

Abstract
Asparagine-linked glycosylation (NLG) plays a significant role in a diverse range of cellular processes, including protein signaling and trafficking, the immunologic response, and immune system evasion by pathogens. A major impediment to NLG-related research is an incomplete understanding of the central enzyme in the biosynthetic pathway, the oligosaccharyl transferase (OTase). Characterization of the OTase is critical for developing ways to inhibit, engineer, and otherwise manipulate the enzyme for research and therapeutic purposes. The minimal understanding of this enzyme can be attributed to its complex, transmembrane structure, and the resulting instability and resistance to overexpression and purification. The following article describes an optimized procedure for recombinant expression and purification of PglB, a bacterial OTase, in a stably active form. The conditions screened at each step, the order of screening, and the method of comparing conditions are described. Ultimately, the following approach increased expression levels from tens of micrograms to several milligrams of active protein per liter of E. coli culture, and increased stability from several hours to greater than six months post-purification. This represents the first detailed procedure for attaining a pure, active, and stable OTase in milligram quantities. In addition to presenting an optimized protocol for expression and purification of PglB, these results present a general guide for the systematic optimization of the expression, purification, and stability of a large, transmembrane protein.
Keywords: Membrane protein, purification protocol, recombinant expression, asparagine-linked, glycosylation, oligosaccharyl transferase, PglB
INTRODUCTION
Biological researchers are invariably familiar with the importance of membrane proteins in therapeutic development and throughout cell biology. Several well-known statistics demonstrate this import; most notably, membrane proteins account for approximately one-third of the human proteome and comprise a majority of current drug targets [1–3]. However, standard overexpression and purification techniques are often unsuitable for this class of proteins, which presents a major obstacle to research progress. There exist promising developments for acquiring large quantities of membrane proteins, including cell-free translation systems, directed evolution of well-expressing bacteria, and the ever-increasing advances in the efficiency of current approaches [4,5]. However, expressing and purifying membrane proteins currently remains a largely empirical, time-consuming, and high-risk endeavor, leaving many important membrane-bound enzymes uncharacterized and presenting significant gaps in the understanding of cellular pathways.
The proteins in the eukaryotic N-linked glycosylation (NLG) biosynthetic pathway provide an example of this phenomenon, as virtually all of the enzymes are membrane-bound. NLG plays a major role in many cell processes, including immune-system response, protein signaling and trafficking, and pathogenic invasion strategies [6–9]. In addition, NLG introduces several prospective tools in therapeutics; N-linked sugars are capable of functioning as indicators of cell state and type [10–12] and represent a novel chemical platform for developing new therapeutics and enhancing efficacy of current drugs [10,13]. Most current studies involving NLG, however, focus on determining the specific effects of the glycan modification on a target of interest, often aimed primarily at establishing the glycosylated sites within a protein and the effects of the modification on function [14–17]. Far less is known about the enzymes comprising the biosynthetic pathway of NLG due to the difficulty of expressing and purifying the involved membrane proteins in a stable and active state. Closing this knowledge gap would allow for enhanced ability to efficiently manipulate, study, and thus control and utilize NLG.
The system of N-linked glycosylation in the bacteria Campylobacter jejuni is now well recognized as an important and tractable model for studying biochemical principles of the pathway, both in bacteria and more broadly (Supplementary Information, Figure 1) [18–20]. The central enzyme in the NLG pathway is the oligosaccharyl transferase (OTase), which catalyzes the transfer of a specific oligosaccharide to asparagine side chains. In yeast, the OTase is a complex composed of eight subunits, all of which have one or more transmembrane domains. In comparison, the OTase in C. jejuni (termed “PglB”) is comprised of a single subunit, which is homologous to the catalytic subunit of the eukaryotic OTase (Figure 1). Thus, PglB presents an exceptional opportunity for learning about the fundamental biochemistry involved in asparagine glycosylation, as well as studying the effects of NLG in bacteria and as a tool for protein engineering and high-level N-glycoprotein synthesis [21,22]. The recent structural and biochemical data published on PglB show the motifs responsible for catalysis are conserved throughout all kingdoms of life, solidifying its role as an important and general mechanistic model for N-linked glycosylation [19,20].
Figure 1.
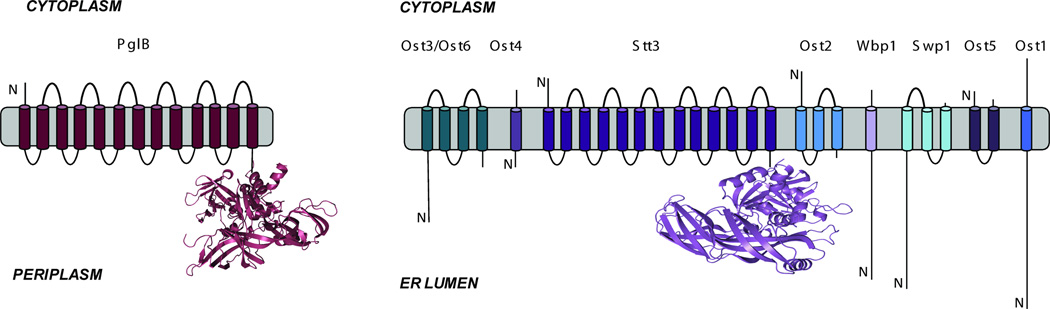
Comparison of the OTases in C. jejuni (bacteria) and S. cerevisiae (eukaryotes). Images highlight the similarity between the S. cerevisiae catalytic subunit ‘STT3’ and PglB.
Although PglB is ostensibly a tractable target relative to the eukaryotic OTase, the enzyme represents a challenge in its own right. PglB has thirteen transmembrane domains and is fairly large (82 kDa), which accounts for poor recombinant expression and instability in E. coli Thus, characterization of PglB has lagged relative to other C. jejuni NLG enzymes, despite the potential of this OTase to reveal fundamental principles about the mechanism of OTases across the evolutionary spectrum. This manuscript describes the systematic approach used to optimize the expression, purification, and stability of active PglB. The conditions screened at each step, the order of screening, and the method of comparison for each condition are described. Specific activity values are used to determine the optimal conditions for balancing protein recovery with activity recovery. This information provides the first available protocol for expressing and purifying milligram quantities of a stable and active OTase. The method is intended to aid researchers interested in C. jejuni N-linked glycosylation, and also to illustrate an activity-guided approach to optimizing expression, purification, and stability of a specific membrane protein of interest.
MATERIALS AND METHODS
Vectors and cloning
The PglB gene was amplified by PCR from the C. jejuni genome NCTC 11168 [23,24]. Primers used in the PCR encoded a BamHI site at the N-terminus prior to the start codon and His10-UGA-XhoI on the C-terminus prior to the native stop codon. The PCR product was purified and digested with BamHI and XhoI (New England Biolabs, NEB) and ligated into the corresponding sites in the pET24a(+) vector (Invitrogen) using T4 DNA ligase (NEB) and standard molecular biology procedures. The resulting vector was sequenced and then transformed using manufacturer-supplied protocols into BL21 (DE3) RIL E. coli competent cells (Agilent) for expression. Additional vectors screened for expression of PglB with alternate fusion tags include pGEX with Glutathione-S-transferase (GST) (GE Healthcare), pMAL-c2X with MBP (NEB), pET SUMO with SUMO (Invitrogen), pET Trx with Thioredoxin (EMD Millipore), and pGBH with GB1 [25] (Supplementary Information, Table I).
Table I.
Values from final optimized purification protocol of PglB on a 3.6- gram cell pellet, approximately 1/7 the weight from 1L culture.
| Fraction | Concentration (mg/mL) a,b |
Total protein (mg) c |
Rate (nmol/min) b,d |
Total activity (nmol/min) c |
Specific activity (nmol/min*mg) |
Percent Yield |
Fold Purification |
|---|---|---|---|---|---|---|---|
| lysate | 12.85 | 515 | 1.0E-05 | 8.4E-02 | 1.6E-04 | 100 | 1.0 |
| cleared lysate | 6.74 | 270 | 9.5E-06 | 7.6E-02 | 2.83E-04 | 91 | 1.7 |
| CEF (unwashed) | 2.25 | 83 | 1.1E-05 | 7.9E-02 | 9.48E-04 | 93 | 5.8 |
| CEF (washed) | 1.6 | 68 | 8.6E-06 | 7.3E-02 | 1.08E-03 | 87 | 6.6 |
| solubilized | 0.59 | 25 | 7.0E-06 | 5.9E-02 | 2.36E-03 | 70 | 15 |
| Ni-NTA purified | 0.2 | 0.6 | 8.5E-05 | 5.4E-02 | 9.06E-02 | 64 | 550 |
Concentration of total protein in fraction.
Measured value (see Methods).
Volume-corrected.
Rate of transfer of radioactive sugar substrate to peptide substrate by active PglB in fraction.
Protein Expression
Pre-optimized expression was carried out according to the following procedure except when specifically noted: 5-mL solutions of LB at 25g/L were at 37°C, until reaching an O.D.= 0.6–0.8. At this point the temperature was turned to 16°C and cultures were induced by adding IPTG to a final concentration of 1 mM. Cultures were left to shake overnight. The following day, cells were harvested and lysed according to the conditions described below. Competent cells screened included BL21 (DE3) RIL, BL21 (DE3) Gold, BL21 (DE3) RP, BL21 (DE3) pLys (all BL21 strains from Agilent), Rosetta 2 (DE3) (Novagen), Rosetta gami-2 (DE3) (Novagen), and C41 (Lucigen). In contrast, the optimized expression procedure involves using autoinduction media ZYM-5052, a high-density growth media [26]. For autoinduction expression, one liter of autoinduction media was made up in a 6-liter flask to allow adequate aeration of the cultures. 10 g tryptone and 5 g yeast extract were combined with 960 mL of deionized water and autoclaved. Once the media had cooled, Kanamycin and Chloramphenicol were added for final concentrations of 100 µg/mL and 170 µg/mL, respectively. Just before inoculation, the following media components were added:
25 mL of 40X Salt Solution: 1 M Na2HPO4, 1 M KH2PO4, 2 M NH4Cl, 0.2 M Na2SO4
20 mL of 50X 5052: 25% glycerol (v/v), 2.5% glucose (w/v), 10% α-lactose monohydrate (w/v)
0.2 mL of 1000X trace metals
2 mL of 1 M MgSO4
This gives the final composition of ZYM-5052: 1% N-Z-amine (tryptone), 0.5% yeast extract,25 mM Na2HPO4, 25 mM KH2PO4, 50 mM NH4Cl, 5 mM Na2SO4, 2 mM MgSO4, 0.2X trace metals, 0.5% glycerol, 0.05% glucose, 0.2% lactose. Optimized expression is carried out by inoculating the 1L autoinduction media with a 5-mL culture grown from a recent transformation of the PglB vector into BL21 (DE3) RIL. The inoculated media is grown at 37°C for 4–5 hours shaking at 200 RPM. The temperature is then turned down to 16°C and cultures are left shaking at 200 RPM overnight. The following day, cultures are harvested and cell pellets are weighed and stored at −80°C until purification.
Activity Assay
The PglB activity assay has been described in detail elsewhere [27]. Briefly, to a tube of radiolabeled substrate, Und-PP-Bac-[3H]GalNAc, DMSO (10 µL), 2X PglB Assay Buffer containing 100 mM HEPES, pH 7.5 / 280 mM sucrose / 2.4% (v/v) Triton X-100 (100 µL), 1 M MnCl2 (2 µL), H20 (73 µL), and 5 µL PglB fraction are combined. The assay is initiated by the addition of 10 µL of a 2 mM stock of the peptide Acetyl-DQNAT-pNF in DMSO [28]. Time points of the reaction are taken by quenching aliquots of the reaction in biphasic solutions of 3:2:1 CHCl3: MeOH: 4 mM MgCl2 (1.2 mL). The aqueous layer is then extracted and the organic layer washed twice with theoretical upper phase (TUP) with salt (2 × 600 µL). The aqueous layer and washes are combined and mixed with 5 mL of scintillation fluid (Ecolite, MP Biomedicals). The organic layer was mixed with 5 mL of scintillation fluid (OptiFluor, Perkin Elmer) and all fractions are subjected to scintillation counting.
Protein Quantification
Total protein in each of the purification fractions is estimated using the Bio-Rad protein assay (Bio-Rad, #500-0006) according to the manufacturer-supplied protocol. Bovine Serum Albumin (BSA) standard (Thermo Scientific Pierce) is used to create a standard curve. Fractions are taken during each step of the purification from a given cell pellet and stored at −80°C. Upon purifying to the desired end-point, the protein quantities for all samples are measured at the same time to minimize error. Pure PglB protein is quantified by measuring the absorbance at 280 nm and using an extinction coefficient of 117,300 M−1 cm−1 (for T7-PglB-His10).
Cell lysis
All steps of purification are performed on ice or at 4°C. A cell pellet of 3–5 g in weight is thawed on ice and resuspended in 50 mM HEPES pH 7.5, 10% glycerol, 100 mM NaCl for 0.1 grams of cell pellet per mL. Protease inhibitor cocktail solution, EDTA-free (Calbiochem) and hen-egg lysozyme powder (Amresco) is added for 1 µL/mL and 1 mg/mL, respectively. Mixture is agitated gently for ~1 hour. Cell lysis is performed by sonication (Sonics Vibracell, VC 505 (500 watts) & VC 750 (750 watts)). Sonication is performed on ice for 3 × 1 min at 50% amplitude, pulsing at 1 s on/1 s off, and with breaks between cycles to prevent warming of the mixture. Lysates are centrifuged at 8000 × g, 4°C for 35 minutes to remove unlysed cells and insoluble cell debris. The supernatant (cleared lysate) is decanted and pellet is discarded.
Isolation of Cell-Envelope Fraction (CEF)
The cleared lysate is centrifuged at 150000 × g, 4°C for 60 minutes. After the spin, the supernatant is discarded. The pelleted fraction (the CEF) is transferred as quantitatively as possible to a Pyrex homogenizer using a small spatula. Two mL of high-salt buffer (50 mM HEPES pH 7.5, 10% glycerol, 250 mM NaCl, 250 mM KCl) is then added to the centrifuge tube and the remaining CEF is resuspended by gently scraping the bottom of the tube with the spatula. This wash solution is added to the homogenizer containing the rest of the CEF. The CEF is homogenized in a final volume of 35 mL of the high-salt buffer (33 mL added, after the 2 mL wash). The homogenized CEF is returned to the centrifuge tube and this solution is centrifuged at 150000 × g, 4°C for 60 minutes. Again, the supernatant is discarded. The pellet, containing the washed CEF, is again transferred and homogenized in 5–10 mL of 50 mM HEPES pH 7.5, 30% glycerol, 20 mM imidazole. This washed CEF fraction is either stored at −80°C or purification is continued.
Extraction of membrane proteins from cell membrane
The optimization procedure for PglB solubilization from the membrane is modeled after one used previously for the yeast oligosaccharyl transferase [29]. Washed CEF is solubilized by homogenizing in 50 mM HEPES pH 7.5, 30% glycerol, 20 mM imidazole in a volume that roughly yields a final concentration of 10 mg total protein per mL. The 5% DDM detergent solution (in 50 mM HEPES pH 7.5) is diluted 1:10 in the homogenized CEF volume for a final concentration of 0.5% detergent for solubilization. This 0.5% (w/v) concentration corresponds to roughly 200X CMC of DDM (100X CMC of Triton X-100, and 9X CMC of OG, also screened at 0.5%) [30]. This solution is re-homogenized thoroughly, and the solution is vortexed at the maximum setting for two minutes. The solution is then diluted 1:3 in 50 mM HEPES pH 7.5, 30% glycerol, 20 mM imidazole, giving a final concentration of 50 mM HEPES pH 7.5, 30% glycerol, 0.17% DDM, 20 mM imidazole, and roughly 3.3 mg/mL protein. The solubilized CEF is centrifuged at 100000 × g, 4°C for 60 minutes. The supernatant is decanted into a clean, pre-chilled 50-mL conical tube for further purification, and the pellet is discarded.
Ni-NTA purification
The supernatant, containing the solubilized membrane-proteins, is added to Ni-NTA agarose resin (Qiagen) that has been pre-equilibrated in 50 mM HEPES pH 7.5, 30% glycerol, 20 mM imidazole, 0.01% DDM. The protein solution is gently agitated overnight with the resin. Ni-NTA purification is carried out with Buffer M (50 mM HEPES pH 7.5, 20% glycerol, 300 mM NaCl, 0.01% DDM) plus the specified amount of imidazole used for washes and Buffer E (50 mM HEPES pH 7.5, 30% glycerol, 100 mM NaCl, 0.01% DDM) plus specified imidazole used for elutions. Batchbinding solution is allowed to flow through a disposable gravity-filtration column (BioRad). The following fractions are collected: Flow through, Wash A: 1 × 25 mL Buffer M + 40 mM imidazole. Wash B: 3 × 1 mL Buffer M + 100 mM imidazole, Elution A: 6 × 0.5 mL fractions of Buffer E + 300 mM imidazole, Elution B: 6 × 0.5 mL Buffer E + 600 mM imidazole. Five microliters of every other elution is removed and used to determine the location of the active PglB using the activity assay. Fractions containing most activity are combined. The combined solution is concentrated and buffer is exchanged using an Amicon Ultra-100K centrifugal filter (Millipore) into Buffer E (no imidazole). SDS-PAGE is used to verify purity and then the concentration of the pure PglB solution is quantified using UV-absorbance at λ = 280 nm. The solution is then divided into aliquots and stored at −80°C.
RESULTS
Part I: Optimization of PglB expression
Optimization of PglB expression in E. coli involved screening many expression constructs and conditions. To facilitate testing the multitude of expression conditions, 5-mL cultures are grown for each condition specified (direct comparisons are always grown simultaneously). Unless specified otherwise, the cultures are grown at 37°C until they reach an O.D. of 0.6–0.8, at which point the temperature is shifted to 16°C for overnight growth. These mini-cultures are then centrifuged to concentrate the cells. The cell pellets are weighed and resuspended in a volume of lysis buffer for 0.1 g/mL and are then lysed using sonication. The activity rates of the lysates are compared, and the conditions of the culture giving the highest level of PglB activity are considered to have the highest level of expression of functional PglB. Growth and expression comparisons are carried out on at least two separate occasions to ensure conclusions drawn are accurate.
Protein Tags and Gene Truncations
Membrane proteins are often expressed as truncations of the native protein to impart higher expression or stability to the protein [31]. It has been shown that the C-terminal, soluble domain of PglB is not functional when expressed on its own (region from residue 420 to C-terminus) [32]. However, an apparent degradation product of PglB is observed via SDS-PAGE or western blot when full-length PglB is over-expressed in E. coli (Figure 2). The degradation product is expected to contain the C-terminal domain of PglB because it is visible on western blots when an anti-His (C-terminal) antibody is used but not when an anti-T7 (N-terminal) antibody is used. It was considered that this degradation product might represent a more stable and easily expressed version of PglB. To test this hypothesis, several truncation constructs were made (Figure 2). Constructs were expressed and normalized for expression via western blot (data not shown). However, it was found that all of the constructs lacked catalytic activity, including those that lacked only approximately 50 residues from the C- or N-terminus (Figure 2). These observations can now be explained by the recent structural and biochemical data, which reveal that a required catalytic motif appears in transmembrane-loop regions close to the N-terminus [19,20].
Figure 2.
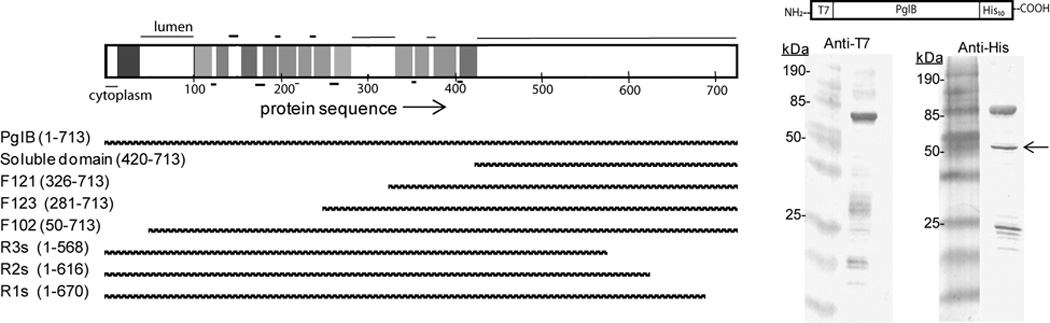
Investigating a natural degradation product of PglB for independent activity and enhanced expression. Top left: topology diagram for PglB based on available structural data (PDB 3RCE). The horizontal bar represents PglB with the protein sequence numbered along the bottom starting with the N-terminus. The location of each transmembrane helix is represented by a vertical bar and the thin black horizontal lines represent the location of the soluble regions (lumen on top, cytoplasm on bottom). Bottom left: Bars representing the various truncated constructs of PglB, with length aligned to X-axis of topology diagram. The name assigned to each construct is listed on the left along with the residues encompassed by that construct. Right: Western blots showing the degradation product of interest (indicated by the arrow) when stained with the anti-His antibody (C-terminal tag) but not when stained with the anti-T7 antibody (N-terminal tag).
Screening of expression tags represents another common approach used to alter expression, solubility, localization, and other functional aspects of proteins [33]. The pglB gene was cloned into several vectors encoding N-terminal fusion tags that have been shown to positively affect expression (Supplementary Information, Table I). These tags include: T7, Glutathione-S-transferase (GST), Maltose Binding Protein (MBP), SUMO, Thioredoxin, and GB1 [25,34–37]. Of course, the tags tested are by no means comprehensive, as the repertoire of expression tags is always expanding. For example the 13-kDa fusion tag, Mistic, represents one of many successful additions to the recent repertoire of expression tags capable of improving expression and solubility of certain intractable proteins [38]. Within those tested, it was estimated that the MBP tag (in the pMAL-c2X vector, New England Biolabs), the T7-tag (pET-24a(+) vector, Novagen) and the GB1 tag (pGBH, [25]) give the highest expression under standard expression conditions, which can easily be discerned from a visual inspection of SDS-PAGE analyses (Supplementary Information, Figure 2). The constructs that clearly expressed in the highest quantities were thereafter used for systematic optimization.
Expression and induction conditions
A variety of E. coli competent cells were screened to determine the highest-yielding strain for PglB expression. Seven types of E. coli expression strains were transformed with the MBP-PglB and T7-PglB vector constructs; these included C41(DE3), Rosetta 2, Rosetta 2 gami, and the following BL21(DE3) strains: RIL, Gold, RP, pLys. The BL21(DE3) RIL cells show the highest level of expression of both MBP-PglB and T7-PglB as measured by activity levels and western blotting (data not shown). This result suggested that this strain is optimal for PglB generally and that the outcome is not tag-specific.
A range of induction parameters were investigated, including IPTG concentration used to induce expression, optical density of cultures at the time of induction, incubation temperature post-induction, and length of time cultures were grown following induction. The effects of each of these variables are interdependent; thus, each combination should ideally be tested. Table II in the Supplementary Information shows the organization scheme for this screening process. Ultimately it was found that when cultures are induced at a very high O.D. (>1.2) rather than the typical O.D. of 0.6–0.8, significant improvement in expression resulted (Figure 3A). The benefits of inducing at this high O.D. indicated autoinduction may be a successful way to express PglB, since the mechanism of autoinduction involves growing the cells to a saturating density, at which point expression is automatically induced [26]. The procedure is similar to IPTG-induced expression, except autoinduction involves using a media specifically developed to couple induction of expression with the uptake of lactose, which occurs only at saturating density (see Methods). This method not only eliminates the need to frequently monitor O.D. of the culture, it also ensures that the expression cultures have the highest beneficial O.D. when induction occurs, and prevents any expression of the target protein prior to induction.
Table II.
Minor change in centrifugal spin significantly affects percent yield of purification step
| Fraction | Concentration (mg/mL) |
Total protein (mg) |
Rate (nmol/min) |
Total activity (nmol/min) |
Specific activity (nmol/min*mg) |
Percent Yield |
Fold Purification |
|
|---|---|---|---|---|---|---|---|---|
| Prep 1a | lysate | 9.5 | 380 | 1.2E-04 | 2.3E-01 | 6.12E-04 | 100 | 1.0 |
| cleared lysate (10000 × g) | 8.0 | 304 | 7.5E-05 | 1.4E-01 | 4.70E-04 | 61 | 0.8 | |
| Prep 2 | lysate | 12.9 | 514 | 351 | 4.7E+06 | 9.1E+03 | 100 | 1 |
| cleared lysate (8000 × g) | 6.7 | 270 | 318 | 4.2E+06 | 1.6E+04 | 91 | 2 |
Numbers are given for two PglB preparations representative of numbers seen in additional preps.
Figure 3.

Improvements in expression due to a) inducing with IPTG at higher O.D and b) using autoinduction. A. Cell cultures of each construct were grown at 37°C to an O.D. of 0.7 and 1.2, at which point the cultures were induced with 1 mM IPTG and the temperature was shifted from 37 to 16°C. Fractions from equal cell-pellet weights were purified over Ni-NTA resin and elutions were compared via SDS-PAGE. B. Cell cultures of each construct were grown in LB using IPTG (induced at O.D. = 1) as well as using autoinduction media. Equal weights of cell pellet were lysed and spun at 10000 × g to remove debris. The cleared lysates were used to measure initial rates of activity for each fraction. C. Graph indicates activity levels for PglB-pET24a(+) expression cultures that were grown under the specified conditions. For the autoinduced cultures, it was of interest to determine the optimal time to grow the cultures at 37°C before the temperature shift to 16 °C; hence, the hours post-inoculation at 37 °C were varied and compared. This is in contrast to the IPTG-induced cultures, which require strict monitoring of the O.D. to determine the induction time. Error bars indicate the standard error of activity measurements. Activity assays were performed as described above using cleared lysate fractions from equal weights of cell pellet from each condition.
Autoinduction involves growing cell cultures to saturation, resulting in dramatically increased cell weight obtained per liter of culture relative to other expression methods. Therefore, when comparing autoinduction expression to IPTG-induced expression, it was important to determine whether improvements in protein yield are due to the greater number of cells produced per liter of culture or to improved expression of PglB per cell. While one liter of IPTG-induced culture (induced at an O.D. of 0.6–0.8) gives roughly 2 grams of cells, one liter of autoinduction culture generally yields at least 25 grams of cell weight (with expression parameters otherwise held constant). Thus, activity per gram of cell weight was measured for PglB in pMAL, pET24a, and pGBH vectors grown with both autoinduction and IPTG induction (Figure 3B).
In order to determine the optimal time of induction (or temperature shift) to use in the comparison, PglB-pET24a(+) was expressed using both IPTG-induction at various O.D.s and autoinduction with varying time spent growing at 37°C before shifting the temperature to 16°C (Figure 3C). Results show that PglB expressed using autoinduction is optimal with a temperature shift at 4.5 hours post-inoculation and that PglB expressed using IPTG induction is optimal when induced at an O.D. ~ 1.6. Furthermore, autoinduction yields significant increases per gram of cell weight over expression using IPTG-induction in addition to yielding more cell weight per liter of culture (Figure 3B). It was additionally shown that with autoinduction, PglB expressed in the pET24a(+) vector gives higher levels of active protein than PglB expressed in the pMAL and pGBH vectors (Figure 3B). For this reason, the PglB-pET24a(+) construct was used for subsequent optimization and experiments. Use of this construct was also advantageous because the T7- tag is significantly smaller than both MBP and GB1, and thus most closely resembles the native (untagged) PglB.
Part II: Optimization of PglB purification
Once expression levels of PglB had been improved using autoinduction, the purification procedure was optimized so that the higher expression would translate into higher levels of pure, active protein. A flow chart of the general procedure for PglB purification is shown in Figure 4A. Optimization begins at the top of the flow chart, and the following steps were not optimized until optimized conditions were established for all previous steps. Optimizing a purification step involved dividing the ‘crude’ fraction into equal parts. Each part was subjected to one of several conditions. The total activity and protein concentration in the fractions before and after the purification step were measured for each condition. These measurements allowed calculation of specific activity, fold purification, and percent yield (see Methods) These values provide a definitive measure by which to judge the optimal condition for each purification step, where optimal is defined as maintaining maximal activity and minimal levels of total protein (Figure 4B). Table I provides the values obtained using the final optimized purification protocol. A stepwise procedure for the purification steps can be found in the Supplementary Information.
Figure 4.

Procedure for optimizing purification protocol for PglB. A. Flow chart of the general procedure for purifying PglB. The boxes contain the fraction acquired at each step, and adjacent to the arrows is the physical procedure performed to get from one fraction to the next. B. The values of total protein and total activity in percent of initial (where ’initial values’ refer to those in lysate) obtained using optimized procedure. Values are shown for each purification step diagrammed in the flow chart in part A and correspond to the values in Table I. Note the steep decrease in total protein concentration associated with each step and the comparatively mild loss of total activity.
Isolation and processing of cell-membrane fraction
Cell lysis was performed with sonication in three sets of two minutes each. Cells were incubated on ice before, during, and post-sonication to prevent the temperature of the cell suspension from rising and potentially inducing cellular stress response and increased protein degradation and denaturation. Sonication is considered a relatively harsh method of lysis, so as a comparison two PglB cell pellets of equal weight were lysed either by French Press or by sonication. Purification and activity results were unaffected by the method of lysis, indicating sonication is not causing significant impairment to protein yields (data not shown).
After lysis of cells, a 10000 × g centrifugal spin is performed to remove unbroken cells and other extraneous cell debris, yielding the ‘cleared lysate’ supernatant. While there are general recommendations for the speed at which to achieve this desired separation, at times it is beneficial to vary the exact speed of the spin to improve retention of the protein of interest. In this case, the first spin was initially performed at the standard speed of 10000 × g. Calculation of the fold purification and the percent yield showed that the step was increasing the specific activity by only a small amount, but 40% of the activity was being lost (Table II). The speed was lowered to 8000 × g, which improved the yield and fold purification (Table II). Particularly, as this is one of the first purification steps, the effects on the final yield of protein are compounded. Thus, a relatively small difference in initial the centrifugal separation speed had considerable effects on the ultimate yield of pure protein.
The supernatant (‘cleared lysate’) is then subjected to a second centrifugal spin at 150000 × g, which pellets the membrane fraction, and soluble proteins remain in the supernatant. This membrane fraction, also referred to as the cell-envelope fraction, or ‘CEF’, is then resuspended as a semi-pure fraction containing only the cell-membrane content (cell-membrane lipids and all membrane-associated proteins). A good percent yield (90–95%) and high fold purification (3–6 fold) were observed with standard parameters; thus, it was not necessary to optimize this step further.
A subsequent salt wash of the membrane fraction involves homogenizing the pellet from the first 150000 × g spin (‘unwashed CEF’) in a buffer containing a high concentration of salt. The high salt presumably disrupts electrostatic interactions between proteins associated with membrane lipids or integral-membrane proteins, such that any non-integral membrane proteins are released into the aqueous surrounding. The CEF is then repelleted in a second 150000 × g spin, which is expected to contain only integral-membrane proteins (‘washed CEF’). It was of interest to determine whether the identity of the salt would play a role in the effectiveness of the wash. To test this possibility, a comparison was made between three solutions with the following salt contents: 500 mM NaCl, 500 mM KCl, 250 mM NaCl + 250 mM KCl (Table III). The results show the fold purification and percent yield are highest for the CEF wash performed using the combination of KCl and NaCl. While the salt wash using the combined salt solution was advantageous, interestingly, the use of solutions of only 500 mM KCl or 500 mM NaCl resulted in both unfavorable yields and fold purification. Consequently, the combination salt wash was used routinely thereafter.
Table III.
Comparison of several salt solutions in CEF salt wash efficiency.
| Fraction | Total Protein (mg) |
Rate (nmol/min) |
Total activity |
Specific activity |
Percent Yield |
Fold Purification |
|---|---|---|---|---|---|---|
| NaCl CEF (pre-wash) | 143 | 5.6E-06 | 7.2E-02 | 5.0E-04 | 100 | 1.0 |
| NaCl CEF (post-wash)a | 75 | 2.5E-06 | 3.6E-02 | 4.7E-04 | 49 | 0.9 |
| KCl CEF (pre-wash) | 152 | 5.2E-06 | 6.8E-02 | 4.5E-04 | 100 | 1.0 |
| KCl CEF (post-wash) | 75 | 1.6E-06 | 2.5E-02 | 3.3E-04 | 36 | 0.7 |
| NaCl + KCl CEF (pre-wash) | 189 | 6.2E-06 | 8.0E-02 | 4.2E-04 | 100 | 1.0 |
| NaCl + KCl CEF (post-wash) | 75 | 3.6E-06 | 5.4E-02 | 7.2E-04 | 68 | 1.7 |
The percent yield and fold-purification values for ‘post-wash’ fractions were determined relative to the pre-washed state only.
It is worth noting that the specific activity measurements were indispensable for determining that this customary purification step is actually obstructive for PglB purification unless it is carried out in specific salt conditions. At this point, the CEF is resuspended in a small volume (less than 10 mL lysis buffer) so that membrane structure remains intact and will provide stability. This concentrated solution can be dispensed into aliquots and stored at −80°C, where it remains stable for a year or more.
Solubilization of membrane proteins
Solubilization of the CEF involves addition of a high concentration of detergent to the membrane fraction, such that the native lipid structure surrounding the membrane proteins is disrupted and the membrane proteins may be released into solution. The lost lipid periphery on these proteins is presumably replaced with stabilizing detergent micelles, or a mixture of lipids and detergent. The identity and concentration of the detergent used imparts highly variable yields of active protein. However, many detergents are very costly and large detergent screens are often time-consuming. Rather than investigating a large range of detergents, four detergent conditions were initially screened to resolve the amount of optimization that needed to be performed. The detergents initially screened were DDM (n-dodecyl-β-D-maltoside), OG (n-octyl-β-D-glucoside), Triton X-100, and an equal combination of the former three. A final concentration of 0.5% (v/v) was used for the four detergent conditions (see Methods). These detergents were chosen for the following reasons: DDM has traditionally been successful in the solubilization of membrane proteins [39,40], OG has a high CMC (critical micelle concentration) which facilitates easy removal of detergent at later points, and Triton X-100 is economical and oft-used in protein biochemistry.
A 5% solution of detergent is diluted 10-fold into the resuspended CEF, which has been diluted to 10 mg/mL total protein. This mixture is homogenized and vortexed vigorously, followed by dilution to lower the detergent concentration (see Methods). Results (Table IV) indicate that DDM is most efficient at solubilizing active PglB relative to the other detergent solutions tested. The use of 0.5% DDM gives a very favorable percent yield, eliminating the need to further optimize this step. In the common case that initial detergent conditions fail to provide satisfactory results, additional detergents and concentrations are generally compared.
Table IV.
Comparison of several detergent solutions in CEF-solubilization efficiency.
| Fraction | Total protein (mg) |
Rate (nmol/min) |
Total activity (nmol/min) |
Specific activity (nmol/min*mg) |
Percent yield |
Fold purification |
|---|---|---|---|---|---|---|
| pre-solubilizeda | 27.9 | 1.4E-05 | 8.3E-03 | 3.0E-04 | 100 | 1.0 |
| post-DDMb | 9 | 1.2E-05 | 7.1E-03 | 7.9E-04 | 86 | 2.7 |
| post-OG | 2.7 | 1.1E-06 | 6.6E-04 | 2.4E-04 | 8 | 0.8 |
| post-Triton | 6.3 | 9.8E-07 | 5.9E-04 | 9.3E-05 | 7 | 0.3 |
| post-combination | 7.2 | 7.0E-06 | 4.2E-03 | 5.8E-04 | 51 | 2.0 |
Percent yield and fold-purification values for ‘post-solubilized’ fractions were determined relative to pre-solubilized fraction.
Fraction after solubilization with DDM. All conditions had a final detergent concentration of 0.5%.
Affinity chromatography
The specificity of PglB affinity purification using Ni-NTA affinity was inefficient under batch binding conditions initially used. Initial concentrations for batchbinding were 50 mM HEPES, pH 7.5, 0.17% DDM, 10 mM imidazole. A high relative concentration of contaminating proteins co-eluted with PglB and recovery of PglB from the column was incomplete. There are several additives which are known to decrease non-specific binding to Ni-NTA resin and between proteins [41]. Figure 5A shows a gel of PglB elutions after batch binding with several conditions. Addition of glycerol to all buffers and increasing imidazole from 10 to 20 mM successfully decreased the amount of the low-molecular weight impurities relative to the full-length PglB. Diluting the solution two-fold during batch binding significantly decreased impurities as well; however, only when batch binding proceeded for a longer duration did these conditions result in increased purity without decreasing yields in elutions relative to flow through. In general, longer batch binding time is required for maximal binding of a more dilute solution of His-tagged proteins. For PglB, batch binding diluted, solubilized CEF to fresh Ni-NTA resin overnight, in the presence of glycerol and increased imidazole, resulted in the highest purity with the least loss of protein in the flow through and washes.
Figure 5.
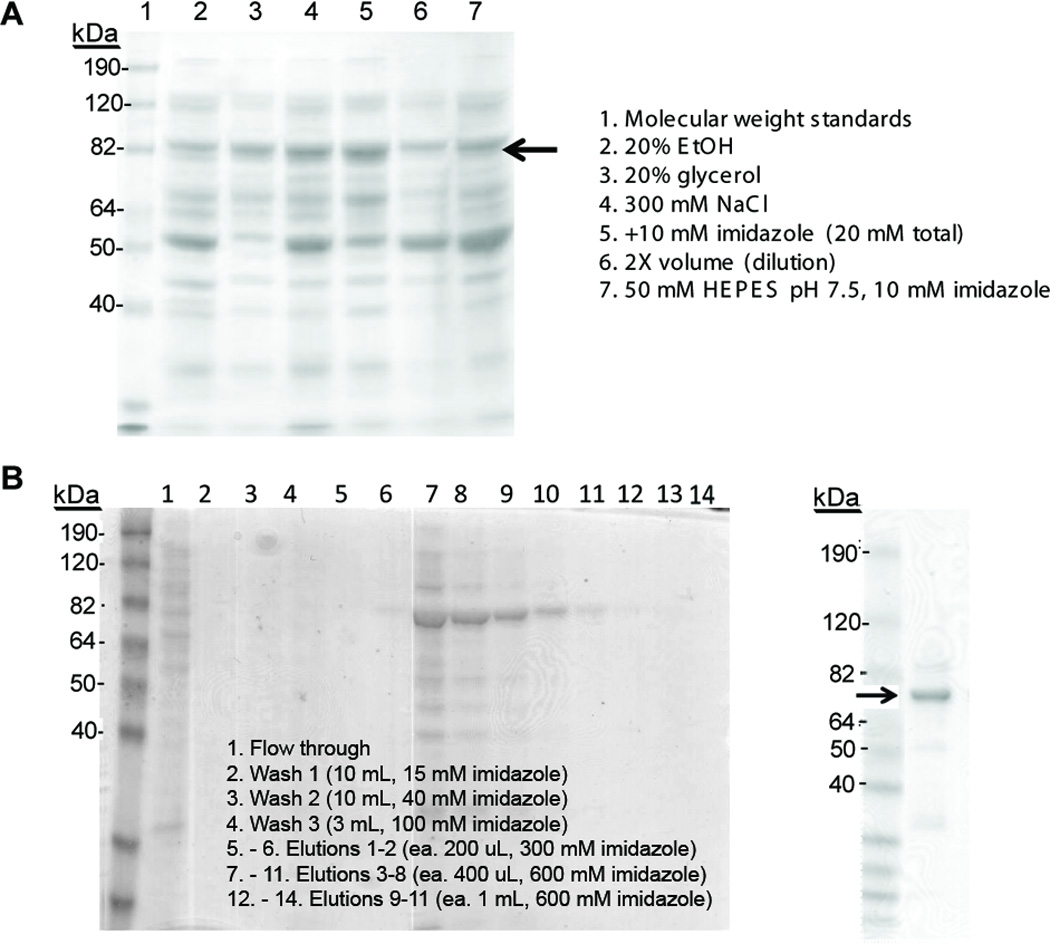
Optimization of Ni-NTA purification. A. Batch binding buffer conditions screened for effect on PglB purity after Ni-NTA column. Lane 1 contains the Benchmark pre-stained protein ladder (Invitrogen), Lanes 2–6 contain standard buffer of 50 mM HEPES, pH 7.5, 10 mM imidazole, 0.17% DDM, plus the additive noted to the right of the gel. Buffer 7 contained only standard buffer. Arrow indicates PglB band. B. Improved purification of PglB over Ni-NTA column. Coomassie-stained SDS-PAGE of flow through, washes, and elutions are shown (bottom left). Based on activity measurements of each fraction, the elution fractions 5–10 (corresponding to lanes 7–12 on the bottom left gel) were combined. The right-most Coomassie-stained SDS PAGE shows the purity of the solution after concentrating the combined elutions. The arrow indicates PglB band. The improvement in purity in the right-most gel can be attributed to (1) the purification accompanying the method of concentration, as the 100-kDa MWCO filter causes many of the smaller remaining impurities to be removed with the filtrate, and (2) homogenization of the PglB dispersity during concentration, eliminating the >82-kDa molecular-weight band, which appears to be a persistent oligomer of PglB.
Another alteration that significantly affected the purification was replacement of the His6 tag (native to the pET24a(+) vector) with a His10 tag. This change was made during expression optimization, and thus all purification optimization heretofore described was performed with the His10 tag. Lengthening of the His-tag causes a clear increase in the affinity of PglB for the Ni-NTA resin and it is noted that much higher imidazole concentrations are required to elute all the bound PglB from the resin (up to 600 mM from 300 mM). In fact, using the typical concentration of 300 mM imidazole for elution was ultimately found to recover only a fraction of the bound PglB. Accordingly, very little protein remained in the flow through and washes based on gel analysis (Figure 5B) and activity assays. These changes resulted in a final value fold-purification of 500 (Table I), relative to the ~50-fold purification values achieved before Ni-NTA-purification optimization.
Buffer Exchange
It has been observed that PglB activity is rapidly lost after elution rom the Ni-NTA column due to prolonged exposure to very high imidazole concentration. Dialysis, which was previously used to exchange the buffer and remove the imidazole after elution, proceeded too slowly for PglB to maintain activity. Therefore, an alternative method was used to exchange buffer: a HiTrap desalting column (G.E. Healthcare) was used immediately after elution to replace the elution buffer with one that lacked imidazole. However, use of the desalting column diluted the protein and caused significant loss of activity. Next, an Amicon Ultra 100-kDa MWCO cellulose filter was used, which concentrates the fractions containing PglB, and also serves as a method to exchange the buffer and remove the imidazole (Figure 5B). While the molecular weight of PglB (82 kDa) is below the molecular-weight cut-off of 100 kDa, the mass is high enough that there is virtually no PglB is lost in the filtrate. This may in part be due to the presence of the DDM detergent micelle surrounding PglB. In contrast, the DDM detergent micelle alone (without PglB bound) flows through the 100 kDa-MWCO filter despite the reasonably large size of the DDM micelle (~ 50 kDa) [30]. This selectivity is very advantageous because it allows the protein to be concentrated without simultaneously concentrating the detergent to a degree that is deleterious to the protein stability. In addition, the loss of lower molecular-weight impurities is consistently observed when using these high molecular-weight cut-off filters presumably due to the fact that these contaminants can pass through the filter.
In summary, this method of buffer exchange results in negligible loss of activity whereas dialysis of a PglB solution overnight at 4°C results in complete loss of enzyme activity. In fact, it was found that PglB solutions lose activity when dialyzed overnight, even in the absence of imidazole. Thus, it is likely that the dialysis procedure was a cause of activity loss in PglB in addition to the high imidazole. Using the 100-kDa MWCO Amicon filter, Ni-NTA-purified PglB is concentrated and buffer-exchanged to roughly 10 µM in 50 mM HEPES pH 7.5, 100 mM NaCl, 30% glycerol, 0.01% DDM. This solution is distributed into aliquots and stored at −80°C and, under these conditions, remains stable for at least two months (Supplementary Information, Figure 4).
DISCUSSION
The purification values in Table I were recorded for a 3.6 g cell pellet, which represents roughly one seventh of the cell weight obtained from one liter of culture using autoinduction. The 0.6 mg protein remaining at this point is > 95% purified PglB as judged by SDS-PAGE (Figure 5B). Thus, we estimate that per liter of culture, using autoinduction expression and the optimized purification conditions, 1–5 mg pure PglB is obtained. This represents a 100-fold increase in levels of pure, stable PglB relative to pre-optimized levels, which were estimated to be ~50 µg per liter of culture. Importantly, multiple PglB preparations have been completed using the current optimized protocol, demonstrating the reproducibility of yields obtained, and indicating that the values shown in Table I are representative of an average purification.
Nonetheless, the optimized procedure for purifying PglB is far more time-consuming than purification of most soluble proteins. The obligatory steps of isolating and processing the E. coli membrane fraction add considerable time and effort to the purification process. Additionally, a greater emphasis is placed on maintaining the protein at a cold temperature, working rapidly, and following protocols strictly. Furthermore, optimized quantities of membrane proteins often seem scant relative to typical yields for soluble proteins. These difficulties, however, are well-recognized by those intending to express and purify an uncharacterized membrane protein, and there are many thorough reviews that provide an overview of these challenges and potential solutions [42–45]. While optimizing PglB expression and purification, several additional obstacles and solutions were encountered, and it is hoped that these may be beneficial for non-specialists to consider when commencing such an effort.
Ironically, it proved challenging to navigate the abundant options available that have potential to improve the expression or stability of a membrane protein. Various cell lines, fusion tags, expression-culture additives, detergent alternatives, protein stabilizers, and countless other methodologies have reported instances of success in handling of certain membrane proteins. Yet, there are very few guarantees, and optimizing most steps remains an empirical process. In the case of PglB, it proved to be more time-efficient and economical to begin optimization of each step with a limited range of commonly used conditions and expand based on those results, when necessary. Deciding on a standard for sufficiency is useful, and when a step is particularly problematic, it may be practical to allot a specific amount of time to the task. If progress is not made by the end of the time period, perhaps it is wiser to try another route or target entirely. It was also found helpful to develop a clear strategy from the onset, and to begin with the first step of the procedure. Once the first step of the procedure has been optimized, the next step of the procedure should only then be addressed, because conditions used at each step in expression and purification can have unpredictable results on those that follow. The importance of designing a systematic procedure for optimization and an objective measure of comparison cannot be underestimated. The inevitably qualitative nature of protein biochemistry—resulting from variability in individual transformation colonies, expression cultures, loading of gels and western-blot transfers—multiplies the uncertainty of qualitative judgments.
The presented optimization of PglB expression and purification was performed with the ultimate purpose of kinetic and mechanistic characterization, rather than crystallographic studies. Implications of this goal include a primary interest in obtaining high quantities of pure, active, and stable protein. In contrast, crystallography-driven optimizations often prioritize obtaining monodisperse, concentrated protein, which are absolute requirements for obtaining a crystal structure. Because the goal of the presented procedure is biochemical, SDS-PAGE and western blots alone represent poor methods for comparing the optimal conditions for expression and purification of the enzyme. While a condition used in a purification step may yield a bright band on a western blot, it may be unfavorable for activity yields. If constructs with tags of various sizes are screened, the differences in molecular weight can be a perplexing factor to account for in gel densitometry measurements. Additionally, expression tags can have effects on structure, activity and monodispersity of the enzyme, which cannot be accounted for using SDS-PAGE. Purifying a protein based on specific activity and yields allows a more rigorous and precise measure of the most efficient purification condition to use for each step.
However, the requirement for a quantitative activity assay can be problematic, as activity assays are protein-specific, and reagents and equipment can make the assays laborious or expensive to perform frequently. Undeniably, in the developing field of membrane-protein biology, most information acquired is important and useful even if lacking in certain methodological precision. Though, while it may be exciting to speculate on results from experiments and techniques available, it is equally important to be conscientious of the questions that these experiments leave unanswered.
Supplementary Material
-
►
A protocol for recombinant production of PglB, a bacterial oligosaccharyl transferase.
-
►
PglB was overexpressed in E. coli using high-density autoinduction.
-
►
An average procedure gives 550-fold purification with 64% yield.
-
►
Pure enzyme catalyzes N-linked glycosylation for over two months with no activity loss.
ACKNOWLEDGMENTS
The authors would like to thank former lab member Dr. Mark Chen for his contributions to PglB expression analysis; specifically, gratitude is expressed for cloning and expression of multiple PglB fusion-tag constructs. The authors are grateful to the full Imperiali group for frequent contributions of invaluable scientific discussions and technical knowledge.
Funding Sources
This work was supported by NIH Grant GM039334 (to B.I.).
ABBREVIATIONS USED
- NLG
asparagine-linked glycosylation
- OTase
oligosaccharyl transferase
- TM
transmembrane
- CEF
cell envelope fraction
- O.D.
Optical Density
- PCR
polymerase chain reaction
- TMHMM
TM Hidden Markov Model
- DMSO
dimethylsulfoxide
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- SDS
sodium dodecyl sulfate
- PAGE
polyacrylamide gel electrophoresis
- LB
Luria-Bertani
- Ni-NTA
Nickel-Nitrilotriacetic acid
- IPTG
isopropyl-β-D-thiogalactoside
- EDTA
ethylenediaminetetraacetic acid
- DDM
n-dodecyl-β-D-maltopyranoside
- OG
n-octyl-β-D-glucoside
- MBP
maltose binding protein
- GB1
G-protein B1 domain
- SUMO
small ubiquitin-like modifier
- Und-PP-Bac-[3H]GalNAc
undecaprenyl diphosphate-linked-α-Bac- α-N-acetylgalactosamine
- Bac
bacillosamine (2,4-diacetamido-2,4,6-trideoxyglucose)
- pF
para-nitrophenylalanine
- CMC
critical micelle concentration
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Ahram M, Litou ZI, Fang R, Al-Tawallbeh G. Estimation of membrane proteins in the human proteome. In Silico Biol. 2006;6:379–386. [PubMed] [Google Scholar]
- 2.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 3.Yildirim MA, Goh KI, Cusick ME, Barabasi AL, Vidal M. Drug-target network. Nat Biotechnol. 2007;25:1119–1126. doi: 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]
- 4.Katzen F, Peterson TC, Kudlicki W. Membrane protein expression: no cells required. Trends Biotechnol. 2009;27:455–460. doi: 10.1016/j.tibtech.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 5.Sarkar CA, Dodevski I, Kenig M, Dudli S, Mohr A, et al. Directed evolution of a G protein-coupled receptor for expression, stability, and binding selectivity. Proc Natl Acad Sci U S A. 2008;105:14808–14813. doi: 10.1073/pnas.0803103105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Q, Miller LJ, Dong M. Role of N-linked glycosylation in biosynthesis, trafficking, and function of the human glucagon-like peptide 1 receptor. Am J Physiol Endocrinol Metab. 2010;299:E62–E68. doi: 10.1152/ajpendo.00067.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luthi F, Leibundgut K, Niggli FK, Nadal D, Aebi C, et al. Serious medical complications in children with cancer and fever in chemotherapy-induced neutropenia: results of the prospective multicenter SPOG 2003 FN study. Pediatr Blood Cancer. 2012;59:90–95. doi: 10.1002/pbc.23277. [DOI] [PubMed] [Google Scholar]
- 8.Ubelhart R, Bach MP, Eschbach C, Wossning T, Reth M, et al. N-linked glycosylation selectively regulates autonomous precursor BCR function. Nat Immunol. 2010;11:759–765. doi: 10.1038/ni.1903. [DOI] [PubMed] [Google Scholar]
- 9.Londrigan SL, Turville SG, Tate MD, Deng YM, Brooks AG, et al. N-linked glycosylation facilitates sialic acid-independent attachment and entry of influenza A viruses into cells expressing DC-SIGN or L-SIGN. J Virol. 2011;85:2990–3000. doi: 10.1128/JVI.01705-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Berkel PH, Gerritsen J, Perdok G, Valbjorn J, Vink T, et al. N-linked glycosylation is an important parameter for optimal selection of cell lines producing biopharmaceutical human IgG. Biotechnol Prog. 2009;25:244–251. doi: 10.1002/btpr.92. [DOI] [PubMed] [Google Scholar]
- 11.Bartusik D, Aebisher D, Lyons AM, Greer A. Bacterial inactivation by a singlet oxygen bubbler: identifying factors controlling the toxicity of (1)O2 bubbles. Environ Sci Technol. 2012;46:12098–12104. doi: 10.1021/es303645n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deniaud A, Bernaudat F, Frelet-Barrand A, Juillan-Binard C, Vernet T, et al. Expression of a chloroplast ATP/ADP transporter in E. coli membranes: behind the Mistic strategy. Biochim Biophys Acta. 2011;1808:2059–2066. doi: 10.1016/j.bbamem.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 13.Lizak C, Fan YY, Weber TC, Aebi M. N-Linked glycosylation of antibody fragments in Escherichia coli. Bioconjug Chem. 2011;22:488–496. doi: 10.1021/bc100511k. [DOI] [PubMed] [Google Scholar]
- 14.Wei Z, Lin T, Sun L, Li Y, Wang X, et al. N-linked glycosylation of GP5 of porcine reproductive and respiratory syndrome virus is critically important for virus replication in vivo. J Virol. 2012;86:9941–9951. doi: 10.1128/JVI.07067-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hurt JK, Fitzpatrick BJ, Norris-Drouin J, Zylka MJ. Secretion and N-linked glycosylation are required for prostatic acid phosphatase catalytic and antinociceptive activity. PLoS One. 2012;7:e32741. doi: 10.1371/journal.pone.0032741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hillaire ML, van Eijk M, Nieuwkoop NJ, Vogelzang-van Trierum SE, Fouchier RA, et al. The number and position of N-linked glycosylation sites in the hemagglutinin determine differential recognition of seasonal and 2009 pandemic H1N1 influenza virus by porcine surfactant protein D. Virus Res. 2012;169:301–305. doi: 10.1016/j.virusres.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 17.Somnuke P, Hauhart RE, Atkinson JP, Diamond MS, Avirutnan P. N-linked glycosylation of dengue virus NS1 protein modulates secretion, cell-surface expression, hexamer stability, and interactions with human complement. Virology. 2011;413:253–264. doi: 10.1016/j.virol.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwarz F, Aebi M. Mechanisms and principles of N-linked protein glycosylation. Curr Opin Struct Biol. 2011;21:576–582. doi: 10.1016/j.sbi.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 19.Lizak C, Gerber S, Numao S, Aebi M, Locher KP. X-ray structure of a bacterial oligosaccharyltransferase. Nature. 2011;474:350–355. doi: 10.1038/nature10151. [DOI] [PubMed] [Google Scholar]
- 20.Jaffee MB, Imperiali B. Exploiting topological constraints to reveal buried sequence motifs in the membrane-bound N-linked oligosaccharyl transferases. Biochemistry. 2011;50:7557–7567. doi: 10.1021/bi201018d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guarino C, DeLisa MP. A prokaryote-based cell-free translation system that efficiently synthesizes glycoproteins. Glycobiology. 2012;22:596–601. doi: 10.1093/glycob/cwr151. [DOI] [PubMed] [Google Scholar]
- 22.Chen MM, Bartlett AI, Nerenberg PS, Friel CT, Hackenberger CP, et al. Perturbing the folding energy landscape of the bacterial immunity protein Im7 by site-specific N-linked glycosylation. Proc Natl Acad Sci U S A. 2010;107:22528–22533. doi: 10.1073/pnas.1015356107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor DE, Eaton M, Yan W, Chang N. Genome maps of Campylobacter jejuni and Campylobacter coli. J Bacteriol. 1992;174:2332–2337. doi: 10.1128/jb.174.7.2332-2337.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karlyshev AV, Henderson J, Ketley JM, Wren BW. An improved physical and genetic map of Campylobacter jejuni NCTC 11168 (UA580) Microbiology. 1998;144(Pt 2):503–508. doi: 10.1099/00221287-144-2-503. [DOI] [PubMed] [Google Scholar]
- 25.Bao WJ, Gao YG, Chang YG, Zhang TY, Lin XJ, et al. Highly efficient expression and purification system of small-size protein domains in Escherichia coli for biochemical characterization. Protein Expr Purif. 2006;47:599–606. doi: 10.1016/j.pep.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 26.Studier FW. Protein production by auto-induction in high-density shaking cultures. Protein Expression and Purification. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 27.Glover KJ, Weerapana E, Numao S, Imperiali B. Chemoenzymatic synthesis of glycopeptides with PglB, a bacterial oligosaccharyl transferase from Campylobacter jejuni. Chem Biol. 2005;12:1311–1315. doi: 10.1016/j.chembiol.2005.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen MM, Glover KJ, Imperiali B. From peptide to protein: comparative analysis of the substrate specificity of N-linked glycosylation in C. jejuni. Biochemistry. 2007;46:5579–5585. doi: 10.1021/bi602633n. [DOI] [PubMed] [Google Scholar]
- 29.Sharma CB, Lehle L, Tanner W. N-Glycosylation of yeast proteins. Characterization of the solubilized oligosaccharyl transferase. Eur J Biochem. 1981;116:101–108. doi: 10.1111/j.1432-1033.1981.tb05306.x. [DOI] [PubMed] [Google Scholar]
- 30.Bhairi SM, Mohan C EMD Biosciences. Detergents Booklet. San Diego, CA: 2007. [Google Scholar]
- 31.Graslund S, Sagemark J, Berglund H, Dahlgren LG, Flores A, et al. The use of systematic N- and C-terminal deletions to promote production and structural studies of recombinant proteins. Protein Expr Purif. 2008;58:210–221. doi: 10.1016/j.pep.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 32.Maita N, Nyirenda J, Igura M, Kamishikiryo J, Kohda D. Comparative structural biology of eubacterial and archaeal oligosaccharyltransferases. J Biol Chem. 2010;285:4941–4950. doi: 10.1074/jbc.M109.081752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie H, Guo XM, Chen H. Making the most of fusion tags technology in structural characterization of membrane proteins. Mol Biotechnol. 2009;42:135–145. doi: 10.1007/s12033-009-9148-x. [DOI] [PubMed] [Google Scholar]
- 34.Davies AH, Jowett JB, Jones IM. Recombinant baculovirus vectors expressing glutathione-S-transferase fusion proteins. Biotechnology (N Y) 1993;11:933–936. doi: 10.1038/nbt0893-933. [DOI] [PubMed] [Google Scholar]
- 35.Pryor KD, Leiting B. High-level expression of soluble protein in Escherichia coli using a His6-tag and maltose-binding-protein double-affinity fusion system. Protein Expr Purif. 1997;10:309–319. doi: 10.1006/prep.1997.0759. [DOI] [PubMed] [Google Scholar]
- 36.Sachdev D, Chirgwin JM. Solubility of proteins isolated from inclusion bodies is enhanced by fusion to maltose-binding protein or thioredoxin. Protein Expr Purif. 1998;12:122–132. doi: 10.1006/prep.1997.0826. [DOI] [PubMed] [Google Scholar]
- 37.Panavas T, Sanders C, Butt TR. SUMO fusion technology for enhanced protein production in prokaryotic and eukaryotic expression systems. Methods Mol Biol. 2009;497:303–317. doi: 10.1007/978-1-59745-566-4_20. [DOI] [PubMed] [Google Scholar]
- 38.Bernaudat F, Frelet-Barrand A, Pochon N, Dementin S, Hivin P, et al. Heterologous expression of membrane proteins: choosing the appropriate host. PLoS One. 2011;6:e29191. doi: 10.1371/journal.pone.0029191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.VanAken T, Foxall-VanAken S, Castleman S, Ferguson-Miller S. Alkyl glycoside detergents: synthesis and applications to the study of membrane proteins. Methods Enzymol. 1986;125:27–35. doi: 10.1016/s0076-6879(86)25005-3. [DOI] [PubMed] [Google Scholar]
- 40.Rosevear P, VanAken T, Baxter J, Ferguson-Miller S. Alkyl glycoside detergents: a simpler synthesis and their effects on kinetic and physical properties of cytochrome c oxidase. Biochemistry. 1980;19:4108–4115. doi: 10.1021/bi00558a032. [DOI] [PubMed] [Google Scholar]
- 41.QIAGEN. N-NTA reagent compatibility table. 2008:1–4. [Google Scholar]
- 42.Rosenbusch JP. Stability of membrane proteins: relevance for the selection of appropriate methods for high-resolution structure determinations. J Struct Biol. 2001;136:144–157. doi: 10.1006/jsbi.2001.4431. [DOI] [PubMed] [Google Scholar]
- 43.Grisshammer R. Understanding recombinant expression of membrane proteins. Curr Opin Biotechnol. 2006;17:337–340. doi: 10.1016/j.copbio.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 44.Aebi A, Neumann P. Endosymbionts and honey bee colony losses? Trends Ecol Evol. 2011;26:494. doi: 10.1016/j.tree.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 45.Lluis MW, Godfroy JI, 3rd, Yin H. Protein engineering methods applied to membrane protein targets. Protein Eng Des Sel. 2012 doi: 10.1093/protein/gzs079. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.