

Abstract
Studies suggest that plasticity in the infralimbic prefrontal cortex (IL) in rodents and its homolog in humans is necessary for inhibition of fear during the recall of fear extinction. The recall of extinction is impaired by locally blocking metabotropic glutamate receptor type 5 (mGluR5) activation in IL during extinction training. This finding suggests that mGluR5 stimulation may lead to IL plasticity needed for fear extinction. To test this hypothesis, we recorded AMPA and NMDA currents, AMPA receptor (AMPAR) rectification, and intrinsic excitability in IL pyramidal neurons in slices from trained rats using whole-cell patch-clamp recording. We observed that fear extinction increases the AMPA/NMDA ratio, consistent with insertion of AMPARs into IL synapses. In addition, extinction training increased inward rectification, suggesting that extinction induces the insertion of calcium-permeable (GluA2-lacking) AMPARs into IL synapses. Consistent with this, selectively blocking calcium-permeable AMPARs with Naspm reduced the AMPA EPSCs in IL neurons to a larger degree after extinction. Extinction-induced changes in AMPA/NMDA ratio, rectification, and intrinsic excitability were blocked with an mGluR5 antagonist. These findings suggest that mGluR5 activation leads to consolidation of fear extinction by regulating the intrinsic excitability of IL neurons and modifying the composition of AMPARs in IL synapses. Therefore, impaired mGluR5 activity in IL synapses could be one factor that causes inappropriate modulation of fear expression leading to anxiety disorders.
Introduction
Some individuals exposed to traumatic experiences develop anxiety disorders such as posttraumatic stress disorder and exhibit deficient regulation of fear responses. Experimental models such as rodent extinction of conditioned fear have been developed to study the mechanisms underlying fear regulation. Extinction involves learning to inhibit fear reactions to a stimulus that has been previously paired with an aversive stimulus such as a mild electrical shock. Studies in both humans and rodents show that interactions between the amygdala, hippocampus, and infralimbic prefrontal cortex (IL) are important for fear extinction learning and memory (Quirk and Mueller, 2008; Pape and Paré, 2010; Milad and Quirk, 2012; Orsini and Maren, 2012). During the recall of fear extinction, it is proposed that IL inhibits fear by reducing activity outflow from the amygdala (Quirk et al., 2003), a structure critical for learning and expressing fear memories (Phelps and LeDoux, 2005; Schafe et al., 2005). In order for IL to inhibit fear after extinction, plasticity must occur in afferents to IL or in IL neurons themselves to allow IL neurons to respond more vigorously to the conditioned stimulus during extinction recall (Milad and Quirk, 2002; Holmes et al., 2012).
Blocking metabotropic glutamate receptor type 5 (mGluR5) pharmacologically (Fontanez-Nuin et al., 2011) or with a knock-out approach (Xu et al., 2009) was shown to impair recall of auditory fear extinction the following day. Furthermore, mice lacking mGluR5 also exhibit impaired synaptic plasticity (Lu et al., 1997) and the activation of mGluR5 can induce synaptic plasticity (Rodrigues et al., 2002; Clem et al., 2008; Ayala et al., 2009) and intrinsic plasticity (Sourdet et al., 2003; El-Hassar et al., 2011). It is therefore possible that mGluR5 activation in IL could mediate extinction-induced synaptic and intrinsic plasticity. Consistent with this possibility, local infusion of the mGluR5 blocker MPEP into IL is sufficient to disrupt recall of fear extinction (Fontanez-Nuin et al., 2011).
In the present study, we investigated whether fear extinction induces synaptic changes in addition to intrinsic changes in IL neurons and whether mGluR5 is required for these changes by examining the ratio of AMPA to NMDA synaptic currents and AMPA receptor (AMPAR) rectification in IL neurons from naive, fear conditioned, and extinguished rats with whole-cell patch-clamp recordings. We found that fear extinction increases the ratio of AMPA to NMDA synaptic currents and AMPA rectification index in IL neurons. These extinction-induced synaptic changes and the previously described extinction-induced intrinsic excitability changes (Santini et al., 2008) were prevented by blocking mGluR5 receptors, indicating that fear extinction induces mGluR5-dependent synaptic and intrinsic plasticity in IL.
Materials and Methods
Subjects.
Male Sprague Dawley rats (30–40 d postnatal) were group housed in transparent polyethylene cages located in a negative pressure clean room (Colorado Clean Room, Ft. Collins, CO) and maintained on a 12 h light/12 h dark schedule. Food (standard laboratory rat chow) and water were available ad libitum. All experiments were approved by the institutional animal care and use committee of the Ponce School of Medicine in compliance with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals.
Behavioral training.
Rats were fear conditioned, extinguished, and tested in a 25 × 29 × 28 cm chamber with aluminum and Plexiglas walls (Coulbourn). The floor consisted of stainless steel bars that could be electrified to deliver a mild shock. A speaker was mounted on the outside wall and illumination was provided by a single overhead light. The chamber was situated inside a sound-attenuating box (Med Associates) with a ventilating fan, which produced an ambient noise level of 60 dB. Between sessions, floor trays and shock bars were cleaned with 70% alcohol solution and the chamber walls were wiped with a damp cloth. Behavior was recorded with digital video cameras (Micro Video Products).
Rats were separated into three experimental groups: Naive, conditioned (Cond), and extinction (Ext). Rats of the Cond and Ext groups received one habituation trial (tone alone) and three conditioning trials (tone-shock pairings) on day one. The conditioned stimulus was a tone (4 kHz, 75 dB, 30 s; 2 min intertrial interval) and the unconditioned stimulus was a mild scrambled foot shock (0.44 mA, 0.5 s) that co-terminated with the tone. On day 2, the Ext group received 15 extinction trials (tone alone) while the Cond group remained in their home cages. On day 3, the Cond and Ext groups were killed immediately after two test trials (tone alone) and prepared for slice recordings. The Naive group was killed immediately after two test trials and prepared for slice recordings. All training was done in the same context, as described previously (Santini et al., 2008). Freezing to the tone (absence of movement except breathing) was quantified during the 30 s of the tone presentation and analyzed using the FreezeScan program (Clever Systems).
Whole-cell recordings.
Immediately after the test, rats were deeply anesthetized with pentobarbital (65 mg/kg) and perfused transcardially with cold artificial CSF (ACSF) with sucrose substituted for the sodium chloride and decapitated. Next, 300 μm coronal slices of the mPFC were cut with a Vibratome as described previously (Santini et al., 2008). The mPFC slices were incubated at room temperature in ACSF for at least 1 h before being transferred to a submersion recording chamber and perfused at 2–3 ml/min with room temperature ACSF with 50 μm picrotoxin to block GABAA postsynaptic currents. The neurons were visualized with infrared video microscopy using a 40× water-immersion objective on an upright E600FN microscope (Nikon Instruments). Whole-cell recordings were done with glass pipettes with a resistance of 2.5–4 MΩ when filled with an internal solution containing the following (in mm): 12 TEA-Cl, 140 CsOH, 10 HEPES, 140 gluconic acid, 10 biocytin, 2 adenosine triphosphate, 3 guanosine triphosphate, and 0.4 cesium-ethyleneglycol-bis(2-aminoethylether)-N,N,N′, N′-tetra acetic acid (Cs-EGTA, 0.4). pH was adjusted to 7.3 with CsOH (300 mOsm). After establishing a whole-cell voltage-clamp recording, the resting membrane potential, membrane resistance, membrane capacitance, and access resistance were measured. Recordings were filtered at 4 kHz, digitized at 10 kHz, and saved to a computer using pCLAMP9 (Molecular Devices).
AMPA and NMDA currents.
EPSCs composed of AMPA- and NMDA-receptor (NMDAR)-mediated currents in IL neurons were measured in response to the stimulation of layer V with a glass microelectrode (Fig. 1B). The EPSC of IL pyramidal neurons of layer V was recorded because they have reciprocal connections with subcortical structures including the amygdala (Vertes et al., 2004; Gabbott et al., 2005). Picrotoxin was added to the bath to block GABAA-mediated currents. AMPAR-mediated EPSCs were measured as the peak of the EPSCs recorded at −60 mV, whereas NMDAR-mediated EPSCs were measured as the amplitude of the EPSC 45 milliseconds after the stimulus at +60 mV (Cabezas and Buño, 2006; Van den Oever et al., 2008; Lin et al., 2009; Amano et al., 2010). In some experiments, the AMPA to NMDA ratio was calculated from isolated AMPA and NMDA EPSCs. In these experiments, AMPA EPSCs were isolated by blocking NMDA currents with 100 μm DL-2-amino-5-phosphonopentanoic acid (AP5), and the NMDA EPSCs were obtained by subtracting the remaining AMPA EPSCs from the original composite EPSCs. The rectification index of the synaptic AMPAR-mediated EPSCs was measured as the ratio of the peak of the EPSCs recorded at −60 mV to the peak of the EPSCs measured at +60 mV at the same time point as the peak at −60 mV. In some experiments, the AMPA EPSCs were isolated by adding 50 μm picrotoxin and 100 μm AP5 to block GABAA and NMDAR currents, respectively (Clem and Barth, 2006).
Figure 1.

Fear extinction increases the AMPA to NMDA current ratio in IL neurons. A, Percentage freezing during tone in rats that received auditory fear conditioning (Cond), conditioning and extinction (Ext), or were untrained (Naive). Rats were killed after 2 test tones on day 3 and prefrontal brain slices were prepared. B, Photomicrograph showing the placement of the stimulation and recording electrodes in layer V of IL. C, Examples of AMPA and NMDA EPSCs recorded at −60 mV and +60 mV, respectively, in each group. D, The Ext group exhibited larger AMPA to NMDA EPSC ratios than the Cond and Naive groups. Naive neurons, n = 24; Cond neurons, n = 22; and Ext neurons, n = 24. E, The average AMPA to NMDA ratio for each rat (Ext, blue circles; Cond, black circles) shows a significant negative correlation with the percentage freezing at test. *p < 0.05.
Relative contribution of (GluA2-lacking) calcium-permeable AMPARs.
The percentage of calcium-permeable AMPAR (CP-AMPAR) current was obtained by the subtraction of the peak amplitude AMPA EPSCs recorded at −60 mV before and after adding the selective CP-AMPAR antagonist Naspm (Vikman et al., 2008; Kott et al., 2009; Clem and Huganir, 2010). The percentage blocking of the EPSCs by Naspm was calculated for neurons from each group.
Intrinsic excitability.
The intrinsic excitability of layer V pyramidal neurons located in IL was measured using whole-cell current-clamp recordings at a holding potential of −70 mV. Action potentials were evoked by injecting 800 ms depolarizing current pulses (40–350 pA at 10 pA increments with an intertrial interval of 5 s), as described previously (Santini et al., 2008). Recordings were filtered at 4 kHz (MultiClamp 700A; Molecular Devices), digitized at 10 kHz, and saved to a computer using pClamp9 (Molecular Devices). Membrane potentials (Vms) were not corrected for the junction potential of 9 mV. The series resistance was equal across groups. The input resistance (Rin) was measured from a 5 mV, 50 ms depolarizing pulse in voltage-clamp mode at a holding potential of −60 mV. The fast afterhyperpolarizing potential (fAHP) of the second evoked spike was measured by subtracting the voltage at the peak of the fAHP from the threshold potential for spike initiation. The fAHP was measured from the trace showing the maximum number of spikes. The medium afterhyperpolarizing potentials (mAHPs) and slow afterhyperpolarizing potentials (sAHPs) were measured after the end of the 800 ms pulse. The mAHP was measured as the peak of the AHP, and the sAHP was measured as the average potential during a 50 ms period beginning 280 ms after the end of the 800 ms depolarizing pulse (Faber and Sah, 2002; Santini et al., 2008). The sAHP was measured from the first trace that showed just two spikes to maintain the spike-induced calcium increases relatively constant between cells. For these experiments, the internal solution contained the following (in mm): 150 K-MeSO4, 10 KCl, 0.1 EGTA, 0.3 GTP, and 0.2 ATP, pH 7.3 (300 mOsm).
Behavioral procedure for mGluR5.
On day 1, rats were exposed to one habituation tone followed by three tones (CS) that coterminated with a mild shock (US; three tone-shock pairings). After matching for equivalent levels of freezing, conditioned rats were divided into a vehicle-treated group (Veh) and a group treated with the mGluR5 antagonist 2-methyl-6-(phenylethynyl)pyridine (MPEP; Ascent Scientific), was systemically injected (10 mg/kg intraperitoneal) 30 min before extinction training as described previously (Fontanez-Nuin et al., 2011). MPEP was dissolved in 0.9% saline. The age of the animals in the systemic experiments ranged between 30 and 35 d. On day 2, rats received extinction training consisting of 15 tone-alone trials. On day 3, rats received two tone-alone trials to test for recall of extinction. All phases of training were done in the same context.
Statistical analysis.
The total time spent freezing during the 30 s tone was measured and converted to percentage freezing. The percentage of time spent freezing (Blanchard and Blanchard, 1972) was used as a measure of conditioned fear. Freezing is the cessation of all movements except respiration. The behavioral data were analyzed using commercial software (FreezeScan; Clever Systems). The electrophysiological data were analyzed using Clampfit (Molecular Devices). Student's t test or one-way ANOVA (STATISTICA; Statsoft) were used to analyze the behavioral and electrophysiological data. After a significant main effect, Tukey HSD or Fisher post hoc tests were performed. Values are reported as the means ± SEM.
Results
Fear extinction increases AMPA/NMDA ratios in IL pyramidal neurons
To investigate whether fear extinction induces synaptic changes in IL pyramidal neurons, three groups of rats were tested. Two groups were exposed to auditory fear conditioning on day 1 consisting of three tone-shock pairings. On day 2, the Ext group (n = 11) received 15 tone-alone trials while the Cond group (n = 9) remained in their home cages. On day 3, Cond and Ext rats were killed immediately after testing for recall of conditioning and extinction (two tone-alone trials), respectively. A third group (Naive, n = 12) remained in their home cages until day 3 when they received two test tones and were killed. As shown in Figure 1A, the Cond and Ext groups conditioned to similar levels on day 1 (Cond, 60% freezing; Ext, 67%). On day 3, the Cond group froze more than either the naive or Ext group. In addition, the Ext group showed good recall of extinction. One-way ANOVA showed a significant main effect (F(2,29) = 97.68; p < 0.001), and post hoc comparisons indicated that Cond group froze more than the Ext group (p < 0.001) or the Naive group (p < 0.001). Immediately after killing, prefrontal brain slices were prepared from all three groups and whole-cell patch-clamp recordings were made from pyramidal neurons of layer V in IL. AMPA and NMDAR-mediated EPSCs were evoked by local extracellular stimulation with a glass microelectrode. Picrotoxin was included in the bath to block GABAA-mediated IPSCs. AMPAR-mediated EPSCs were measured as the peak of the EPSCs recorded at −60 mV, whereas NMDAR-mediated EPSCs were measured as the amplitude of the EPSC 45 milliseconds after the stimulus at +60 mV when AMPARs had already closed (Fig. 1C). A change in input resistance of the neurons could also affect the AMPA to NMDA ratios by disproportionally filtering the faster AMPA EPSCs more than the slower NMDA EPSCs. To minimize the effects of input resistance on the measurements of the EPSCs, all synaptic measurements were done using an intracellular solution that contained Cs+2 (to block various potassium channels and hyperpolarization-activated cation channels), QX314 (to block voltage-gated sodium channels), and TEA (to block various potassium channels). Under these conditions, there was no difference in input resistance among the groups (Table 1).
Table 1.
Electrophysiological properties of IL neurons in synaptic plasticity experiments
| Naive | Cond | Ext | |
|---|---|---|---|
| Decay Tau of composite EPSCs at +60 mV | 177 ± 52 | 124 ± 7 | 136 ± 15 |
| Decay Tau of isolated AMPA EPSCs at −60 mV | ND | 9.1 ± 0.5 | 8.7 ± 0.6 |
| Input resistance of composite EPSCs (MΩ) | 256 ± 23 | 259 ± 16 | 227 ± 24 |
| Input resistance of isolated AMPA EPSCs (MΩ) | ND | 290 ± 25 | 288 ± 35 |
One-way ANOVA showed no difference between the groups for any measure (p > 0.05). Recordings were done with a Cs2+-based intracellular solution.
ND indicates not determined.
We examined IL neurons for changes in the ratio of AMPAR to NMDAR synaptic currents. As shown in Figure 1D, neurons from the Ext group had larger AMPA to NMDA EPSC ratios than either the Cond or Naive groups. A one-way ANOVA showed a significant main effect (F(2,63) = 8.71; p < 0.001), and post hoc comparisons found that the Ext group had greater AMPA/NMDA current ratios than the Cond (p < 0.001) and Naive (p = 0.004) groups. These results suggest that fear extinction induced synaptic plasticity in IL neurons. In addition, rats that expressed less fear at test had greater AMPA to NMDA ratios than rats that expressed high fear (Fig. 1E). Pearson's analysis showed a significant negative correlation between the average AMPA/NMDA ratio for each rat and the percentage freezing at test (Pearson = −0.49; p = 0.03). As shown in Table 1, all three groups exhibited equivalent decay kinetics at +60 mV, indicating that a change in decay kinetics could not account for the increase in the AMPA to NMDA ratio.
Fear extinction increases AMPAR rectification in IL pyramidal neurons
In addition to simply inserting more AMPARs into synapses, experience and learning can modify the subunit composition of AMPARs, which is reflected by changes in the rectification of synaptic AMPARs (Clem and Barth, 2006; Xu et al., 2008; Amano et al., 2010; Clem and Huganir, 2010; Clem et al., 2010). The insertion of AMPARs lacking the GluA2 subunit increases the rectification and increases the calcium permeability of the AMPARs (Cull-Candy et al., 2006). Therefore, to evaluate whether fear conditioning or extinction modifies the subunit composition of the synaptic AMPARs, we measured the rectification of AMPARs in IL synapses. AMPAR-mediated EPSCs were measured as the peak of the composite EPSCs recorded at −60 mV and +60 mV (Fig. 2A). These measurements were used to calculate the rectification index (Xu et al., 2008; Clem and Huganir, 2010), the ratio of the AMPA EPSCs at −60 mV to the EPSCs at +60 mV. As illustrated in Figure 2B, neurons from the Ext group had significantly larger rectification indexes than neurons from either the Cond group or the Naive group. A one-way ANOVA showed a significant main effect (F(2,67) = 3.76; p = 0.03), and post hoc comparisons found that the Ext group had larger rectification indexes compared with the Cond group (p = 0.04) and showed a trend to be larger than the Naive group (p = 0.08). In addition, rats that expressed less fear at test had greater rectification indexes than rats that expressed high fear (Fig. 2C). Pearson's analysis showed a significant negative correlation between average rectification index for each rat and the percentage freezing at test (Pearson = −0.59; p = 0.01). Furthermore, the AMPA/NMDA ratio for each neuron showed a significant positive correlation with the AMPA rectification index (Pearson = 0.87; p = 0.01; Fig. 2D).
Figure 2.
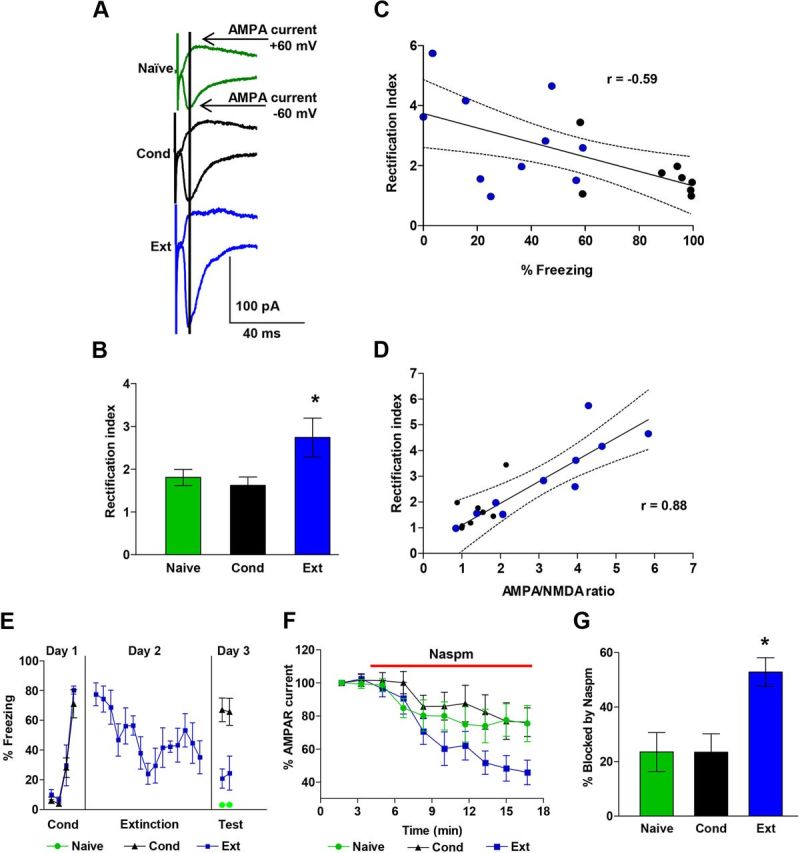
Fear extinction increases CP-AMPARs in IL synapses. A, Examples of EPSCs recorded at −60 mV and +60 mV in neurons from each group. Data are from the same neurons as in Figure 1. Vertical line indicates when AMPA EPSCs were measured. B, Average AMPAR rectification index of EPSCs from Naive (n = 24 neurons), Con (n = 22 neurons), and Ext (n = 24 neurons) groups. C, The average AMPAR rectification index for each rat (Ext, blue circles; Cond, black circles) showed a significant negative correlation with the percentage freezing at test. D, The AMPA to NMDA ratio for each neuron showed a significant positive correlation with the AMPAR rectification index. E, Behavior of rats used to test percentage blockade by Naspm. F, Time course showing that Naspm (red bar) reduced the AMPAR EPSCs recorded at −60 mV in the Ext, Naive, and Cond groups. G, Group data showing percentage blockade of AMPAR EPSCs by Naspm (50 μm) in Ext (n = 10), Cond (n = 7), and Naive (n = 11) groups. *p < 0.05.
The increase in AMPA rectification after extinction suggests that extinction induces the insertion of CP-AMPARs into IL synapses. If IL synapses contain more CP-AMPARs after extinction, then AMPA EPSCs in IL neurons after extinction should be more sensitive to blockade by Naspm, which selectively blocks CP-AMPARs (Vikman et al., 2008; Kott et al., 2009; Clem et al., 2010). As shown in Figure 2E, F, application of 50 μm Naspm produced a gradual inhibition of AMPA EPSCs in IL neurons that was greatest in the Ext group. A one-way ANOVA of the average of the last two time points for each group showed a significant main effect (F(2,25) = 3.42; p = 0.04), and post hoc comparisons found that the Ext group had a larger percentage block by Naspm compared with the Cond (p = 0.04) and Naive (p = 0.02) groups (Fig. 2G).
Pharmacological isolation of AMPA and NMDA currents gives similar results
To extend our findings, we also calculated the AMPA-NMDA ratio and AMPA rectification index by pharmacologically isolating the AMPA and NMDA EPSCs in IL neurons from additional Cond and Ext groups (Fig. 3). The NMDA component was blocked with the NMDAR blocker AP5 to isolate the AMPA EPSCs, and the NMDA EPSCs were obtained by subtraction of the AMPA EPSCs from the combined EPSCs (Fig. 3B,C). Consistent with our initial findings, IL neurons from the Ext group had larger AMPA-NMDA ratios than the Cond group (Fig. 3D; T = 2.04, p = 0.01). To determine whether our original measurements of NMDA current from the composite EPSCs were affected by any remaining AMPA currents, we compared the NMDA currents obtained from the composite EPSCs with those obtained by pharmacologically isolating the NMDA EPSCs in the same neurons. The correlation between NMDA current of composite EPSCs and pharmacologically isolated NMDA EPSCs was 0.97, indicating that our measurements of the NMDA currents from the composite EPSCs were not affected by AMPA currents at that point. We also calculated the rectification indexes from traces in which the AMPA EPSCs were isolated by blocking the NMDA component with AP5 (Fig. 3E). Calculating the rectification index in this manner also showed that AMPA EPSCs in neurons of the Ext group had more rectification than those of the Cond group (Fig. 3F; T = 2.03, p = 0.04).
Figure 3.
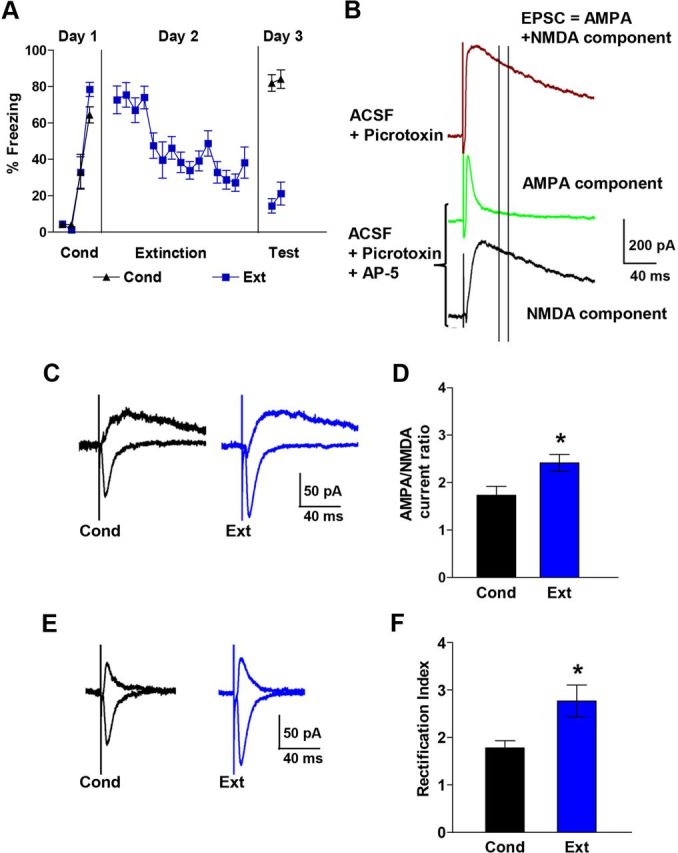
Pharmacologically isolated AMPA and NMDA EPSCs in IL neurons also show that fear extinction induces an increase in the AMPA to NMDA ratio and AMPAR rectification index. A, Behavior of rats used to measure pharmacologically isolated AMPA and NMDA EPSCs. B, Examples of EPSCs recorded at +60 mV before (brown) and after (green) blocking NMDARs with AP-5 (100 μm). After blocking NMDAR with AP-5, the number of NMDA EPSCs was calculated by subtracting the AMPA component from the composite EPSCs. Two vertical lines indicate when NMDA EPSCs were measured in Figure 1. Note that at this point the majority of the EPSCs were mediated by NMDARs. C, Examples traces of isolated AMPA and NMDA EPSCs in each group. D, AMPA to NMDA ratio of Ext (n = 17) and Cond (n = 14) groups. E, Example traces of isolated AMPA EPSCs recorded at −60 mV and +60 mV. F, Average AMPAR rectification index of isolated AMPA EPSCs in the Ext (n = 17) and Cond (n = 16) groups.
Blocking mGluR5 prevents extinction-induced synaptic plasticity in IL
To determine whether mGluR5 activation mediates extinction-induced synaptic changes, we systemically injected the mGluR5 antagonist MPEP 30 min before extinction. As shown in Figure 4A, MPEP-injected rats showed impaired recall of extinction memory on day 3 compared with rats that received saline before extinction or conditioning only (Sal-Ext: 18 ± 6% freezing, MPEP-Ext: 51 ± 8% freezing, Sal-Cond: 67 ± 4% freezing). One-way ANOVA showed a significant main effect (F(2,16) = 10.5; p < 0.001) and post hoc comparisons indicated that Sal-Cond and MPEP-Ext groups froze more than the Sal-Ext group (p < 0.05). After testing for the recall of extinction on day 3, rats were killed and the IL neurons were examined for changes in the ratio of AMPA to NMDAR synaptic currents and AMPAR rectification index (Fig. 4B). Figure 4C shows that IL neurons from the MPEP-Ext rats showed significantly smaller AMPA to NMDA EPSC ratios than neurons from the Sal-Ext rats (Sal-Cond: 1.6 ± 0.3, Sal-Ext: 4.4 ± 0.8, MPEP-Ext: 1.8 ± 0.2). A one-way ANOVA showed a significant main effect (F(2,33) = 6.34; p = 0.005) and post hoc comparisons found that the MPEP-Ext group had smaller AMPA to NMDA ratios than the Sal-Ext group (p = 0.01), but not the Sal-Cond group (p = 0.96). In addition, IL neurons from the MPEP-Ext group exhibited smaller AMPA rectification indexes compared with the Sal-Ext group (Fig. 4D; Sal-Ext: 2.7 ± 0.3, Sal-Cond: 1.7 ± 0.3, MPEP-Ext: 1.6 ± 0.1). A one-way ANOVA showed a significant main effect (F(2,33) = 5.71; p = 0.007) and post hoc comparisons found that the MPEP-Ext group had smaller AMPA rectification indexes compared with the Sal-Ext group (p = 0.009), but not the Sal-Cond group (p = 0.95). These results demonstrate that mGluR5 activation is needed for the extinction-induced synaptic changes in IL.
Figure 4.

Blockade of mGluR5 impairs extinction recall and prevents the extinction-induced increase in AMPA to NMDA ratio and AMPAR rectification index. A, Behavior of groups. Before extinction, rats were injected intraperitoneally with 10 mg/kg MPEP (MPEP-Ext; n = 6) or saline (Sal-Ext; n = 6). MPEP-Ext rats showed more freezing on the test day than the Sal-Ext rats. Sal-Cond rats were injected with saline on day 2 and returned to their home cages. B, Example traces of EPSCs recorded at −60 mV and +60 mV for each group. C, Average AMPA to NMDA ratio for each group. D, Average AMPAR rectification index in IL neurons for each group. *p < 0.05.
Blocking mGluR5 prevents extinction-induced intrinsic plasticity in IL
Knowing that inhibition of mGluR5 receptors disrupts fear extinction memory and prevents synaptic plasticity in IL, we next evaluated whether mGluR5 receptor blockade also disrupts extinction-induced intrinsic plasticity in IL neurons (Santini et al., 2008). To determine whether mGluR5 receptor activation mediates extinction-induced intrinsic changes, we examined the intrinsic excitability of IL neurons in slices from the same rats used to examine the effects of MPEP on extinction-induced synaptic changes in IL (Fig. 5A). Four measurements of neuronal excitability were examined: the number of evoked spikes, the first interspike interval (ISI), the sAHP, and the fAHP. Replicating our previous findings (Santini et al., 2008), IL neurons from the Sal-Ext group fired more spikes in response to depolarizing current steps than neurons from the Sal-Cond group (Fig. 5A–C). In contrast, IL neurons from the MPEP-Ext rats fired the same number of spikes as the Sal-Cond group, indicating that blocking mGluR5 had prevented the extinction-induced increase in number of evoked spikes. Repeated-measures ANOVA across all three groups showed a main effect of group (F(2,34) = 6.07; p = 0.005) and a significant current by group interaction (F(29,58) = 3.70; p < 0.001). Post hoc comparisons indicated that neurons in the Sal-Ext group generated significantly more spikes than both Sal-Cond and MPEP-Ext groups at all intensities between 260 and 350 pA (p < 0.01). The maximum number of spikes that could be evoked in IL neurons in the MPEP-Ext group was less than that in the Sal-Ext group and equivalent to that in the Sal-Cond group (Sal-Cond: 3.8 ± 0.6, Sal-Ext: 6.9 ± 1.4, MPEP: 2.8 ± 0.4). One-way ANOVA showed a main effect of group (F(2,34) = 6.44; p = 0.004), and post hoc comparisons indicated that the maximum number of evoked spikes in the MPEP-Ext group was significantly lower than in the Sal-Ext (p = 0.001) group. The MPEP-Ext and Sal-Cond groups were not different from each other (p = 0.35). There was no difference in input resistance, the amount of depolarizing current necessary to evoke an action potential (rheobase), or in resting membrane potential among the groups (Table 2).
Figure 5.

Blockade of mGluR5 prevents extinction-induced intrinsic excitability changes in IL pyramidal neurons. A, Example traces from saline Cond (Sal-Cond; n = 15), saline Ext (Sal-Ext; n = 9), and MPEP-Ext (n = 13) groups. Neurons were recorded from slices taken from the same rats used in Figure 4. B, Number of spikes evoked by depolarizing steps of increasing current in different groups. C, Group data of the maximum number of spikes that could be evoked by current steps in neurons from Sal-Cond, MPEP-Ext, and Sal-Ext groups. D, Group data of the duration of the first ISI from traces showing the maximum number of spikes in each neuron. E, Group data of the sAHP. Examples are shown in traces at the bottom. F, Group data of the fAHP measured after the second evoked spike. Examples are shown in the traces at the bottom. *p < 0.05 one-way ANOVA; #p < 0.05; Student's t test.
Table 2.
Electrophysiological properties of IL neurons in intrinsic plasticity experiments
| Property | Saline-Cond | Saline-Ext | MPEP-Ext |
|---|---|---|---|
| Vm (mV) | −59 ± 1 | −54 ± 2 | −54 ± 1 |
| Input resistance (mΩ) | 252 ± 18 | 254 ± 30 | 253 ± 14 |
| Rheobase (pA) | 105 ± 9 | 111 ± 16 | 117 ± 8 |
| mAHP (mV) | −6 ± 0.5 | −7.2 ± 0.5 | −6.2 ± 0.7 |
One-way ANOVA showed no difference between the groups for any measure (p > 0.05). Recordings were done with a KMeSO4-based intracellular solution.
Because extinction also reduced the first ISI, the sAHP, and the fAHP (Santini et al., 2008), we also determined whether blocking mGluR5 receptors also prevented these extinction-induced intrinsic changes. As shown in Figure 5D, neurons from the Sal-Ext group fired spikes with a smaller first ISI than neurons from the MPEP-Ext group (T = 1.72, p = 0.02), suggesting that MPEP had prevented extinction training from inducing a reduction in the first ISI. We were unable to determine whether MPEP completely blocked the reduction in the first ISI because one-way ANOVA showed no difference between the three groups when the Sal-Cond group was included in the analysis (F(2,34) = 49.70; p = 0.16). Because the amplitude of the sAHP depends on the number of spikes (Abel et al., 2004), we measured the sAHP in IL neurons from traces in which cells fired only two spikes (Santini et al., 2008). MPEP also appeared to prevent the extinction-induced reduction in the sAHP (Fig. 5E), because the sAHP was smaller in the Sal-Ext group than in the MPEP-Ext group (T = 1.72, p = 0.017). After inclusion of the Sal-Cond group in the analysis, one-way ANOVA showed no difference between the three groups (F(2,34) = 2.14; p = 0.13), precluding any further comparisons. Because the passive change in membrane potential from the current injection can contaminate the fAHP after the first evoked spike (Duvarci and Paré, 2007), we measured the fAHPs evoked by the second spike (Fig. 5F). Neurons from the Sal-Ext group had smaller fAHPs than neurons from either the Sal-Cond or the MPEP-Ext groups. A one-way ANOVA revealed a main effect of group (F(2,20) = 10.75; p = 0.0007) and post hoc comparisons indicated that the Sal-Ext group had significantly reduced fAHP amplitudes compared with both the MPEP-Ext group and the Sal-Cond group (both p < 0.01). In contrast to the sAHP and the fAHP, the mAHP measured as the negative peak at the end of the current pulse showed no difference between groups (Table 2). These results show that blocking mGluR5 prevented the extinction-induced changes in the number of evoked spikes, first ISI, sAHP, and fAHP.
Discussion
We examined the synaptic and intrinsic properties of IL neurons after fear conditioning and extinction to investigate the role of mGluR5 activation in extinction-induced synaptic and intrinsic plasticity in IL. The main findings in this study were as follows: (1) fear extinction induced an increase in the AMPA to NMDA ratio and AMPA rectification index, (2) fear extinction increased the contribution of CP-AMPARs in IL excitatory synapses, (3) blocking mGluR5 prevented these extinction-induced synaptic changes, and (4) blocking mGluR5 also prevented extinction-induced intrinsic plasticity. Our results demonstrate that fear extinction induces synaptic and intrinsic changes in IL that depend on mGluR5 activation. Although we used adolescent animals, our results appear to be applicable to adults. Similar extinction-induced synaptic changes were found previously in IL in adult animals (Pattwell et al., 2012). Furthermore, infusion of the mGluR5 blocker MPEP into IL blocks the recall of fear extinction in adult animals (Fontanez-Nuin et al., 2011), suggesting that mGluR5 activation in IL also induces extinction-related plasticity in adults.
Fear extinction induces synaptic plasticity in IL
Previous studies have suggested that fear extinction memory is encoded by IL neurons (Quirk et al., 2006) that show enhanced responses to extinguished cues during extinction recall (Milad and Quirk, 2002). Although several experimental manipulations suggest that fear extinction induces synaptic plasticity in IL (Herry et al., 1999; Herry and Garcia, 2002; Burgos-Robles et al., 2007), a direct demonstration of extinction-induced synaptic changes in IL was lacking until a recent study showed that fear extinction induces IL synaptic plasticity in preadolescent and adult mice (Pattwell et al., 2012). Consistent with the slower extinction learning found in adolescent rats (McCallum et al., 2010; Kim et al., 2011), the adolescent mice did not show evidence of fear extinction or IL synaptic plasticity after five extinction trials (Pattwell et al., 2012), suggesting that fear extinction is less robust during the adolescent period that extends from approximately postnatal day 28 to 42 in rodents (Spear, 2000).
However, when sufficient extinction is given to adolescent rats, they do show extinction and activation of IL (Kim et al., 2007b; Santini et al., 2008; Amano et al., 2010; McCallum et al., 2010; Kim et al., 2011). In the present study, we exposed adolescent rats to a longer extinction protocol than Pattwell et al., and found that neurons from the Ext group had significantly larger AMPA to NMDA EPSC ratios than either the Cond or Naive group, indicating that fear extinction induced synaptic plasticity in IL. Moreover, rats that expressed less fear at test had greater AMPA to NMDA ratios compared with rats that expressed high fear. The increased AMPA to NMDA ratio found in both mice (Pattwell et al., 2012) and rats (present study) after fear extinction is consistent with the insertion of AMPARs into the postsynaptic membrane and/or an increase in the open-time or conductance of synaptic AMPARs.
We have extended the recent findings of Pattwell et al. (2012) by showing that fear extinction also increases AMPAR rectification and blockade of AMPAR-mediated EPSCs by Naspm, suggesting that fear extinction induces the insertion of GluA2-lacking CP-AMPARs into IL synapses (Clem and Barth, 2006; Lee et al., 2006; Van den Oever et al., 2008; Vikman et al., 2008; Kott et al., 2009; Clem and Huganir, 2010; Lee et al., 2010; Wiltgen et al., 2010). Because CP-AMPARs have a larger single channel conductance (Swanson et al., 1997), the increase in the AMPA to NMDA ratio is likely mediated by the insertion of CP-AMPARs into IL synapses. Consistent with this, the AMPA to NMDA ratio was correlated with the AMPA rectification in individual IL neurons. The trafficking of CP-AMPARs seems to be a key mechanism for strengthening and modifying emotional memories. CP-AMPARs are inserted into lateral amygdala synapses after fear conditioning and are removed when the fear memory is erased by giving extinction during reconsolidation of fear memory (Clem and Huganir, 2010). The larger single channel conductance (Swanson et al., 1997), enhancement of EPSCs at higher frequencies (Rozov and Burnashev, 1999), and increased calcium permeability (Cull-Candy et al., 2006) provided by CP-AMPARs appear to facilitate the strengthening of synapses needed for the formation of emotional memories.
Critical role of group I mGluRs in extinction-induced synaptic and intrinsic plasticity
Studies indicate that group I mGluRs (mGluR1 and mGluR5) are important for extinction of conditioned fear (Kim et al., 2007a; Xu et al., 2009; Clem and Huganir, 2010; Fontanez-Nuin et al., 2011). Activation of mGluR1 seems to mediate reversal of conditioning-induced synaptic changes in the amygdala, including conditioning-induced potentiation of AMPA currents (Kim et al., 2007b) and conditioning-induced insertion of CP-AMPARs (Clem and Huganir, 2010). Therefore, mGluR1 reduces conditioned fear by weakening lateral amygdala synapses to depress amygdala responses to the conditioned stimulus.
In contrast, mGluR5 contributes to inhibition of conditioned fear by strengthening IL synapses and increasing the intrinsic excitability of IL neurons. Our results suggest that fear extinction activates mGluR5 receptors, which increase the intrinsic excitability of IL neurons and strengthen the synaptic activation of IL neurons via an increase in CP-AMPARs. Our results do not rule out the possibility that other receptors also modulate fear extinction-induced synaptic and intrinsic plasticity in IL. Because blocking muscarinic (Santini et al., 2012), β adrenergic (Mueller et al., 2008), NMDA (Burgos-Robles et al., 2007), or D2 (Mueller et al., 2010) or D4 (Pfeiffer and Fendt, 2006) dopamine receptors in IL also disrupts recall of fear extinction, these receptors may also be involved synergistically or in parallel with mGluR5 activation in the induction of synaptic or intrinsic plasticity in IL.
Although the intracellular mechanism by which mGluR5 induces these changes remains to be determined, previous studies suggest potential mechanisms. The stimulation of mGluR5 receptors activates phospholipase C, leading to the production of inositol trisphosphate and the release of intracellular calcium (Power and Sah, 2007; El-Hassar et al., 2011). Eventually, this results in CREB phosphorylation (Wang et al., 2008; Verpelli et al., 2011), perhaps by activating calcium/calmodulin-activated adenylate cyclases and protein kinase A (PKA; Wang and Storm, 2003). Fear extinction increases CREB phosphorylation in IL (Mamiya et al., 2009), and blocking PKA in IL during extinction learning impairs extinction recall (Mueller et al., 2008), suggesting that PKA-mediated phosphorylation of CREB leads to the formation of extinction memory in IL. Because increasing CREB activity increases the intrinsic excitability of neurons (Viosca et al., 2009; Zhou et al., 2009; Benito and Barco, 2010) and PKA activation leads to the synaptic incorporation of CP-AMPARs (Esteban, 2003; Boehm, 2006), mGluR5 activation could induce intrinsic and synaptic plasticity in IL via downstream stimulation of PKA and CREB. In addition, mGluR5 can also activate extracellular signal-regulated kinase (ERK) to increase neuronal excitability (Hu et al., 2007). Given that fear extinction increases ERK activation in IL (Kim et al., 2011), which is needed for extinction recall (Hugues et al., 2006), mGluR5 activation of ERK could mediate the extinction-induced plasticity in IL. Therefore, downstream activation of PKA, ERK, and CREB are likely candidates for mediating mGluR5's induction of synaptic and intrinsic plasticity in IL during extinction.
In conclusion, our results suggest that glutamatergic afferents from the amygdala, ventral hippocampus, or other structures (Sierra-Mercado et al., 2010), which provide information about the lack of association between the conditioned and unconditioned stimulus, activate mGluR5 on IL neurons during fear extinction learning. The activation of mGluR5 induces synaptic and intrinsic plasticity in IL neurons. During the recall of extinction memory, these changes would allow IL neurons to respond more robustly to the conditioned stimulus (Milad and Quirk, 2002) and inhibit fear expression via increased activation of inhibitory networks in the amygdala (Quirk et al., 2003; Ehrlich et al., 2009; Amano et al., 2010). The development of pharmacological agonists that promote these synaptic and/or intrinsic changes in IL could be combined with extinction-based exposure therapy to facilitate fear extinction learning and treat posttraumatic stress disorder patients.
Footnotes
The authors declare no competing financial interests.
This work was supported by the National Science Foundation (Grant # IOS 0842159 to J.T.P.) and the Research Centers in Minority Institutions Behavioral Core Facility, which is supported by grants from the National Center for Research Resources (Grant #5G12RR003050-26) and the National Institute on Minority Health and Health Disparities (Grant #8G12MD007579-27). M.T.S.-O. and O.S.-C. were supported by the Ponce School of Medicine and Health Sciences–Research Initiative for Scientific Enhancement program (supported by National Institutes of Health–National Institute of General Medical Sciences Grant #GM082406). We thank Maria Colon and Eliezer Ruiz for their assistance with the behavioral studies and Drs. Gregory J. Quirk and Mohammed R. Milad for their comments on the manuscript.
References
- Abel HJ, Lee JC, Callaway JC, Foehring RC. Relationships between intracellular calcium and afterhyperpolarizations in neocortical pyramidal neurons. J Neurophysiol. 2004;91:324–335. doi: 10.1152/jn.00583.2003. [DOI] [PubMed] [Google Scholar]
- Amano T, Unal CT, Paré D. Synaptic correlates of fear extinction in the amygdala. Nat Neurosci. 2010;13:489–494. doi: 10.1038/nn.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala JE, Chen Y, Banko JL, Sheffler DJ, Williams R, Telk AN, Watson NL, Xiang Z, Zhang Y, Jones PJ, Lindsley CW, Olive MF, Conn PJ. mGluR5 positive allosteric modulators facilitate both hippocampal ltp and ltd and enhance spatial learning. Neuropsychopharmacology. 2009;34:2057–2071. doi: 10.1038/npp.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito E, Barco A. CREB's control of intrinsic and synaptic plasticity: implications for CREB-dependent memory models. Trends Neurosci. 2010;33:230–240. doi: 10.1016/j.tins.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Blanchard DC, Blanchard RJ. Innate and conditioned reactions to threat in rats with amygdaloid lesions. J Comp Physiol Psychol. 1972;81:281–290. doi: 10.1037/h0033521. [DOI] [PubMed] [Google Scholar]
- Boehm J, Kang MG, Johnson RC, Esteban J, Huganir RL, Malinow R. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51:213–225. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Burgos-Robles A, Vidal-González I, Santini E, Quirk GJ. Consolidation of Fear Extinction Requires NMDA Receptor-Dependent Bursting in the Ventromedial Prefrontal Cortex. Neuron. 2007;53:871–880. doi: 10.1016/j.neuron.2007.02.021. [DOI] [PubMed] [Google Scholar]
- Cabezas C, Buño W. Distinct transmitter release properties determine differences in short-term plasticity at functional and silent synapses. J Neurophysiol. 2006;95:3024–3034. doi: 10.1152/jn.00739.2005. [DOI] [PubMed] [Google Scholar]
- Clem RL, Barth A. Pathway-specific trafficking of native AMPARs by in vivo experience. Neuron. 2006;49:663–670. doi: 10.1016/j.neuron.2006.01.019. [DOI] [PubMed] [Google Scholar]
- Clem RL, Huganir RL. Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science. 2010;330:1108–1112. doi: 10.1126/science.1195298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem RL, Celikel T, Barth AL. Ongoing in vivo experience triggers synaptic metaplasticity in the neocortex. Science. 2008;319:101–104. doi: 10.1126/science.1143808. [DOI] [PubMed] [Google Scholar]
- Clem RL, Anggono V, Huganir RL. PICK1 regulates incorporation of calcium-permeable AMPA receptors during cortical synaptic strengthening. J Neurosci. 2010;30:6360–6366. doi: 10.1523/JNEUROSCI.6276-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy S, Kelly L, Farrant M. Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Curr Opin Neurobiol. 2006;16:277–280. doi: 10.1016/j.conb.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Duvarci S, Paré D. Glucocorticoids enhance the excitability of principal basolateral amygdala neurons. J Neurosci. 2007;27:4482–4491. doi: 10.1523/JNEUROSCI.0680-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Lüthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. doi: 10.1016/j.neuron.2009.05.026. [DOI] [PubMed] [Google Scholar]
- El-Hassar L, Hagenston AM, D'Angelo LB, Yeckel MF. Metabotropic glutamate receptors regulate hippocampal CA1 pyramidal neuron excitability via Ca(2)(+) wave-dependent activation of SK and TRPC channels. J Physiol. 2011;589:3211–3229. doi: 10.1113/jphysiol.2011.209783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Faber ES, Sah P. Physiological role of calcium-activated potassium currents in the rat lateral amygdala. J Neurosci. 2002;22:1618–1628. doi: 10.1523/JNEUROSCI.22-05-01618.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanez-Nuin DE, Santini E, Quirk GJ, Porter JT. Memory for fear extinction requires mGluR5-mediated activation of infralimbic neurons. Cereb Cortex. 2011;21:727–735. doi: 10.1093/cercor/bhq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbott PL, Warner TA, Jays PR, Salway P, Busby SJ. Prefrontal cortex in the rat: projections to subcortical autonomic, motor, and limbic centers. J Comp Neurol. 2005;492:145–177. doi: 10.1002/cne.20738. [DOI] [PubMed] [Google Scholar]
- Herry C, Garcia R. Prefrontal cortex long-term potentiation, but not long-term depression, is associated with the maintenance of extinction of learned fear in mice. J Neurosci. 2002;22:577–583. doi: 10.1523/JNEUROSCI.22-02-00577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Vouimba RM, Garcia R. Plasticity in the mediodorsal thalamo-prefrontal cortical transmission in behaving mice. J Neurophysiol. 1999;82:2827–2832. doi: 10.1152/jn.1999.82.5.2827. [DOI] [PubMed] [Google Scholar]
- Holmes A, Fitzgerald PJ, MacPherson KP, DeBrouse L, Colacicco G, Flynn SM, Masneuf S, Pleil KE, Li C, Marcinkiewcz CA, Kash TL, Gunduz-Cinar O, Camp M. Chronic alcohol remodels prefrontal neurons and disrupts NMDAR-mediated fear extinction encoding. Nat Neurosci. 2012;15:1359–1361. doi: 10.1038/nn.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu HJ, Alter BJ, Carrasquillo Y, Qiu CS, Gereau RW., 4th Metabotropic glutamate receptor 5 modulates nociceptive plasticity via extracellular signal-regulated kinase-Kv4.2 signaling in spinal cord dorsal horn neurons. J Neurosci. 2007;27:13181–13191. doi: 10.1523/JNEUROSCI.0269-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugues S, Chessel A, Lena I, Marsault R, Garcia R. Prefrontal infusion of PD098059 immediately after fear extinction training blocks extinction-associated prefrontal synaptic plasticity and decreases prefrontal ERK2 phosphorylation. Synapse. 2006;60:280–287. doi: 10.1002/syn.20291. [DOI] [PubMed] [Google Scholar]
- Kim JH, Li S, Richardson R. Immunohistochemical analyses of long-term extinction of conditioned fear in adolescent rats. Cereb Cortex. 2011;21:530–538. doi: 10.1093/cercor/bhq116. [DOI] [PubMed] [Google Scholar]
- Kim J, Lee S, Park H, Song B, Hong I, Geum D, Shin K, Choi S. Blockade of amygdala metabotropic glutamate receptor subtype 1 impairs fear extinction. Biochem Biophys Res Commun. 2007a;355:188–193. doi: 10.1016/j.bbrc.2007.01.125. [DOI] [PubMed] [Google Scholar]
- Kim J, Lee S, Park K, Hong I, Song B, Son G, Park H, Kim WR, Park E, Choe HK, Kim H, Lee C, Sun W, Kim K, Shin KS, Choi S. Amygdala depotentiation and fear extinction. Proc Natl Acad Sci U S A. 2007b;104:20955–20960. doi: 10.1073/pnas.0710548105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kott S, Sager C, Tapken D, Werner M, Hollmann M. Comparative analysis of the pharmacology of GluR1 in complex with transmembrane AMPA receptor regulatory proteins gamma2, gamma3, gamma4, and gamma8. Neuroscience. 2009;158:78–88. doi: 10.1016/j.neuroscience.2007.12.047. [DOI] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, He K, Song L, Huganir RL. Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J Neurophysiol. 2010;103:479–489. doi: 10.1152/jn.00835.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JL, Milton AL, Everitt BJ. Reconsolidation and extinction of conditioned fear: inhibition and potentiation. J Neurosci. 2006;26:5881–5887. doi: 10.1523/JNEUROSCI.0323-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HC, Mao SC, Su CL, Gean PW. Alterations of excitatory transmission in the lateral amygdala during expression and extinction of fear memory. Int J Neuropsychopharmacol. 2009:1–11. doi: 10.1017/S1461145709990678. [DOI] [PubMed] [Google Scholar]
- Lu YM, Jia Z, Janus C, Henderson JT, Gerlai R, Wojtowicz JM, Roder JC. Mice lacking metabotropic glutamate receptor 5 show impaired learning and reduced CA1 long-term potentiation (LTP) but normal CA3 LTP. J Neurosci. 1997;17:5196–5205. doi: 10.1523/JNEUROSCI.17-13-05196.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamiya N, Fukushima H, Suzuki A, Matsuyama Z, Homma S, Frankland PW, Kida S. Brain region-specific gene expression activation required for reconsolidation and extinction of contextual fear memory. J Neurosci. 2009;29:402–413. doi: 10.1523/JNEUROSCI.4639-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum J, Kim JH, Richardson R. Impaired extinction retention in adolescent rats: effects of D-cycloserine. Neuropsychopharmacology. 2010;35:2134–2142. doi: 10.1038/npp.2010.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ. Fear extinction as a model for translational neuroscience: ten years of progress. Annu Rev Psychol. 2012;63:129–151. doi: 10.1146/annurev.psych.121208.131631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller D, Porter JT, Quirk GJ. Noradrenergic signaling in infralimbic cortex increases cell excitability and strengthens memory for fear extinction. J Neurosci. 2008;28:369–375. doi: 10.1523/JNEUROSCI.3248-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller D, Bravo-Rivera C, Quirk GJ. Infralimbic D2 Receptors Are Necessary for Fear Extinction and Extinction-Related Tone Responses. Biol Psychiatry. 2010;68:1055–1060. doi: 10.1016/j.biopsych.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsini CA, Maren S. Neural and cellular mechanisms of fear and extinction memory formation. Neurosci Biobehav Rev. 2012;36:1773–1802. doi: 10.1016/j.neubiorev.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC, Paré D. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev. 2010;90:419–463. doi: 10.1152/physrev.00037.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattwell SS, Duhoux S, Hartley CA, Johnson DC, Jing D, Elliott MD, Ruberry EJ, Powers A, Mehta N, Yang RR, Soliman F, Glatt CE, Casey BJ, Ninan I, Lee FS. Altered fear learning across development in both mouse and human. Proc Natl Acad Sci U S A. 2012;109:16318–16323. doi: 10.1073/pnas.1206834109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer UJ, Fendt M. Prefrontal dopamine D4 receptors are involved in encoding fear extinction. Neuroreport. 2006;17:847–850. doi: 10.1097/01.wnr.0000220142.29413.6f. [DOI] [PubMed] [Google Scholar]
- Phelps EA, LeDoux JE. Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron. 2005;48:175–187. doi: 10.1016/j.neuron.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Power JM, Sah P. Distribution of IP3-mediated calcium responses and their role in nuclear signalling in rat basolateral amygdala neurons. J Physiol. 2007;580:835–857. doi: 10.1113/jphysiol.2006.125062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology. 2008;33:56–72. doi: 10.1038/sj.npp.1301555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Likhtik E, Pelletier JG, Paré D. Stimulation of medial prefrontal cortex decreases the responsiveness of central amygdala output neurons. J Neurosci. 2003;23:8800–8807. doi: 10.1523/JNEUROSCI.23-25-08800.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Garcia R, González-Lima F. Prefrontal mechanisms in extinction of conditioned fear. Biol Psychiatry. 2006;60:337–343. doi: 10.1016/j.biopsych.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Rodrigues SM, Bauer EP, Farb CR, Schafe GE, LeDoux JE. The group I metabotropic glutamate receptor mGluR5 is required for fear memory formation and long-term potentiation in the lateral amygdala. J Neurosci. 2002;22:5219–5229. doi: 10.1523/JNEUROSCI.22-12-05219.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozov A, Burnashev N. Polyamine-dependent facilitation of postsynaptic AMPA receptors counteracts paired-pulse depression. Nature. 1999;401:594–598. doi: 10.1038/44151. [DOI] [PubMed] [Google Scholar]
- Santini E, Quirk GJ, Porter JT. Fear conditioning and extinction differentially modify the intrinsic excitability of infralimbic neurons. J Neurosci. 2008;28:4028–4036. doi: 10.1523/JNEUROSCI.2623-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Sepulveda-Orengo M, Porter JT. Muscarinic receptors modulate the intrinsic excitability of infralimbic neurons and consolidation of fear extinction. Neuropsychopharmacology. 2012;37:2047–2056. doi: 10.1038/npp.2012.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, Doyère V, LeDoux JE. Tracking the fear engram: the lateral amygdala is an essential locus of fear memory storage. J Neurosci. 2005;25:10010–10014. doi: 10.1523/JNEUROSCI.3307-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra-Mercado D, Padilla-Coreano N, Quirk GJ. Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology. 2011;36:529–538. doi: 10.1038/npp.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sourdet V, Russier M, Daoudal G, Ankri N, Debanne D. Long-term enhancement of neuronal excitability and temporal fidelity mediated by metabotropic glutamate receptor subtype 5. J Neurosci. 2003;23:10238–10248. doi: 10.1523/JNEUROSCI.23-32-10238.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear LP. The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav Rev. 2000;24:417–463. doi: 10.1016/S0149-7634(00)00014-2. [DOI] [PubMed] [Google Scholar]
- Swanson GT, Kamboj SK, Cull-Candy SG. Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation, and subunit composition. J Neurosci. 1997;17:58–69. doi: 10.1523/JNEUROSCI.17-01-00058.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Oever MC, Goriounova NA, Li KW, Van der Schors RC, Binnekade R, Schoffelmeer AN, Mansvelder HD, Smit AB, Spijker S, De Vries TJ. Prefrontal cortex AMPA receptor plasticity is crucial for cue-induced relapse to heroin-seeking. Nat Neurosci. 2008;11:1053–1058. doi: 10.1038/nn.2165. [DOI] [PubMed] [Google Scholar]
- Verpelli C, Dvoretskova E, Vicidomini C, Rossi F, Chiappalone M, Schoen M, Di Stefano B, Mantegazza R, Broccoli V, Böckers TM, Dityatev A, Sala C. Importance of Shank3 protein in regulating metabotropic glutamate receptor 5 (mGluR5) expression and signaling at synapses. J Biol Chem. 2011;286:34839–34850. doi: 10.1074/jbc.M111.258384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertes RP, Hoover WB, Viana Di Prisco G. Theta rhythm of the hippocampus: subcortical control and functional significance. Behav Cogn Neurosci Rev. 2004;3:173–200. doi: 10.1177/1534582304273594. [DOI] [PubMed] [Google Scholar]
- Vikman KS, Rycroft BK, Christie MJ. Switch to Ca2+-permeable AMPA and reduced NR2B NMDA receptor-mediated neurotransmission at dorsal horn nociceptive synapses during inflammatory pain in the rat. J Physiol. 2008;586:515–527. doi: 10.1113/jphysiol.2007.145581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viosca J, Lopez de Armentia M, Jancic D, Barco A. Enhanced CREB-dependent gene expression increases the excitability of neurons in the basal amygdala and primes the consolidation of contextual and cued fear memory. Learn Mem. 2009;16:193–197. doi: 10.1101/lm.1254209. [DOI] [PubMed] [Google Scholar]
- Wang H, Storm DR. Calmodulin-regulated adenylyl cyclases: cross-talk and plasticity in the central nervous system. Mol Pharmacol. 2003;63:463–468. doi: 10.1124/mol.63.3.463. [DOI] [PubMed] [Google Scholar]
- Wang H, Wu LJ, Zhang F, Zhuo M. Roles of calcium-stimulated adenylyl cyclase and calmodulin-dependent protein kinase IV in the regulation of FMRP by group I metabotropic glutamate receptors. J Neurosci. 2008;28:4385–4397. doi: 10.1523/JNEUROSCI.0646-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiltgen BJ, Royle GA, Gray EE, Abdipranoto A, Thangthaeng N, Jacobs N, Saab F, Tonegawa S, Heinemann SF, O'Dell TJ, Fanselow MS, Vissel B. A role for calcium-permeable AMPA receptors in synaptic plasticity and learning. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Wu LJ, Wang H, Zhang X, Vadakkan KI, Kim SS, Steenland HW, Zhuo M. Presynaptic and postsynaptic amplifications of neuropathic pain in the anterior cingulate cortex. J Neurosci. 2008;28:7445–7453. doi: 10.1523/JNEUROSCI.1812-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zhu Y, Contractor A, Heinemann SF. mGluR5 has a critical role in inhibitory learning. J Neurosci. 2009;29:3676–3684. doi: 10.1523/JNEUROSCI.5716-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Won J, Karlsson MG, Zhou M, Rogerson T, Balaji J, Neve R, Poirazi P, Silva AJ. CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nat Neurosci. 2009;12:1438–1443. doi: 10.1038/nn.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]