patients with multiple mutations have a significantly increased risk for life threatening cardiac events (including ICD shocks, ACA, or SCD) as compared to patients with single mutations; 2) Among patients with multiple mutations, double hit in a single gene was associated with a greater risk for life threatening cardiac events than double hit in different genes; and 3) most patients with multiple mutations who experience fatal/near-fatal events experience prior syncope during adolescence or early adulthood.
Several studies have investigated the risk for cardiac events among LQTS patients with multiple mutations. Westenskow et al.3 reported on 20 patients (8% of probands) with compound mutations who had a more severe phenotype than probands with one or no identified mutations. Functional studies in Xenopus oocytes showed that coexpression of 2 mutant subunits was associated with a reduction in current density that was equivalent to the additive effects of the single mutations.3 Tester et al.4 reported on 29 patients (10.8% of probands) with compound mutations and found that these patients experience cardiac events earlier in life than patients with a single mutation, but their QTc was not significantly prolonged nor did they have more frequent cardiac events than patients with a single mutation. Most recently, Itoh et al.5 reported on 26 patients (8% of probands) with compound mutations in a multicenter Japanese population. Patients with compound mutations were found to have a prolonged QTc and earlier onset of cardiac events. These studies, however, were limited by the small number of patients and therefore focused primarily on the endpoint of cardiac event of any type (primarily syncope). In addition they did not compare the risk between subsets of patients with compound mutations.
The present study confirms previous findings regarding the risk associated with a prolonged QTc and the earlier onset of cardiac events among patients with multiple mutations. Furthermore, we have shown that LQTS patients with multiple mutations have a 2.3-fold increased risk for life threatening cardiac events (comprising ICD shocks, ACA or SCD), and that patients with multiple mutations in a single LQTS gene (comprising 42% of patients with multiple mutations) had the highest risk for life threatening cardiac events, exhibiting more than 3-fold increased risk as compared to patients with a single mutation.
The presence of two mutations in either the LQT1 or LQT5 gene may result in the Jervell and Lange-Nielsen syndrome (JLNS), a severe form of LQTS with sensory hearing loss due to involvement of the auditory nerves11–12. The JLNS is a relatively rare disease, comprising less than 1% of affected patients with LQTS, and is associated with high rates of syncope and life-threatening cardiac events.13 It should be noted that in the present study we excluded patients with the JLNS. The pathogenic mechanism among patients with multiple mutations in a single LQTS gene may be similar to the pathogenic mechanism among patients with the JLNS, increasing the mutation burden on the same ion channel current and thus resulting in a very high risk for fatal or near fatal events.
Finally, the present study showed that the strongest predictor of life threatening cardiac events among the high risk subgroup of patients with multiple mutations is occurrence of syncope. Patients with multiple mutations who experienced syncope had a cumulative risk for fatal or near fatal cardiac events of 48% during follow-up. Furthermore, 44% of the patients who experienced fatal/near-fatal events experienced the event during beta-blocker therapy and 89% experienced prior syncope during adolescence or early adulthood, suggesting that more aggressive therapies (including ICD implantation) should be recommended in patients with multiple mutations who experience symptoms during follow-up.
Study Limitations
The current study included patients who were thoroughly tested for multiple mutations; we did not include proband patients who were tested for fewer than 5 LQTS-associated genes or individuals who were found positive for the LQTS-causing family mutation but were tested for only that single mutation as we could not rule out that they harbor an additional LQTS mutation. Therefore, we could not determine the prevalence of multiple mutations in the international LQTS registry. We could not discern among patients with multiple mutations in the same gene, whether the two mutations reside on the same allele, or whether each allele contains a separate mutation. One of the mutations included in this study (R1047L) is controversial due to a reported frequency of 2–4%. However, results were similar when this mutation was excluded from the analysis. Patients with multiple mutations in different LQTS genes exhibited a somewhat higher risk for life threatening cardiac events, which was not significantly different as compared with single mutations. However, this analysis may be limited by sample size and should therefore be interpreted with caution. Thus, it is possible that inclusion of a larger cohort of patients will show a significant increased risk for life threatening cardiac events even among patients with digenic mutations as compared with single mutations. Due to sample size limitation, we should use caution before extrapolating the present study results to clinical practice. Nevertheless, this study is the largest study to date exploring the effects of multiple LQTS mutations on clinical outcome.
Summary and clinical implications
Advances in genetic testing technology have led to a proliferation of new genetic tests and more cost effective genetic testing that can establish a definitive molecular diagnosis for symptomatic patients suspected to have inherited arrhythmias. Specifically, genetic testing and genotype-phenotype correlations can add important information for predicting outcome and for selection of treatment among patients with the congenital long QT syndrome.14–17 The presence of multiple mutations is not uncommon among LQTS patients (8–11%) and is associated with worse outcome as compared to a single mutation. Herein, we have shown that LQTS patients with multiple mutations have a greater risk for life-threatening cardiac events as compared to patients with a single mutation and have found a subset of patients with multiple mutations that is associated with worse outcome.
Should our findings be confirmed in a larger cohort, we would recommend using a lower threshold for ICD implantation among patients with multiple mutations, particularly among patients who had experienced syncope or those with multiple mutations in the same LQTS gene.
Supplementary Material
Figure 2.
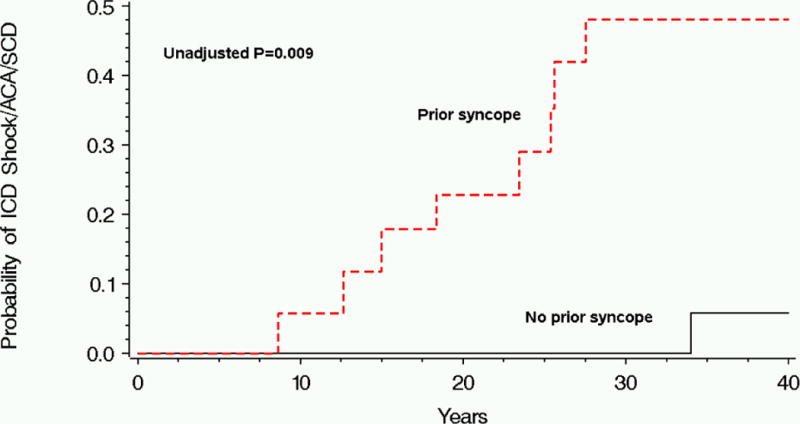
Cumulative probability of life threatening cardiac events by history of syncope among patients with multiple mutations.
ACA = aborted cardiac arrest; ICD = implantable cardioverter defibrillator; SCD = sudden cardiac death.
Table 2.
Multivariate analysis: Predictors of life threatening cardiac events.
| Hazard ratio | 95% CI | P Value | |
|---|---|---|---|
| Multiple mutations vs. single mutation* | 2.28 | 1.17–4.42 | 0.015 |
| Multiple mutations in single gene vs. single mutation | 3.15 | 1.30–7.61 | 0.010 |
| Multiple mutations involving different genes vs. single mutation | 1.68 | 0.69–4.11 | 0.256 |
| Female | 2.25 | 1.01–5.02 | 0.048 |
| QTc ≥ 500msec | 2.22 | 1.04–4.74 | 0.041 |
Models were adjusted also for time dependent beta blockers, and QTc missing.
The effects of multiple mutations in single gene and in multiple genes were assessed in a separate model.
Table 3.
Multivariate analysis: Clinical predictors of life threatening cardiac events among patients with multiple mutations.
| Hazard Ratio | 95% CI | P Value | |
|---|---|---|---|
| Syncope | 15.83 | 4.89–51.19 | < 0.001 |
| QTc ≥ 500msec | 4.45 | 1.09–18.18 | 0.038 |
| Female | 5.94 | 0.65–54.48 | 0.115 |
Models were adjusted also for time dependent beta blockers, and QTc missing.
List of abbreviations
- ACA
Aborted cardiac arrest
- ECG
Electrocardiogram
- ICD
Implantable Cardioverter Defibrillator
- LQTS
Long QT Syndrome
- QTc
Corrected QT interval
- SCD
Sudden Cardiac Death
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest disclosure: This work was supported in part by research grantsHL-33843 and HL-51618 to the University of Rochester Medical Center from the National Institutes of Health, Bethesda, Maryland. Dr. Moss reports receiving a research grant from GeneDx; Dr. Kaufman research grant from CardioDx and St. Jude Medical. Dr. Ackerman has a consulting relationship and license agreement/royalty arrangement with Transgenomic and received consultant fees from Medtronic, Biotronik, Boston Scientific, and St Jude Medical.
This research was carried out while Dr. Alon Barsheshet was a Mirowski-Moss Career Development Awardee at the University of Rochester Medical Center, Rochester, NY.
References
- 1.Goldenberg I, Moss AJ. Long QT syndrome. J Am Coll Cardiol. 2008;51:2291–300. doi: 10.1016/j.jacc.2008.02.068. [DOI] [PubMed] [Google Scholar]
- 2.Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866–74. doi: 10.1056/NEJMoa022147. [DOI] [PubMed] [Google Scholar]
- 3.Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Compound mutations: a common cause of severe long-QT syndrome. Circulation. 2004;109:1834–41. doi: 10.1161/01.CIR.0000125524.34234.13. [DOI] [PubMed] [Google Scholar]
- 4.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–17. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 5.Itoh H, Shimizu W, Hayashi K, et al. Long QT syndrome with compound mutations is associated with a more severe phenotype: a Japanese multicenter study. Heart Rhythm. 2010;7:1411–8. doi: 10.1016/j.hrthm.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 6.Wilde AA. Long QT syndrome: a double hit hurts more. Heart Rhythm. 2010;7:1419–20. doi: 10.1016/j.hrthm.2010.06.027. [DOI] [PubMed] [Google Scholar]
- 7.Moss AJ, Schwartz PJ, Crampton RS, et al. The long QT syndrome. Prospective longitudinal study of 328 families Circulation. 1991;84:1136–44. doi: 10.1161/01.cir.84.3.1136. [DOI] [PubMed] [Google Scholar]
- 8.Mantel N, Byar D. Evaluation of response-time data involving transient states: an illustration using heart transplant data. J Am Stat Assoc. 1974;69:81–6. [Google Scholar]
- 9.Lin DY, Wei LJ. The Robust Inference for the Proportional Hazards Model. J Am Stat Assoc. 1989;84:1074–8. [Google Scholar]
- 10.Therneau TM, Grambsch PM. Modeling Survival Data: Extending the Cox Model. New York, NY: Springer-Verlag; 2000. [Google Scholar]
- 11.Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J. 1957;54:59–68. doi: 10.1016/0002-8703(57)90079-0. [DOI] [PubMed] [Google Scholar]
- 12.Schwartz PJ, Spazzolini C, Crotti L, et al. The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation. 2006;113:783–90. doi: 10.1161/CIRCULATIONAHA.105.592899. [DOI] [PubMed] [Google Scholar]
- 13.Goldenberg I, Moss AJ, Zareba W, et al. Clinical course and risk stratification of patients affected with the Jervell and Lange-Nielsen syndrome. J Cardiovasc Electrophysiol. 2006;17:1161–8. doi: 10.1111/j.1540-8167.2006.00587.x. [DOI] [PubMed] [Google Scholar]
- 14.Zareba W, Moss AJ, Schwartz PJ, et al. Influence of genotype on the clinical course of the long-QT syndrome. International Long-QT Syndrome Registry Research Group. N Engl J Med. 1998;339:960–5. doi: 10.1056/NEJM199810013391404. [DOI] [PubMed] [Google Scholar]
- 15.Moss AJ, Shimizu W, Wilde AA, et al. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation. 2007;115:2481–9. doi: 10.1161/CIRCULATIONAHA.106.665406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldenberg I, Zareba W, Moss AJ. Long QT Syndrome. Curr Probl Cardiol. 2008;33:629–94. doi: 10.1016/j.cpcardiol.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 17.Shimizu W, Horie M, Ohno S, et al. Mutation site-specific differences in arrhythmic risk and sensitivity to sympathetic stimulation in the LQT1 form of congenital long QT syndrome: multicenter study in Japan. J Am Coll Cardiol. 2004;44:117–25. doi: 10.1016/j.jacc.2004.03.043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.