

Rhodium-stabilized carbenes possess diverse reactivity in a variety of organic transformations, such as addition to C–C and C–heteroatom multiple bonds, insertion into C–H and C–heteroatom bonds, as well as ylide formation.[1] However, this powerful chemistry is mostly limited to rhodium carbenes derived from the corresponding diazo compounds. Obviously, development of new and efficient methods to access Rh carbenoids from alternative and stable precursors could expand the scope of this chemistry.
It is known that the stability of the 1,2,3-triazole ring is affected by the substituents at the N1, C4, and C5 atoms of the heterocycle.[2] For example, triazoles 1 that bear a sulfonyl group at the N1 atom exist in equilibrium with diazoimine tautomer 2.[3] Gevorgyan and Fokin took advantage of this process by trapping diazoimine 2 with a RhII catalyst to produce the putative RhII–iminocarbene 3 (Scheme 1).[4] This intermediate possessed reactivity inherent for RhII carbenoids. For example, it reacted with alkenes to form cyclopropane derivatives 4.[4] Later, this process was performed by Fokin and co-workers in a highly enantioselective manner.[5] On the other hand, the presence of the imino group at the α-position of 3 opens opportunities for novel heterocyclizations. Thus, the transannulation[6] reaction of N-sulfonyl triazoles with nitriles produced imidazoles 5,[4] whereas the reaction with alkynes[7a] led to pyrroles 6.[7b] As shown by Fokin and co-workers, Rh–iminocarbene 3 could also undergo insertion into a secondary or tertiary C–H bond of alkanes (as a solvent) to produce valuable β-chiral amines 7 in high yields and enantioselectivities.[8a] In 2012, Murakami reported an efficient RhII-catalyzed hydration of triazoles 1 to form α-aminoketones 8, proceeding through insertion of the RhII–iminocarbene intermediate into the O–H bond of water.[8b] Shortly after, Fokin and co-workers disclosed the RhII-catalyzed reaction of N-sulfonyl triazoles 1 with arylboronic acids, thus stereoselectively furnishing enamines 9.[8c] Note-worthy, the N-sulfonyl triazole precursors are easily available by Cu-catalyzed alkyne–azide cycloaddition (CuAAc) reaction,[9] which makes this approach to Rh-stabilized carbenes attractive for the synthesis of valuable carbo- and heterocyclic molecules (Scheme 1).
Scheme 1.

Use of N-sulfonyl triazole to form Rh–iminocarbene.
Very recently, the groups of Murakami[10] and Fokin[11] independently reported the RhII-catalyzed denitrogenative rearrangement of 1-(N-sulfonyl-triazol-4-yl)alkanols 10, proceeding through migration of different groups to the Rh–carbene center of 12. The subsequent elimination of rhodium from the 12 produces iminoenol 14, which is converted to Z-substituted enaminone 11 upon facile proton transfer (Scheme 2).
Scheme 2.

The RhII-catalyzed rearrangement of triazolyl alcohols. Conditions A (Miura and Murakami[10]): CHCl3, 140°C, microwave, 15 min. Conditions B (Fokin[11]): CHCl3, 70°C, 5–60 min. Oct=octanoate, Ts=4-toluenesulfonyl.
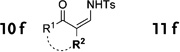
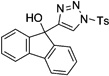
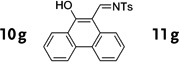
In general, the migratory aptitude of different groups to the Rh–carbene center derived from a diazo compound follows the common tendency: hydride > phenyl > primary alkyl > secondary alkyl groups.[12] Likewise, in the transformation 12→13, the 1,2-migration of hydride is favored over the 1,2-shift of alkyl and phenyl groups (Table 1, entries 1 and 2, respectively). The phenyl group, in turn, migrates more easily than the methyl group (entry 3), whereas the methyl group migrates more easily than the isopropyl group (entry 4). Furthermore, cyclic 1-triazolylalkanols 10 f underwent efficient ring expansion to produce the corresponding cyclic enaminones 11 f (entries 7 and 8). The reaction of fluorenol derivative 10g led almost quantitatively to the corresponding hydroxyphenanthrene derivative 11 g (entries 9 and 10). In most cases, a predominant formation of the Z-stereoisomer was observed, which can be attributed to the concerted transformation 14→11 (kinetic control), and to the higher stability of the Z-stereoisomer as a result of an additional stabilization through intramolecular hydrogen bonding (thermodynamic control).
Table 1.
Migrations of different groups to the metal–carbene center of RhII–iminocarbenes (see Scheme 2).
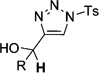
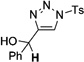
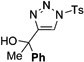
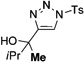
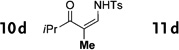
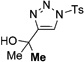
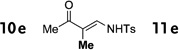
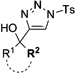
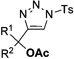
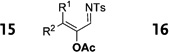
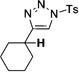
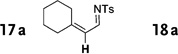
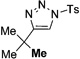
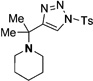
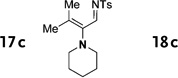
Interestingly, protection of the hydroxy group as acetate enabled its selective migration to the carbene center to form products 16 (entry 11). It was also shown that 4-cyclohexyl and 4-tert-butyl triazoles gave the products of hydride and methyl group migration (18a and 18b, respectively) under these reaction conditions (entries 12 and 13). Moreover, Fokin and co-workers reported the first example of amine migration to the RhII carbenoid center. Thus, the reaction of 4-alkylamino triazole 17c produced the corresponding enamine 18c in good yields (entry 14).[11]
In conclusion, the RhII–iminocarbenes, derived from the corresponding N-sulfonyl 1,2,3-triazoles, could be used in several transformations inherent for metal carbenoids. Thus, cyclopropanation of alkenes, reactions with alkynes, nitriles, and boronic acids, as well as insertion into C–H and O–H bonds were impressively developed. In addition, the recent reports also disclosed migrations of different groups to the RhII–carbene center of imino carbenoids. The N-sulfonyl 1,2,3-triazole precursors are easily available by CuAAc reaction of alkynes with azides, which makes this approach very useful for straightforward generation of RhII carbenoids. Some transformations could even be efficiently performed in a one-pot manner starting from alkynes and sulfonyl azides. Therefore, the reactivity of RhII–iminocarbenes can be tuned easily by variation of substituents in the parent triazole through the simple CuAAc approach. Moreover, a natural low concentration of diazoimine, which exists in equilibrium with triazoles, maintains a low concentration of the reactive RhII carbenoid, which obviates the necessity of slow-addition techniques that are often required in the reactions of diazo compounds.
Footnotes
The support of the National Institutes of Health (GM-64444) is gratefully acknowledged.
References
- 1. For selected reviews on the reactivity of Rh carbenoids, see: Timmons DJ, Doyle MP. J. Organomet. Chem. 2001;617–618:98–104. Davies HML, Manning JR. Nature. 2008;451:417–424. doi: 10.1038/nature06485. Evans PA, editor. Modern Rhodium Catalyzed Organic Reactions. Weinheim: Wiley-VCH; 2005.
- 2.See, for example: Gilchrist TL, Gymer GE. Adv. Heterocycl. Chem. 1974;16:33.
- 3. For earlier reports on the ring-opening of N-sulfonyl 1,2,3-triazoles, see: Harmon RE, Stanley F, Gupta SK, Johnson J. J. Org. Chem. 1970;35:3444–3448. Himbert G, Regitz M. Chem. Ber. 1972;105:2963–2974. Himbert G, Regitz M. Synthesis. 1972:571–573.
- 4.Horneff T, Chuprakov S, Chernyak N, Gevorgyan V, Fokin VV. J. Am. Chem. Soc. 2008;130:14972–14974. doi: 10.1021/ja805079v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Chuprakov S, Kwok SW, Zhang L, Lercher L, Fokin VV. J. Am. Chem. Soc. 2009;131:18034–18035. doi: 10.1021/ja908075u. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Grimster N, Zhang L, Fokin VV. J. Am. Chem. Soc. 2010;132:2510–2511. doi: 10.1021/ja910187s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. For recent review on transannulation reactions of triazoles, see: Chattopadhyay B, Gevorgyan V. Angew. Chem. 2012;124:886–896. doi: 10.1002/anie.201104807. Angew. Chem. Int. Ed. 2012;51:862–872. doi: 10.1002/anie.201104807.; for the first example of a transannulation reaction of pyridotriazoles, see: Chuprakov S, Hwang FW, Gevorgyan V. Angew. Chem. 2007;119:4841–4843. doi: 10.1002/anie.200700804. Angew. Chem. Int. Ed. 2007;46:4757–4759. doi: 10.1002/anie.200700804.
- 7.a) Miura T, Yamauchi M, Murakami M. Chem. Commun. 2009:1470–1471. doi: 10.1039/b819162j. [DOI] [PubMed] [Google Scholar]; b) Chattopadhyay B, Gevorgyan V. Org. Lett. 2011;13:3746–3749. doi: 10.1021/ol2014347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Chuprakov S, Malik JA, Zibinsky M, Fokin VV. J. Am. Chem. Soc. 2011;133:10352–10355. doi: 10.1021/ja202969z. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Miura T, Biyajima T, Fujii T, Murakami M. J. Am. Chem. Soc. 2012;134:194–196. doi: 10.1021/ja2104203. [DOI] [PubMed] [Google Scholar]; c) Selander N, Worrell BT, Chuprakov S, Velaparthi S, Fokin VV. J. Am. Chem. Soc. 2012;134:14670–14673. doi: 10.1021/ja3062453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fokin VV, Raushel J. Org. Lett. 2010;12:4952–4955. doi: 10.1021/ol102087r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miura T, Funakoshi Y, Morimoto M, Biyajima T, Murakami M. J. Am. Chem. Soc. 2012;134:17440–17443. doi: 10.1021/ja308285r. [DOI] [PubMed] [Google Scholar]
- 11.Selander N, Worrell BT, Fokin VV. Angew. Chem. 2012;124:13231–13234. doi: 10.1002/anie.201207820. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2012;51:13054–13057. doi: 10.1002/anie.201207820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. For migration of different groups to Rh–carbene centers, see Ref. [10] and references therein.