

Abstract
Visceral afferents expressing transient receptor potential (TRP) channels TRPV1 and TRPA1 are thought to be required for neurogenic inflammation and development of inflammatory hyperalgesia. Using a mouse model of chronic pancreatitis (CP) produced by repeated episodes (twice weekly) of caerulein-induced AP (AP), we studied the involvement of these TRP channels in pancreatic inflammation and pain-related behaviors. Antagonists of the two TRP channels were administered at different times to block the neurogenic component of AP. Six bouts of AP (over 3 wks) increased pancreatic inflammation and pain-related behaviors, produced fibrosis and sprouting of pancreatic nerve fibers, and increased TRPV1 and TRPA1 gene transcripts and a nociceptive marker, pERK, in pancreas afferent somata. Treatment with TRP antagonists, when initiated before week 3, decreased pancreatic inflammation and pain-related behaviors and also blocked the development of histopathological changes in the pancreas and upregulation of TRPV1, TRPA1, and pERK in pancreatic afferents. Continued treatment with TRP antagonists blocked the development of CP and pain behaviors even when mice were challenged with seven more weeks of twice weekly caerulein. When started after week 3, however, treatment with TRP antagonists was ineffective in blocking the transition from AP to CP and the emergence of pain behaviors. These results suggest: (1) an important role for neurogenic inflammation in pancreatitis and pain-related behaviors, (2) that there is a transition from AP to CP, after which TRP channel antagonism is ineffective, and thus (3) that early intervention with TRP channel antagonists may attenuate the transition to and development of CP effectively.
Introduction
Chronic pancreatitis (CP) is a debilitating disease characterized by persistent inflammation, pain, and irreversible morphological changes often accompanied by partial or total loss of function. Pain in CP may initially be episodic, but increases in occurrence and intensity as the disease develops. In contrast, acute pancreatitis (AP) is defined as an inflammatory event from which the pancreas recovers. Although some argue that AP and CP represent a continuous spectrum of the same disease (Dimcevski et al., 2007), AP and CP have distinct histopathologies, etiologies, and time courses (Dimcevski et al., 2007; Demir et al., 2010). It is also widely accepted that recurrent bouts of AP (RAP) increase the likelihood of developing CP (Demir et al., 2010; Puylaert et al., 2011).
Pain in CP is common and reflects sensitization of pancreatic afferent (sensory) neurons and development of neurogenic inflammation (Liddle and Nathan, 2004; Anaparthy and Pasricha, 2008). Inflammation exposes pancreatic afferents to inflammatory mediators, endogenous neuropeptides, and immune-competent cells and their released cytokines. Unchecked, this process causes destruction of ducts and eventually nerve damage and hyperexcitability. Pain and inflammation associated with pancreatitis has been shown to require transient receptor potential (TRP) channel TRPV1- and TRPA1-expressing afferents, which, when targeted, attenuate the development of experimental AP in mice (Nathan et al., 2001; Schwartz et al., 2011). Based on these results, it has been proposed that activity in this population of pancreatic afferents is responsible for the neurogenic inflammation that causes tissue damage and exacerbation of the initial pancreatic insult. We recently reported a significant increase in TRPV1 and TRPA1 mRNA expression and function in pancreatic afferents in a model of caerulein-induced AP (Schwartz et al., 2011). These changes were correlated with leukocyte infiltration of the pancreas that resolved within 7 d. These changes in afferent function were responsible for at least a portion of the inflammatory reaction, as evidenced by their reversal using TRPV1 or TRPA1 antagonists. Application of these antagonists reduced caerulein-induced AP and pain-related behaviors significantly, and combining the two antagonists produced a greater than additive effect (Schwartz et al., 2011).
The present study used a model of RAP (two episodes/week for up to 10 weeks) that over time developed hallmarks of CP, including pain, fibrosis, and persistent immune cell infiltration of the pancreas. To evaluate the relative contribution of the two sources of pancreatic afferent innervation, we studied spinal and vagal pancreatic sensory neurons in dorsal root ganglia (DRG) and nodose ganglia (NG), respectively. We identified a critical period in the third week of RAP during which a combination of TRPV1 and TRPA1 antagonists prevented RAP from developing into CP. If combination TRP antagonist treatment was initiated after the third week of RAP, however, blockage of TRP channel function was no longer able to reverse inflammation-induced changes in the pancreas, suggesting that TRPV1- and TRPA1-dependent neurogenic inflammation is required for the transition from AP to CP and pain-related behaviors. Once the transition occurs and CP is established, progression of the disease becomes independent of the activity of the two channels.
Materials and Methods
Animals.
Experiments were performed on 6- to 14-week-old male C57BL/6 mice (The Jackson Laboratory) housed in the Association for Assessment and Accreditation of Laboratory Animal Care-accredited facility at the University of Pittsburgh. Mice had access to water and food ad libitum. All protocols were approved by the institutional animal care and use committee.
Surgical procedures and cell labeling.
All surgeries were performed under aseptic conditions. Anesthesia was initiated by inhaled 4% isoflurane and maintained with 2% isoflurane. For retrograde labeling of pancreatic DRG and NG neurons, a laparotomy was performed to expose the pancreatic head into which three injections of Fast Blue (FB; 1% in sterile saline; total volume injected ∼5 μl) were made using a microsyringe. The abdominal muscles and overlying skin were sutured separately and mice were allowed to recover for at least 4 d before any other procedures were performed.
Caerulein-induced CP.
CP was induced by repeated injections of the cholecystokinin analog caerulein (Sigma-Aldrich). Caerulein was dissolved in 0.01 m phosphate-buffer (PB) and administered intraperitoneally (50 μg/kg in 100 μl/injection) hourly for eight doses twice weekly (Tuesdays and Fridays) for 10 weeks (Fig. 1). Control (vehicle) mice received injections of PB. After the last caerulein injection (i.e., 72 h after 1, 3, 6, or 10 weeks of twice weekly injections), mice were killed and pancreata quickly removed, rinsed in saline, blotted, and divided for histological grading, myeloperoxidase quantification, and immunohistochemistry to assess the extent of inflammation. Spinal T9–12 DRG and NG were also harvested, acutely dissociated, cultured (see below), and analyzed for TRPV1 and TRPA1 gene expression by single-cell reverse transcriptase (RT)-PCR assays or used for Ca2+ imaging experiments.
Figure 1.

Time course (10 weeks) and treatment schedule for caerulein (caer) and TRP antagonists. CP was induced by repeated injections of caer administered intraperitoneally (50 mg/kg in 100 ml/injection) hourly for eight doses twice weekly for 10 weeks. The TRPA1 antagonist A-967079 and/or TRPV1 antagonist A-889425 or vehicle (veh) was administered intraperitoneally twice weekly: 1 h before the first caer or veh injection and 4 h later. Antagonist treatment was initiated either 3 weeks before or 3 weeks after the beginning of caer treatment.
Myeloperoxidase assay and histology.
The myeloperoxidase (MPO) assay was performed as described previously (Schwartz et al., 2011). Mice were overdosed with inhaled isoflurane and pancreata were dissected, weighed, added to a beaker containing 1.0 ml 0.5% hexadecyltrimethylammonium bromide (HTAB; Sigma-Aldrich), and finely minced. The contents were transferred to a 15 ml centrifuge tube with another 2 ml of HTAB and sonicated for 10 s before being homogenized for 30 s. Another 2 ml of HTAB was added and the tube was placed on dry ice until all samples were similarly prepared. The samples underwent three freeze–thaw cycles, were centrifuged twice, and then loaded, along with MPO standards (Calbiochem), onto a 96-well plate. The samples and standards were reacted with O-dianisidine dihydrochloride (Sigma-Aldrich) and read on a plate reader at 460 nm every 20 s for 15 min. The slope for each standard was calculated, plotted, and used to calculate units of MPO activity/tissue weight for each sample.
For histopathological assessment, tissues were embedded in paraffin, sectioned (4 μm), stained with hematoxylin and eosin, and coded for examination by a pathologist blinded to treatment group. The severity of pancreatitis was scored using criteria shown in Table 1, and results are expressed as a score from 0 to 3 for inflammatory infiltrate and atrophy. The fibrosis score was subdivided into scores for intralobular fibrosis, perilobular fibrosis, and interlobular fibrosis, each ranging from 0 to 3. Total histological score was the combined scores of inflammatory infiltrate, atrophy, and fibrosis.
Table 1.
Effects of caerulein treatment on pancreatic histology
| Vehicle | Caerulein |
|||
|---|---|---|---|---|
| 3 weeks | 6 weeks | 10 weeks | ||
| Inflammatory infiltrate | 0.00 | 1.00* | 2.00* | 2.00* |
| Atrophy | 0.00 | 2.00* | 3.00* | 3.00* |
| Fibrosis | 0.00 | 3.00* | 3.00* | 3.00* |
Results are expressed as medians (n = 6). Histological parameters were scored in a blinded fashion. Data were analyzed by Kruskal-Wallis one-way ANOVA.
*p < 0.05 for caerulein versus vehicle.
Immunohistochemistry.
Mice were deeply anesthetized (isoflurane), perfused via the right atrium with ice-cold 4% paraformaldehyde in 0.1 m PB, and the pancreas and ganglia (DRG and NG) removed as above. After cryoprotection in 20% sucrose, fixed tissue was embedded in optimal cutting temperature compound (Sakura Finetek), frozen, and sectioned at 20 μm (pancreas) and 14 μm (DRG/NG) thickness. Some pancreatic tissue sections were incubated with rabbit anti-calcitonin gene-related peptide (CGRP) antibody (1:2000; Sigma-Aldrich) followed by Cy3-conjugated anti-rabbit IgG (1:200; Jackson Immunoresearch) to immunolabel sensory fibers. Antibody specificity was confirmed by omission of the primary antibody. Monocytes and macrophages were stained in other pancreatic tissue sections using rat anti-F4/80 (1:1000; Abcam) and Cy3-conjugated anti-rat IgG (1:200). DRG and NG sections were incubated with rabbit anti-pERK (1:600; Cell Signaling Technology) and then with Cy3-conjugated anti-rabbit IgG (1:200). F4/80 specificity was verified by the absence of immunostaining with the isotype control antibody (rat κ monoclonal IgG2b, 1:1000; Abcam), indicating that staining with the F4/80 antibody was not due to nonspecific binding of immunoglobulins to Fc receptors expressed on immune cells. Immunostained tissue sections were photographed using a microscope-mounted digital camera (DFC340FX; Leica). Immunostaining was quantified by setting the brightness threshold to the 8-bit gray scale images using ImageJ and calculating the area (proportion) of immunoreactivity relative to pancreatic area. To determine the gross distribution of sensory fibers within pancreatic sections, the density of nerve fibers (CGRP) and monocytes (F4/80) per unit area was determined in each observation field (500 μm × 500 μm). To reduce variation between tissue slides, control and caerulein-treated pancreata were mounted on the same slides.
Cell culture.
Mice were deeply anesthetized (isoflurane) and perfused with ice-cold Ca2+/Mg2+-free Hanks balanced salt solution (Invitrogen). T9–12 DRG and NG were dissected rapidly and prepared separately for culture, as described previously (Schwartz et al., 2011). Dissociated cells were resuspended in DMEM/F12 medium (Invitrogen) containing 10% fetal calf serum and antibiotics (penicillin/streptomycin, 50 U/ml), and plated onto poly-d-lysine (5 mg/ml)-coated glass coverslips. No additional growth factors were added to the culture medium. Cells were incubated overnight at 37°C and only FB-labeled pancreatic T9–12 DRG and NG neurons were studied.
Calcium imaging.
Neurons were incubated with a 2.5 μm concentration of the Ca2+ indicator fura-2 AM ester with 0.025% Pluronic F-127 for 20 min at room temperature. FB-labeled neurons were placed in a recording chamber and superfused continuously with normal bath solution containing the following (in mm): 130 NaCl, 3 KCl, 2.5 CaCl2, 0.6 MgCl2, 10 HEPES, 10 glucose, pH 7.4, osmolality 325 mOsm at a rate of 1 ml/min. Fluorescence data were acquired on a PC running Metafluor software (Molecular Devices) via a CCD camera (RTE/CCD 1300; Roper Scientific). The ratio (R) of fluorescence emission (510 nm) in response to 340/380 nm excitation (controlled by a λ 10-2 filter changer (Sutter Instrument) was acquired at 1 Hz during drug application. All drugs were applied using a computer-controlled perfusion fast-step system (switching time <20 ms; SF-77B perfusion system; Warner Instruments). The intracellular Ca2+ concentration ([Ca2+]i) was determined from the fura-2 ratio after in situ calibration experiments according to the following equation:
 |
Where Kd is the dissociation constant for fura-2 for Ca2+ at room temperature, Sf2/Sb2 is the fluorescence ratio of the emission intensity excited by 380 nm signal in the absence of Ca2+, and Rmin and Rmax are the minimal and maximal fluorescence ratios, respectively. Determination of these variables has been described in detail previously (Grynkiewicz et al., 1985; Kao et al., 1994; Lu et al., 2006). A brief (4 s) application of 50 mm KCl (high K+) was used to evoke an increase in [Ca2+]i, subsequently referred to as a Ca2+ transient. The magnitude and decay of evoked Ca2+ transient was analyzed as the peak evoked Ca2+ and the time to 50% of the peak, respectively. Cells unresponsive to high K+ were not studied further. Following a 10 min recovery period, cells were exposed to either the TRPV1 agonist capsaicin (CAP; 500 nm; Sigma-Aldrich) for 1 s or the TRPA1 agonist mustard oil (MO; 100 μm; Calbiochem) for 20 s. Stock solutions (10 mm CAP in ethanol or 100 mm MO in 1-methyl-2-pyrrolidinone) and working dilutions (in bath solution) were made fresh daily. The prevalence of CAP- or MO-responsive pancreatic afferents was determined as a percentage of total healthy (high-K+ responsive) FB-positive cells.
Single-cell RT-PCR and qRT-PCR.
Single-cell RT-PCR and qRT-PCR assays were performed as described previously (Schwartz et al., 2011). Briefly, individual FB-labeled pancreatic neurons were collected with large-bore (∼50 μm) glass pipettes and expelled into microcentrifuge tubes containing the RT mixture. RT-PCR was performed with 20 U of Superscript II (Invitrogen). RT reactions were incubated at 65°C for 1.5 min and room temperature for 2 min, and then at 37°C for 20 min and 65°C for 10 min after adding 20 U of Superscript II. For each experiment, negative controls consisted of omitting RT or using a cell-free bath aspirate as template. The first-strand cDNA from a pancreatic sensory neuron was used as template in a PCR containing 1× GoTaq reaction buffer (Promega), 20 mm outer primers, 0.2 mm dNTPs, and 0.2 ml of GoTaq DNA polymerase (Promega). Reactions were incubated at 95°C for 10 min and then cycled 35 times at 94°C/30 s, 52°C/30 s, and 72°C/30 s before a final extension step at 72°C for 10 min. Each initial PCR product served as template in a subsequent PCR using a nested primer pair, the products of which were electrophoresed on 2% agarose–ethidium bromide gels and photographed. Only neurons producing detectable amplification of a housekeeping gene (GAPDH) were analyzed further. For qRT-PCR, the first-strand cDNA of cells expressing the target genes was preamplified (26 cycles) using the PCR condition described above. The products were used as template for real-time PCR using ABsolute QPCR SYBR Green ROX mix (ABgene) in an Applied Biosystems 5700 real-time thermal cycler. Threshold cycle (Ct) values were recorded as a measure of initial template concentration and relative fold changes in RNA levels were calculated by the ΔΔCt method using GAPDH as a reference standard. The validity of the ΔΔCt method and the uniformity of the preamplification was tested by comparing PCR efficiencies calculated directly from individual amplification curves within the window of linearity (GAPDH 1.77, TRPV1 1.79, TRPA1 1.83) and preserved ΔCt values in samples preamplified to different extents (ΔCt difference ± 0.08 cycle), respectively.
TRP receptor antagonists.
The TRPV1 antagonist A-889425 and/or TRPA1 antagonist A-967079 (50–300 mg/kg; Abbott Laboratories) or vehicle was administered intraperitoneally twice weekly (Tuesday and Friday): 1 h before the first caerulein or vehicle injection and 4 h later (Fig. 1).
Behavioral testing.
To assess pain-related behaviors, vehicle- and caerulein-treated mice were placed in Plexiglas boxes 25 cm square by 40 cm high, and their exploratory behaviors were monitored photoelectrically for 15 min periods every week in the morning hours. Photoelectric beams were spaced 1.5 cm apart, providing 0.75 cm spatial resolution. TruScan software (Coulbourn Instruments) were used to analyze the time spent in different parts of the box, path information, distance traveled, and total movements simultaneously in the X–Y plane. The software also analyzed the amount of time each mouse spent in the “vertical plane” or standing position that required stretch of the abdominal muscles, a position that is assumed to be uncomfortable in the presence of abdominal hypersensitivity. Modulation of exploratory behavior was evaluated after combined TRPV1 and TRPA1 antagonist treatment chronically and, in different mice, also after administration of morphine (2.0 mg/kg, subcutaneous) 30 min before the posttreatment time point.
Data presentation and analysis.
Data are presented as mean ± SEM and were analyzed using SigmaStat software (version 3.1; Systat Software). Statistical analyses for differences in changes over time in multiple groups were performed using two-way ANOVA followed by the Holm-Sidak post hoc test. Differences between groups were tested using Student's t test (when comparing two groups) or one-way ANOVA (when comparing more than two groups) followed by post hoc Bonferroni-protected pairwise comparisons. Statistical significance was set at p ≤ 0.05.
Results
Pancreatic inflammation
Biweekly administration of caerulein produced evidence of CP, as demonstrated histologically by extensive inflammatory intralobular, perilobular, and interlobular infiltrate, glandular atrophy, and pancreatic fibrosis that was evident beginning at week 3 (Fig. 2A–D). These morphological changes were greatest at 6 and 10 weeks of caerulein treatment, as indicated by blinded histological scoring (Table 1). MPO activity was significantly increased in pancreata starting at week 3 in caerulein-treated mice and remained significantly elevated through week 10 (Fig. 2E; F(3,24) = 4.72, p < 0.05). MPO activity remained elevated at 10 weeks in pancreata from mice injected with caerulein for 6 weeks despite a 4 week period without caerulein challenge (Fig. 2E, filled squares). MPO activity remained elevated in parallel with histological changes, suggesting that irreversible damage resulted from 6 weeks of repeated caerulein treatment (Fig. 2E). No significant changes in morphology or MPO activity were observed at any time in vehicle-treated mice, which also were not distinguishable from naive mice (data not shown). Consistent with changes in morphology and MPO activity, Figure 3A,C illustrates significant accumulation of F4/80-positive monocytes starting at week 3 that persisted through 10 weeks (F(4,30) = 4.86, p < 0.01).
Figure 2.
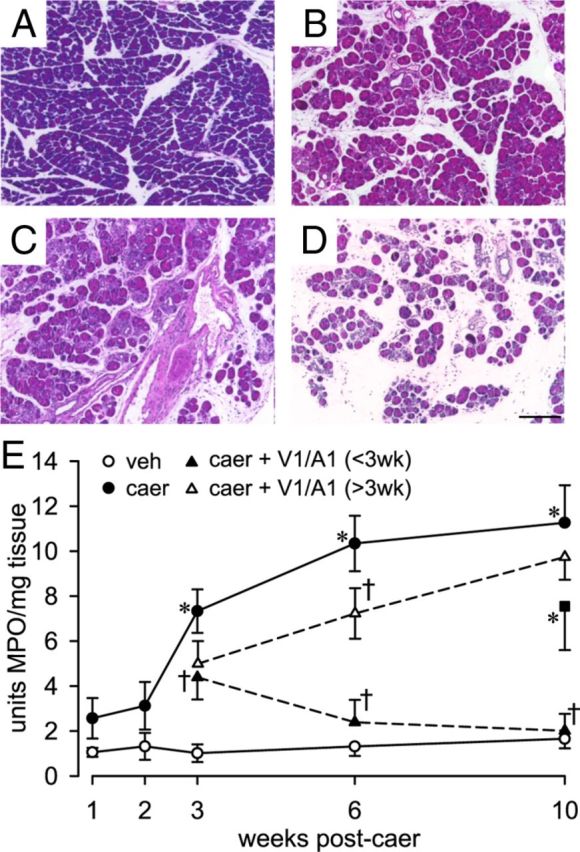
Pancreatic histology examined 10 weeks after repeated injections of vehicle (veh; A) or 3 (B), 6 (C), or 10 (D) weeks after repeated injections of caerulein (caer; n = 6/group) revealed significant inflammatory infiltrate, atrophy, and necrosis. Scale bar, 40 μm. E, MPO activity in pancreata of veh- (n = 6/group) and caer (n = 8/group)-treated mice. MPO activity (units per milligram of tissue) increased significantly starting at week 3 of biweekly caer injections and remained elevated throughout the 10 weeks of caer treatment; 10 weeks of veh injections did not increase MPO activity. Combined TRP antagonists (100 mg/kg each of the V1 and A1 antagonist; V1/A1; n = 6/treatment group) treatment initiated before week 3 (<3wk) decreased MPO activity by 40% at wk 3 and MPO activity returned to control values. Combined TRP antagonists treatment initiated after week 3 (V1/A1 > 3wk) did not prevent the caer-produced increase in MPO activity. These values were significantly different from vehicle-treated mice. In pancreata from a group of mice injected with caer for 6 weeks, MPO activity remained significantly elevated at 10 weeks (n = 6) despite a 4 week period without caer challenge. *p < 0.05 for caer vs veh; †p < 0.05 for caer vs caer + V1/A1 <3 wk or >3 wk, two-way ANOVA with Bonferroni post hoc test.
Figure 3.

Pancreatic monocyte infiltration and innervation density. A, Caerulein (caer) treatment led to infiltration of monocytes (F4/80-immunoreactive round, red cells) when assessed at 3 weeks (caer-3wk) and 6 weeks (caer-6wk) of biweekly caer injections. Monocyte infiltration was diminished by combined TRP antagonists (100 mg/kg each of the V1 and A1 antagonist; V1/A1) treatment initiated before week 3 (<3wk), but not when treatment was initiated after week 3 (>3wk). Treatment effects are quantified in C. B, Caer treatment also increased pancreatic CGRP-immunoreactive nerve fibers (arrows). Combined TRP antagonists treatment (as above) initiated before week 3, but not after week 3, prevented the increase in CGRP-immunoreactive fiber density, quantified in D. *p < 0.05 for caer vs vehicle [veh]); †p < 0.05 for caer vs caer + V1/A1 <3 wk or >3 wk, one-way ANOVA with a Bonferroni post hoc test. n = 6/treatment group. Scale bar, 100 μm.
Pancreas fiber density
To examine whether the progression of CP altered innervation of the pancreas, afferent fibers were immunolabeled with a CGRP antibody. In vehicle-treated mice, CGRP immunostaining was sparse within the pancreatic parenchyma (Fig. 3B). In contrast, CGRP immunostaining density (primarily associated with large- and medium-sized blood vessels) was significantly increased after 3, 6, and 10 weeks of biweekly caerulein treatment (F(3,24) = 4.21, p < 0.05), suggesting that nerve hypertrophy accompanies the onset and progression of CP, which is consistent with other studies showing an increase in pancreatic innervation in a diseased state (Lindsay et al., 2006).
Pancreatitis increases neuronal TRP expression
The majority of pancreatic DRG neurons from vehicle-treated mice expressed mRNA for TRPV1 and TRPA1, whereas significantly fewer NG neurons expressed these transcripts (Table 2). There was no significant increase in the number of pancreatic DRG or NG neurons expressing either TRPV1 or TRPA1 transcripts at any time after caerulein treatment (Table 2). Real-time quantitative PCR on pancreatic neurons that expressed these channels, however, revealed that on a per-cell basis, caerulein treatment increased the relative content of both TRPV1 and TRPA1 mRNA significantly in pancreatic DRG and NG neurons (relative to vehicle treatment) after 3, 6, and 10 weeks of treatment (Table 3).
Table 2.
Expression of TRP transcripts in FB-labeled pancreatic neurons before and after caerulein-induced pancreatitis
| Vehicle | Caerulein |
Caerulein + TRPV1/TRPA1 |
||||
|---|---|---|---|---|---|---|
| 3 weeks | 6 weeks | 10 weeks | <3 weeks | >3 weeks | ||
| TRPV1 | ||||||
| DRG | 69% | 78% | 80% | 78% | 71% | 69% |
| NG* | 38% | 47% | 45% | 42% | 43% | 43% |
| TRPA1 | ||||||
| DRG | 68% | 77% | 77% | 81% | 65% | 62% |
| NG* | 38% | 47% | 45% | 45% | 43% | 38% |
Data were analyzed by χ2 test. In all groups, n ≥ 40 cells.
*p < 0.05. TRPV1 and TRPA1 transcripts were significantly less in NG versus DRG in all groups.
Table 3.
Relative quantity (fold change from vehicle) of neuronal TRPV1 and TRPA1 gene transcripts in CP
| DRG |
NG |
|||
|---|---|---|---|---|
| TRPV1 | TRPA1 | TRPV1 | TRPA1 | |
| Caerulein | ||||
| 3 weeks | 2.03 (48)* | 2.13 (49)* | 2.65 (33)* | 2.40 (34)* |
| 6 weeks | 2.62 (45)* | 2.58 (42)* | 3.08 (29)* | 3.32 (28)* |
| 10 weeks | 2.86 (44)* | 2.83 (40)* | 3.39 (30)* | 3.31 (29)* |
| Caerulein + TRPV1/TRPA1 | ||||
| <3 weeks | 1.21 (42)† | 1.16 (41)† | 1.34 (28)† | 1.11 (31)† |
| >3 weeks | 1.99 (40)* | 1.91 (43)* | 2.26 (32)* | 1.68 (26)* |
The numbers of cells used for mRNA quantification are indicated in parentheses. Data were analyzed by one-way ANOVA.
*p < 0.05 versus vehicle;
†p < 0.05 versus caerulein for 6 weeks with a Holm-Sidak post hoc test to compare groups.
Attenuation of pancreatic changes by TRP antagonists
To determine whether TRPV1 and TRPA1 channel activity contributed to the development of the CP phenotype, selective antagonists of the two TRP channels were used. When treated before week 3 of RAP (and continuing during weeks 4–10), administration of doses of 100 or 300 mg/kg, but not 50 mg/kg, of either the TRPV1 or TRPA1 antagonist significantly and dose dependently attenuated inflammation, as indicated by a decrease in MPO activity assessed at 6 weeks (Fig. 4A). In vehicle-treated mice, neither morphology (data not shown) nor MPO activity was affected by TRPV1 or TRPA1 antagonist treatments (data shown only for greatest dose; Fig. 4A). Combined treatment with a 100 mg/kg concentration of both antagonists was more effective in decreasing MPO activity than 100 or 300 mg/kg concentrations of either antagonist given alone, indicating greater than additive effects of these antagonists. In addition, combined antagonist treatment (100 mg/kg each) initiated before week 3 ameliorated morphological changes in the pancreas (Fig. 4B), reduced nerve hypertrophy (Fig. 3B,D; F(1,12) = 6.01, p < 0.05), and reduced monocyte infiltration (Fig. 3A,C; F(1,12) = 5.22, p < 0.01). TRP channel antagonists also decreased the caerulein-produced increase in TRPV1 and TRPA1 mRNA expression significantly in pancreatic DRG and NG neurons at 6 and 10 weeks (Table 3). Therefore, TRP antagonist treatment started before week 3 and continuing during the subsequent weeks prevented the development of CP as assessed by inflammation, monocyte infiltration, and pancreatic hyperinnervation despite continuing biweekly caerulein treatment.
Figure 4.

TRP antagonists attenuate inflammatory and morphological changes in CP. A, Neither TRPV1 (V1) nor TRPA1 (A1) antagonist treatment affected MPO activity in vehicle (veh)-treated mice (n = 6), but each antagonist dose-dependently attenuated MPO activity in caerulein (caer)-treated mice when administered before week 3. Combined antagonists treatment initiated before week 3 [caer + V1/A1 (<3 wk)] but not after week 3 [caer + V1/A1 (>3 wk)] significantly attenuated MPO activity (doses in milligrams per kilogram given within bars; n = 5–8/treatment group; F > 7.8 for all groups). *p < 0.01 for caer vs veh; †p < 0.01 for caer vs V1 or A1 alone, one-way ANOVA with a Bonferroni post hoc test. B, Representative pancreas histology after injection of veh for 10 weeks or caer (caer-6wk) for 6 weeks without or with combined V1/A1 antagonist treatment (100 mg/kg each) initiated before week 3 (<3wk) or after week 3 (>3wk) of biweekly caer treatment. Scale bar, 100 μm.
In contrast, when treatment was initiated after week 3 (and continuing through week 10), neither single nor combined TRP antagonist treatment influenced the progression of RAP to CP, suggesting that a critical period exists between weeks 2 and 3 during which the transition to CP (and associated pain-related behaviors; see below) can be prevented by reducing or blocking the neurogenic component of RAP. To further examine this apparent critical period, combined TRP antagonists treatment was given only during weeks 2 and 3 and MPO activity was monitored thereafter through week 10. Figure 2E shows that MPO activity was not increased after either 1 or 2 weeks of biweekly caerulein treatment, but was significantly increased thereafter (weeks 3–10). Combined TRP antagonist treatment during weeks 2–3 reduced MPO activity by 40% when assayed after week 3 and, in the absence of further caerulein treatment, MPO activity returned to control values.
Attenuation of pain behaviors by TRP antagonists
Unlike assessment of hypersensitivity of hollow organs that can be mechanically distended, assessment of pancreatic hypersensitivity is indirect, relying on evaluation of open-field behavior (Schwartz et al., 2011). Repeated caerulein injections reduce open field activity and vertical rearing (compared with vehicle injections), behaviors consistent with pain and discomfort (Fig. 5). These behavioral changes were attenuated significantly by coadministration of TRPV1 and TRPA1 antagonists (100 mg/kg each; F(2,18) = 5.22, p < 0.05) if treatment was started before week 3, but not if started after week 3 (Fig. 5). Blocking TRPV1 and TRPA1 channels after the critical period did not attenuate nociceptive behavior, which is consistent with the findings presented above (e.g., inflammation, hyperinnervation). To confirm that pain was indeed blocked by TRP antagonism, the opioid analgesic morphine (2.0 mg/kg) was given 30 min before testing at 10 weeks. Morphine restored open-field activity to a level similar to that in mice treated with TRP antagonists.
Figure 5.
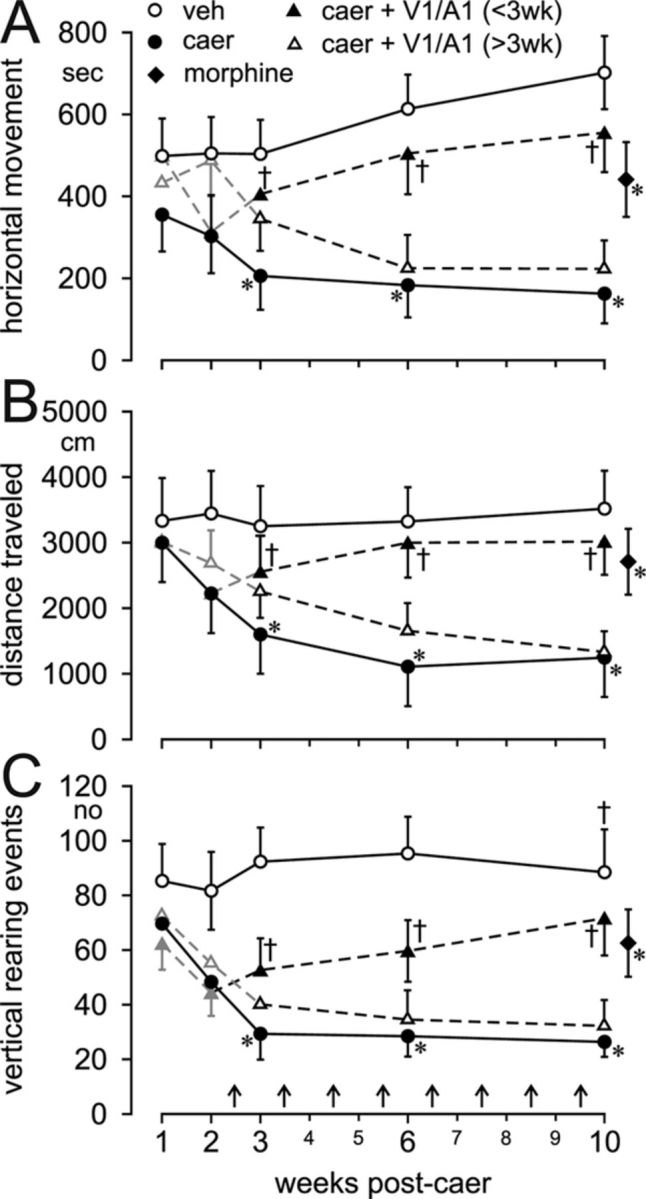
TRP antagonists reverse pain behavior in CP. Open-field activity monitoring revealed that the time (in seconds) spent moving in the horizontal plane (A), the total distance (in centimeters) traveled (B), and the number (n) of rearing events in the vertical plane (C) were significantly reduced in vehicle (veh)- versus caerulein (caer)-treated mice. Administration of TRP antagonists (100 mg/kg both TRPV1 and TRPA1) before week 3 [caer +V1/A1 (<3 wk)] of caer biweekly treatment and continuing through week 10 reversed caer-produced pain behaviors, whereas the same treatment started after 3 weeks [caer +V1/A1 (> 3wk)] did not reverse caer-produced behaviors. Morphine (2.0 mg/kg s.c.) given 30 min before testing significantly attenuated pain-related behavioral parameters in caer-treated mice. All data were analyzed by two-way ANOVA with a Holm-Sidak post hoc test and in all cases F > 11.3. *p < 0.05 caer vs veh; †p < 0.05 caer vs caer + V1/A1 <3 wk or >3 wk. n = 6/treatment group.
Inflammation increases [Ca2+]i and evoked transients in pancreatic afferents
Cytoplasmic calcium critically regulates many cellular functions, including neurotransmitter release, signaling cascades, and membrane channel function (Berridge et al., 2000). [Ca2+]i was increased significantly during the progression of CP (Table 4). Further, depolarization-evoked Ca2+ transients produced by application of 50 mm KCl (high K+) were significantly greater in peak response and decayed more slowly (i.e., time to 50% of the peak) in pancreatic DRG neurons from caerulein-treated mice relative to vehicle-treated mice, suggesting that persistent pancreatitis influences the regulation of sensory neuron [Ca2+]I, as has also been reported for other inflammatory pain models (Lu et al., 2008, 2010).
Table 4.
Effects on [Ca2+]i and evoked transients in pancreatic DRG somata
| Resting [Ca2+]i (nm) | Peak response (nm) 50 mm KCl | T50 (sec) 50 mm KCl | |
|---|---|---|---|
| Vehicle (30) | 59.2 ± 3.8* | 313.1 ± 17.1* | 47.6 ± 7.3* |
| Caerulein | |||
| 3 weeks (30) | 94.4 ± 7.7* | 426.1 ± 31.7* | 86.9 ± 9.9* |
| 6 weeks (21) | 81.0 ± 6.4* | 417.5 ± 24.7* | 98.6 ± 11.7* |
| Caerulein + TRPV1/TRPA1 | |||
| <3 weeks (28) | 48.3 ± 3.0† | 275.6 ± 23.6† | 60.8 ± 11.6† |
| >3 weeks (14) | 95.4 ± 5.2* | 398.3 ± 38.4* | 56.1 ± 10.1† |
Results are expressed as mean ± SEM. The numbers of cells used for quantification are indicated in parentheses. Data were analyzed by one-way ANOVA.
*p < 0.05 versus vehicle;
†p < 0.05 versus caerulein for 6 weeks with a Holm-Sidak post hoc test to compare groups.
When combined TRP antagonist treatment was initiated before week 3 n caerulein-treated mice, neither resting nor high-K+-evoked Ca2+ transients in pancreatic DRG neurons differed from their vehicle-treated counterparts (Table 4). In contrast, when combined TRP antagonist treatment was initiated after week 3 in caerulein-treated mice, pancreatic DRG neurons (harvested at week 6) exhibited both significantly elevated resting [Ca2+]i and amplitude of high-K+-evoked Ca2+ transients relative to vehicle-treated mice, revealing that combined TRP antagonist treatment was unable to reverse these consequences of careulein-induced pancreatitis.
Consistent with the increase in TRPV1 mRNA caused by caerulein treatment, the percentage of DRG neurons that responded to CAP (500 mm) was significantly increased after caerulein treatment (Table 5), whereas the percentage of DRG neurons that responded to MO (100 μm) was not increased, possibly due to a ceiling effect produced by the concentration of MO tested. Unexpectedly. combined administration of TRP antagonists decreased the percentage of pancreatic DRG neurons from careulein-treated mice that responded to CAP, regardless of when administered, relative to vehicle treatment.
Table 5.
Changes in percentage of responders before and after CP
| Percentage of responders |
||
|---|---|---|
| 500 nm CAP | 100 mm MO | |
| Vehicle | 20.0% (6/30)* | 30.0% (9/30) |
| Caerulein | ||
| 3 weeks | 47.1% (16/34)* | 55.9% (19/34) |
| 6 weeks | 57.1% (12/21)* | 42.9% (9/21) |
| Caerulein + TRPV1/TRPA1 | ||
| <3 weeks | 10.3% (3/28)† | 32.1% (9/28) |
| >3 weeks | 7.1% (1/14)†* | 25.0% (7/14) |
All data were analyzed by χ2 test.
*p < 0.05 versus vehicle;
†p < 0.05 versus caerulein for 6 weeks with a postresidual correction.
We also attempted to assess changes in pancreatic vagal afferent neuron somata, but failed to obtain reproducible results. In most instances, a high K+ challenge before application of a TRP agonist resulted in unstable basal [Ca2+]i fluctuation and often cell death; low K+ challenge produced weak, unreliable responses. We attribute this to the greater excitability of pancreatic vagal afferents (multiple action potential discharge) upon depolarization compared with their DRG counterparts (single action potential discharge; Schwartz et al., 2011).
Pancreatitis increases ERK activation in DRG and NG
Phospho-ERK (pERK) was increased significantly in pancreatic DRG neurons at week 3 of RAP in association with the overt caerulein-induced anatomical and inflammatory changes reported above. Interestingly, NG neurons exhibited an increase in the number of pERK-immunoreactive neuron profiles as early as 1 week after initiating caerulein treatment, which increased through week 10 (Fig. 6). Combined administration of TRP antagonists (100 mg/kg each) attenuated the caerulein-produced increase in pERK-immunoreactive neurons significantly when administration was initiated before week 3 of RAP, but not when started after week 3, further supporting the critical period of transition to CP in this model. These data illustrate an increase in pancreatic afferent neuronal excitability over the progression of CP that can be prevented by inhibiting TRPV1 and TRPA1 channels before the transition to CP.
Figure 6.

Neuronal pERK immunoreactivity (IR) in CP. Immunofluorescence images of FB (green), pERK (red), and merged pERK/FB (yellow) in pancreatic T11 DRG (A) and NG neurons (B) from vehicle (veh)- and caerulein (caer-6wk)- treated mice without and after combined TRPV1 and TRPA1 antagonists treatment (100 mg/kg each) started before week 3 [caer + V1/A1 (<3 wk)] or after week 3 [caer +V1/A1 (>3 wk)]. Immunofluorescence images of FB-labeled T11 and NG neurons from veh-treated mice exhibit no/rare expression of pERK, whereas the number of pERK/FB-IR neuron profiles was significantly increased after caer treatment. TRP antagonist treatment started before week 3, but not after week 3, reduced the number of pERK/FB-IR neuron profiles significantly, which is quantified in C for DRG and in D for NG. All data were analyzed by two-way ANOVA with a Holm-Sidak post hoc test (n = 6/treatment group). *p < 0.05 for caer vs veh; †p < 0.05 for caer vs caer + V1/A1 <3 wk or >3 wk. Scale bar, 50 μm.
Discussion
The present study reveals a pivotal role of TRPV1- and TRPA1-expressing pancreatic afferents in the transition from RAP to chronic pancreatic inflammation and pain-related behaviors in a caerulein-induced model of experimental pancreatitis in the mouse. Pancreatic inflammation (MPO and monocyte infiltration), pancreatic fibrosis, neuronal sprouting, and TRPA1 and TRPV1 gene expression were all reduced or blocked significantly by TRPV1 and TRPA1 channel antagonists, particularly when given in combination. In addition, we observed changes in pain-related behaviors and functional changes in the kinetics of depolarization-evoked Ca2+ transients in DRG pancreatic afferents, consistent with what has been reported in other inflammatory models (Lu et al., 2008, 2010). Nociceptive behavior and afferent excitability also were reduced significantly or prevented by TRPV1 and TRPA1 antagonists. Ongoing pain in CP is thought to be maintained by activity in sensitized pancreatic afferents (Zhu and Roper, 2001; Xu et al., 2007). Previous reports suggest that activation of ERK1/2 (via phosphorylation) in DRG neurons contributes to hypersensitivity by increasing gene transcription and/or by posttranslational modification of target proteins (Aley et al., 2001; Averill et al., 2001; Obata et al., 2003; Dai et al., 2004). pERK expression in pancreatic DRG and NG neurons was correspondingly upregulated in the critical period during which initiation of TRP antagonists treatment was effective. These antagonists were only effective in blocking changes when treatment was initiated before week 3 of RAP. These results indicate that, when unaided, the pancreas can recover from repeated injury for a limited time (2 weeks or 4 episodes of AP in this model), and that additional protection can be afforded if the progression of inflammation is blocked by TRPV1 and TRPA1 antagonists.
Previously (Schwartz et al., 2011), we induced AP with 8 hourly injections of caerulein and noted acute changes in pancreatic morphology that resolved relatively quickly; marked edema and inflammation (determined by MPO) was apparent at 24 h, but decreased by half at day 3 and fully recovered by day 7. Interestingly, even a single episode of AP produced an increase in pancreatic afferent TRPV1 and TRPA1 expression and function that lasted up to 3 d (Schwartz et al., 2011). The present results reveal that biweekly caerulein administration over weeks induces CP in an environment of heightened afferent sensitivity (after the first caerulein treatment) that fosters escalating neurogenic inflammation, amplifying the damage done by each subsequent episode of RAP.
TRP antagonists are presumed to target pancreatic afferents that produce neurogenic inflammation in response to pancreatic insult (Liddle, 2007; Lieb and Forsmark, 2009). RAP damages acinar cells that release protons, bradykinin, hydrogen sulfide, serotonin, and Ca2+ that can sensitize pancreatic afferents (Detlefsen et al., 2008; Lieb and Forsmark, 2009). These inflammatory mediators can directly or indirectly upregulate TRPV1 and TRPA1 channels that are expressed in the majority of pancreatic afferents. Therefore, RAP may generate a priming, feedforward pancreatic environment in which sensitization of pancreatic afferents leads to the upregulation of genes that further increase afferent sensitivity, driving additional neurogenic inflammation. This may explain the existence of a critical period during which the TRP antagonists were effective; indeed, the sustained increase in TRP channel expression in afferents started at 3 weeks in this model. Once these channels are upregulated, it may be difficult to limit neurogenic inflammation and prevent permanent damage to the pancreas.
The present study used calcium imaging to study functional changes in the kinetics of depolarization-evoked Ca2+ transients in DRG pancreatic afferents and to assess TRPV1 and TRPA1 receptor function during the progression of CP. Our results showed a significant increase in all tested parameters (resting [Ca2+]i levels, peak response, and decay time) at both 3 and 6 weeks after caerulein treatment, with a significant decrease in the group treated for 3 weeks with caerulein plus TRPV1 and TRPA1 compared with the group given caerulein for 6 weeks. However, treatment for 3 weeks with caerulein plus TRPV1 and TRPA1 resulted in an increase in peak response and no significant change in decay time to high K+ relative to vehicle treatment; furthermore, the resting [Ca2+]i level was significantly different from vehicle and similar to 6 weeks of caerulein treatment. These results prompt a series of interesting questions as to what Ca2+ regulation mechanisms were affected by TRP antagonist treatment and how. In terms of the kinetics of the evoked calcium transient, the peak and the decay are mediated by different mechanisms. The peak response is a combination of influx through voltage-gated calcium channels and input via calcium-induced calcium release from internal stores. The decay kinetics are initially buffered through mitochondria and reuptake into the internal store by sarcoplasmic/endoplasmic reticulum Ca2+-ATPases (fast decay phase) and then further extruded out of the cell via Na+-Ca2+ exchangers and plasma membrane Ca2+-ATPases (slow decay phase). Calcium regulation is highly controlled and fairly well separated. An increase in the magnitude of response is not reflected directly in the time of decay. Using pharmacology, one can manipulate the peak and decay kinetics independently (Scheff et al., 2011). Seeing an inflammation-induced change in both the peak and the decay at both 3 and 6 weeks after caerulein treatment is indicative of at least two different changes that have occurred in Ca2+ dysregulation: (1) an increase in influx/CICR mechanisms and (2) a change in reuptake/extrusion. The selective restoration of Ca2+-transient decay time to high K+, but not the peak and resting [Ca2+]i level, by the TRP antagonists treatment initiated after 3 weeks suggests its selective effects on the cellular machinery for Ca2+ reuptake and extrusion in pancreatic DRG neurons.
Three weeks of RAP also produced a significant increase in the number of pancreatic afferents that expressed pERK, the activated form of kinases regulating gene transcription that contributes to afferent hypersensitivity. CP and pain-related behaviors in this model may have been associated with ERK modulation of TRPV1 and TRPA1 channel activity/function. In support of this possibility, ERK activation was shown to be required for capsaicin-induced facilitation of heat-induced currents in dissociated DRG neurons (Firner et al., 2006) and to mediate NGF- and capsaicin-induced heat hyperalgesia in behavioral studies (Zhuang et al., 2004, 2005). Further, ERK activation mediates the upregulation of TRPV1 expression induced by NGF (Bron et al., 2003; Zhu and Oxford, 2011), supporting a key role for NGF in pancreatic pain and highlighting its modulation of TRPV1 expression and activity (Zhu and Oxford, 2011). In preliminary experiments (unpublished), blockade of ERK1/2 signaling by the MAPK1/2 inhibitor U0126 has been found to reduce inflammation and pain behaviors in CP.
Activated ERK may also contribute to CP and pain behavior via non-TRP-channel-related modification of pancreatic afferents. For example, in DRG neurons, ERK activation has been shown to be responsible for the upregulation of BDNF and NPY in models of peripheral inflammation and neuropathic pain. Inhibition of ERK phosphorylation downregulates BDNF and NPY and is correlated with a reduction in hypersensitivity (Obata and Noguchi, 2004). Furthermore, ERK phosphorylation has also been shown to regulate morphine-induced upregulation of neuropeptides SP and CGRP (Ma and Bisby, 2000; Ma et al., 2001).
In summary, we document here that RAP leads to irreversible morphological changes in pancreatic tissue and robust pain-related behaviors, both of which parallel documented clinical changes seen in CP patients (Liddle and Nathan, 2004; Liddle, 2007). These changes can be blocked in this model within a critical period that occurs after the fourth, but before the seventh experimentally induced episode of AP (i.e., during week 3 of biweekly caerulein treatments). The transition from AP to CP and pain-related behaviors is accompanied by changes in pancreatic afferents, including increased expression of TRPV1, TRPA1, and pERK, all of which have been shown previously to increase afferent sensitivity. The increased expression of these molecules as part of the mechanism responsible for the transition to CP in this model is supported by the efficacy of TRPV1 and TRPA1 antagonists in preventing histological, immunological, and behavioral changes. Accordingly, in patients with RAP, early and aggressive treatment targeting afferent excitability may inhibit progression to CP.
Footnotes
This work was supported by the National Institutes of Health (NIH; Grants T32 DK063922 to E.S.S., UL1 RR024153 to E.S.S. and G.F.G., R01 NS33730 to K.M.A., and R01 DK093525 and NS035790 to G.F.G.) and the National Center for Research Resources, a component of the NIH and the NIH Roadmap for Medical Research (Grants R01 NS035790, R01 DK093525, and R01 NS033730, T32 DK063922, and UL1 RR024153). We thank Michael Burcham for preparation of figures, Pam Cornuet and Timothy McMurray for technical assistance, Michael S. Gold for use of his calcium imaging equipment, and Abbott Laboratories for their generous gift of the TRP antagonists A-967079 and A-889425.
The authors declare no competing financial interests.
References
- Aley KO, Martin A, McMahon T, Mok J, Levine JD, Messing RO. Nociceptor sensitization by extracellular signal-regulated kinases. J Neurosci. 2001;21:6933–6939. doi: 10.1523/JNEUROSCI.21-17-06933.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anaparthy R, Pasricha PJ. Pain and chronic pancreatitis: is it the plumbing or the wiring? Curr Gastroenterol Rep. 2008;10:101–106. doi: 10.1007/s11894-008-0029-4. [DOI] [PubMed] [Google Scholar]
- Averill S, Delcroix JD, Michael GJ, Tomlinson DR, Fernyhough P, Priestley JV. Nerve growth factor modulates the activation status and fast axonal transport of ERK 1/2 in adult nociceptive neurones. Mol Cell Neurosci. 2001;18:183–196. doi: 10.1006/mcne.2001.1015. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- Bron R, Klesse LJ, Shah K, Parada LF, Winter J. Activation of Ras is necessary and sufficient for upregulation of vanilloid receptor type 1 in sensory neurons by neurotrophic factors. Mol Cell Neurosci. 2003;22:118–132. doi: 10.1016/s1044-7431(02)00022-2. [DOI] [PubMed] [Google Scholar]
- Dai Y, Fukuoka T, Wang H, Yamanaka H, Obata K, Tokunaga A, Noguchi K. Contribution of sensitized P2X receptors in inflamed tissue to the mechanical hypersensitivity revealed by phosphorylated ERK in DRG neurons. Pain. 2004;108:258–266. doi: 10.1016/j.pain.2003.12.034. [DOI] [PubMed] [Google Scholar]
- Demir IE, Tieftrunk E, Maak M, Friess H, Ceyhan GO. Pain mechanisms in chronic pancreatitis: of a master and his fire. Langenbecks Arch Surg. 2010;396:151–160. doi: 10.1007/s00423-010-0731-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detlefsen S, Sipos B, Zhao J, Drewes AM, Klöppel G. Autoimmune pancreatitis: expression and cellular source of profibrotic cytokines and their receptors. Am J Surg Pathol. 2008;32:986–995. doi: 10.1097/PAS.0b013e31815d2583. [DOI] [PubMed] [Google Scholar]
- Dimcevski G, Staahl C, Andersen SD, Thorsgaard N, Funch-Jensen P, Arendt-Nielsen L, Drewes AM. Assessment of experimental pain from skin, muscle, and esophagus in patients with chronic pancreatitis. Pancreas. 2007;35:22–29. doi: 10.1097/mpa.0b013e31805c1762. [DOI] [PubMed] [Google Scholar]
- Firner M, Greffrath W, Treede RD. Phosphorylation of extracellular signal-related protein kinase is required for rapid facilitation of heat-induced currents in rat dorsal root ganglion neurons. Neuroscience. 2006;143:253–263. doi: 10.1016/j.neuroscience.2006.07.047. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Kao CH, Wang SJ, Chen CY, Yeh SH. Detection of esophageal carcinoma using Tc-99m MIBI SPECT imaging. Clin Nucl Med. 1994;19:1069–1074. doi: 10.1097/00003072-199419120-00007. [DOI] [PubMed] [Google Scholar]
- Liddle RA. The role of Transient Receptor Potential Vanilloid 1 (TRPV1) channels in pancreatitis. Biochim Biophys Acta. 2007;1772:869–878. doi: 10.1016/j.bbadis.2007.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddle RA, Nathan JD. Neurogenic inflammation and pancreatitis. Pancreatology. 2004;4:551–559. doi: 10.1159/000082180. discussion 559–560. [DOI] [PubMed] [Google Scholar]
- Lieb JG, 2nd, Forsmark CE. Review article: pain and chronic pancreatitis. Aliment Pharmacol Ther. 2009;29:706–719. doi: 10.1111/j.1365-2036.2009.03931.x. [DOI] [PubMed] [Google Scholar]
- Lindsay TH, Halvorson KG, Peters CM, Ghilardi JR, Kuskowski MA, Wong GY, Mantyh PW. A quantitative analysis of the sensory and sympathetic innervation of the mouse pancreas. Neuroscience. 2006;137:1417–1426. doi: 10.1016/j.neuroscience.2005.10.055. [DOI] [PubMed] [Google Scholar]
- Lu SG, Gold MS. Inflammation-Induced Increase in Evoked Calcium Transients in Subpopulations of Rat Dorsal Root Ganglion Neurons. Neuroscience. 2008;153:279–288. doi: 10.1016/j.neuroscience.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SG, Zhang X, Gold MS. Intracellular calcium regulation among subpopulations of rat dorsal root ganglion neurons. J Physiol. 2006;577:169–190. doi: 10.1113/jphysiol.2006.116418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SG, Zhang XL, Luo ZD, Gold MS. Persistent inflammation alters the density and distribution of voltage-activated calcium channels in subpopulations of rat cutaneous DRG neurons. Pain. 2010;151:633–643. doi: 10.1016/j.pain.2010.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Bisby MA. Calcitonin gene-related peptide, substance P and protein gene product 9.5 immunoreactive axonal fibers in the rat footpad skin following partial sciatic nerve injuries. J Neurocytol. 2000;29:249–262. doi: 10.1023/a:1026519720352. [DOI] [PubMed] [Google Scholar]
- Ma W, Zheng WH, Powell K, Jhamandas K, Quirion R. Chronic morphine exposure increases the phosphorylation of MAP kinases and the transcription factor CREB in dorsal root ganglion neurons: an in vitro and in vivo study. Eur J Neurosci. 2001;14:1091–1104. doi: 10.1046/j.0953-816x.2001.01731.x. [DOI] [PubMed] [Google Scholar]
- Nathan JD, Patel AA, McVey DC, Thomas JE, Prpic V, Vigna SR, Liddle RA. Capsaicin vanilloid receptor-1 mediates substance P release in experimental pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1322–1328. doi: 10.1152/ajpgi.2001.281.5.G1322. [DOI] [PubMed] [Google Scholar]
- Obata K, Noguchi K. MAPK activation in nociceptive neurons and pain hypersensitivity. Life Sci. 2004;74:2643–2653. doi: 10.1016/j.lfs.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Dai Y, Tachibana T, Fukuoka T, Tokunaga A, Yoshikawa H, Noguchi K. Differential activation of extracellular signal-regulated protein kinase in primary afferent neurons regulates brain-derived neurotrophic factor expression after peripheral inflammation and nerve injury. J Neurosci. 2003;23:4117–4126. doi: 10.1523/JNEUROSCI.23-10-04117.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puylaert M, Kapural L, Van Zundert J, Peek D, Lataster A, Mekhail N, van Kleef M, Keulemans YC. Pain in chronic pancreatitis. Pain Pract. 2011;11:492–505. doi: 10.1111/j.1533-2500.2011.00474.x. [DOI] [PubMed] [Google Scholar]
- Scheff NN, Lu S, Gold MS. Mechanisms underlying inflammation-induced increase in evoked Ca2+ transients in rat DRG neurons. Soc Neurosci Abstr. 2011;37:380.29. [Google Scholar]
- Schwartz ES, Christianson JA, Chen X, La JH, Davis BM, Albers KM, Gebhart GF. Synergistic role of TRPV1 and TRPA1 in pancreatic pain and inflammation. Gastroenterology. 2011;140:1283, 1291.e1–2. doi: 10.1053/j.gastro.2010.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu GY, Winston JH, Shenoy M, Yin H, Pendyala S, Pasricha PJ. Transient receptor potential vanilloid 1 mediates hyperalgesia and is up-regulated in rats with chronic pancreatitis. Gastroenterology. 2007;133:1282–1292. doi: 10.1053/j.gastro.2007.06.015. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Xu H, Clapham DE, Ji RR. Phosphatidylinositol 3-kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J Neurosci. 2004;24:8300–8309. doi: 10.1523/JNEUROSCI.2893-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Zhu W, Oxford GS. Differential gene expression of neonatal and adult DRG neurons correlates with the differential sensitization of TRPV1 responses to nerve growth factor. Neurosci Lett. 2011;500:192–196. doi: 10.1016/j.neulet.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu WJ, Roper SN. Brain-derived neurotrophic factor enhances fast excitatory synaptic transmission in human epileptic dentate gyrus. Ann Neurol. 2001;50:188–194. doi: 10.1002/ana.1074. [DOI] [PubMed] [Google Scholar]