

Abstract
Increasing attention has been given to the anti-cancer effects of curcumin and the ability of this natural product to inhibit cancer cell proliferation. New curcumin analogs have been developed to optimize the in vitro and in vivo activity of the parent compound yet retain the same safety profile. EF24, a fluorinated synthetic analog, surpasses curcumin in its ability to inhibit cancer cell viability and down-regulate TNFα-induced NF-κB activation. Here we report a critical role of the p38-mediated signaling pathway in the determination of lung cancer cell’s sensitivity to EF24. We have found that EF24-induced decease of lung cancer cell viability was accompanied by upregulated mitogen-activated protein kinases (MAPK) as evidenced by increased phosphorylation of ERK1/2, JNK, and p38. Pharmacological investigation led to our suggestion that EF24 triggers a negative feedback loop through p38 activation. In support of this model, inhibition of p38, either by small molecule inhibitors or through an RNAi-mediated knockdown approach, enhanced the EF24 induced apoptotic death of A549 cells. Thus, inhibition of p38 may boost the EF24 anticancer effect. Indeed, a combination of EF24 and SB203580, a p38 inhibitor, synergistically inhibited clonogenic activity of A549 lung cancer cells and induced their apoptosis as reflected by poly(ADP-ribose) polymerase cleavage, the accumulation of the sub-G1 fraction of cells, and apoptotic cell staining. These studies offer a novel strategy that combines the curcumin analog EF24 with a p38 inhibitor for potentially enhanced therapy in the treatment of lung cancer.
Keywords: curcumin analogs, EF24, p38, p38 inhibitors, lung cancer
1. Introduction
Curcumin has demonstrated great potential as a chemopreventative and therapeutic agent due to its ability to negatively modulate cancer-related biomarkers and inhibit the proliferation of tumor cells but retain pharmacological safety profile in vivo [1–4]. However, clinical studies have shown that curcumin is less efficacious in vivo because over 80% of this compound does not reach systemic circulation, but rather is rapidly excreted [4–5]. This prompted the design of analogs, including the fluorinated analog, EF24 (Fig. 1A), which are more biologically active in inducing apoptosis in vitro assays and also more potent in vivo [6–7]. Extensive studies are being conducted into the mechanism of action of these analogs, in particular EF24, to advance the clinical development of this agent as a promising new therapeutic candidate.
Figure 1. Effect of curcumin and EF24 on A549 lung cancer cell viability.
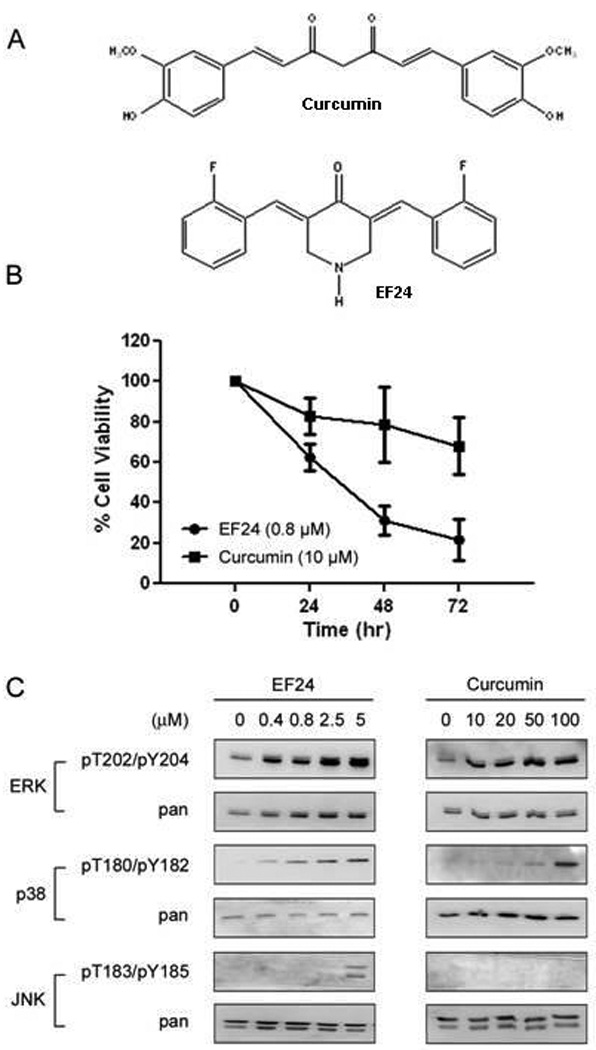
(A) Structures of curcumin and the novel fluorinated curcumin analog, EF24. (B) A549 cells were grown in a 96 well plate and treated with EF24 (0.4 µM or 0.8 µM) or curcumin (10 µM) for indicated times. Cell viability was assessed by the SRB method and is expressed as percentage of vehicle-treated control (0.5% DMSO) (n = 3). The error bars indicate the standard deviation of the mean. (C) A549 cells were treated with EF24 or curcumin for 30 min before the status of ERK, p38, and JNK were determined using phospho-specific antibodies for the Thr/Tyr activation motifs.
Like curcumin, EF24 inhibits the NF-κB signaling pathway [8–9]. The interplay between the NF-κB pathway and other intracellular pathways has been extensively studied. NF-κB appears to be mechanistically linked to the mitogen-activated protein kinase (MAPK) pathways [10–11]. The MAPK pathways are three-tiered kinase regulatory systems, which are activated upon stimulation with extracellular signals such as growth factors and ultimately elicit a corresponding biological response. The three major MAPK family members include the extracellular-regulated kinase/mitogen activated protein kinase (ERK/MAPK), the c-jun N-terminal kinase (JNK), and p38 MAPK, Activation of ERK has been associated with cell growth and differentiation, while JNK and are often activated by stress signals. JNK and p38 not only can respond to mitogens but also a variety of cellular stresses including inflammatory cytokines [12–12]. Recently, p38 has also been implicated in the transcriptional up-regulation of NF-κB, further suggesting a connection between inflammation, cancer, and NF-κB [13–14].
Like other MAPKs, p38 are activated by MAP kinase kinases (MAPKKs/MKKs) with MKK3 and MKK6 being the two main upstream MAPKKs [15]. p38 in turn activates a number of downstream substrates, including MAP kinase-activated protein kinase-2 (MAPKAPK-2/MK2), transcription factor-2 (ATF-2), mitogen- and stress-activated kinase (MSK), and p53 [16–17]. The role of p38 in human cancers appears to be complex. Published data present arguments for both pro-survival and pro-apoptotic functions of p38. However, there are reports that p38 is selectively activated in homogenates of non-small cell lung tumors compared with normal tissue. Thus, it may be involved in malignant cell growth or transformation [18].
Intense efforts in MAPK inhibitor development have led to the evaluation of p38 inhibitors for the treatment of rheumatoid arthritis, skin disorders, and other inflammatory diseases [19–20]. p38 inhibitors have been found to inhibit the production of pro-inflammatory cytokines and therefore inhibit the propagation of the inflammatory response. Interestingly, the pyridinyl imidazole class of compounds, which has long been recognized for its ability to suppress cytokine biosynthesis, has been used as a template for the design of new p38 inhibitors presently used in clinical studies [21]. The pyridinyl imidazoles, which include SB203580 and SB202190, selectively target the ubiquitously expressed p38α and p38β isoforms by competing with ATP for its binding site [20, 22–23].
How susceptible cancer cells are to chemotherapeutic-induced cell death is dependent on a balance between cell death and survival mechanisms. Increased survival signaling could possibly counteract drug efficacy. For example, the induction of NF-κB by Taxol has been well documented and has garnered much attention [24]. Taxol has been documented to induce the phosphorylation and degradation of IκB, leading to the nuclear translocation of the NF-κB heterodimer. This increased NF-κB activation may lead to Taxol-resistance [25–26]. In addition, recent reports indicate that NF-κB inhibitors increased the efficacy of paclitaxel in ovarian cancer [27–28]. In our mode of action studies on EF24 as an anticancer agent, we noticed that treatment of cells with EF24 drastically induced the upregulation of three major MAPK pathways mediated by ERK, JNK, and p38. Examination with pharmacological agents suggests a critical role of p38 in determining cell response to EF24. Indeed, the combination of EF24 and p38 inhibitors exhibits synergistic cytotoxicity against the lung cancer cell line A549. Thus, blocking the p38 pathway may offer a promising strategy to enhance the efficacy of EF24-based therapy for the treatment of cancer and possibly various inflammatory diseases.
2. Materials and Methods
2. 1. Chemicals and antibodies
Curcumin and the analog EF24 were synthesized as previously described [6]. Stock solutions of each (10 µM) were made in DMSO and stored in aliquots at −20°C. The compounds were diluted in incubation media immediately prior to each experiment. Most chemicals are from Sigma-Aldrich (St. Louis, MO) otherwise as stated. The pyridinyl imidazole compounds SB203580 (Cat. No. V1161; Promega; Madison, WI) and SB202190 (Cat. No. S7067; Biosource; Camarillo, CA) were used as p38α/β inhibitors. Antibodies against total and phospho-JNK (Thr183/Tyr185), total and phospho-p38 MAPK (Thr180/Tyr182), total and phospho-ERK (Thr202/Tyr204), PARP (full length and cleaved fragments) were all purchased from Cell Signaling (Cat. No. 4668, 9215S, 4377, 9542; Beverly, MA).
2. 2. Cell Culture
The human non-small cell lung cancer (NSCLC) cell line A549 were grown in RPMI-1640, 10% CellGro FBS (Fisher Scientific; Pittsburgh, PA) and maintained at 37°C in an atmosphere containing 10% CO2. The media was supplemented with 1% penicillin/streptomycin (Fisher Scientific; Pittsburgh, PA).
2.3. Immunoblot analysis
Cells were lysed in 1% NP-40 buffer (50mM Tris-HCl, 150 mM NaCl, 1% NP-40, pH 8,0) supplemented with one complete protease inhibitor cocktail tablet (Cat. No. 04693159001; Roche, Indianapolis, IN) per 10 ml solution and protein from the whole cell extracts were resolved by 12.5% SDS-PAGE (25 – 40 µg/lane), electrotransferred to nitrocellulose membranes, and blocked with 5% nonfat dry milk in TBS-Tween 20 (Fisher Scientific; Pittsburgh, PA). The blots were then incubated with the indicated primary antibodies, followed by horseradish peroxidase-conjugated secondary antiserum from Santa Cruz Biotech. (Santa Cruz, CA). Immunoreactivity was visualized by enhanced chemiluminescence reagent (Cat. No. RPN2106; Amersham Biosciences, Piscataway, New Jersey). For sequential blotting with additional antibodies, the membranes were stripped using a strip buffer solution (2% SDS, 50 mM Tris-HCl, pH 6.8) with 2-mercaptoethanol (1:1000; Fisher Scientific; Pittsburgh, PA) and reprobed with the indicated antibodies.
2.4. In Vitro Viability Assay
Cells were plated at a density of 5,000 cells/well in a 96-well plate, or 1,000 cells/well in a 384-well plate, and allowed to adhere overnight. The following day the cells were treated with drug for 48 hours, or 72 hours, in triplicate. Similar IC50 values were obtained from either a 48 hr or 72 hr assay with A549 cells (Fig. S1). Cell viability was measured with the sulforhodamine B (SRB; ACROS Organics, Geel, Belgium) assay. Cells were fixed with 50 µl of 10% TCA at 4°C for 1hr. The plates were washed with water and stained with 4% SRB (100 µl). Acetic acid (1%) was then used to wash the plates. Lastly, 10 mM unbuffered Tris was added to solubilize the dye. Absorbance at 490 nm was recorded using a 96-well plate reader. The mean value and standard error for each treatment were determined and the % cell viability relative to control (0.5% DMSO) was calculated. The IC50 is defined as the concentration of drug that kills 50% of the total cell population as compared to control cells at the end of the incubation period.
2.5. Clonogenic assay
A549 cells were plated in low density (450 cells/well) in a 12-well plate and were allowed to adhere overnight. The cells were treated with test compounds the following day and every 3 days thereafter. On day 10 after colonies were formed, cells were fixed using 10% TCA for 30 min at 4°C. The wells were washed with water, stained with sulforhodamine B, and then washed with 1% acetic acid. An image of each well was taken. Colonies were counted using Image Processing and Analysis in Java (Image J, Research Service Branch, NIH). Large colonies were defined as colonies with a diameter greater than or equal to 2 mm. Small colonies were defined as though <2 mm in diameter.
2.6. Flow cytometry analysis
Apoptosis was examined by measuring DNA content with flow cytometry for propidium iodide (PI; Invitrogen, Carlsbad, CA) -stained cells. A549 cells were treated with a single agent or combination for 48 hr. Attached and unattached cells were then collected, washed with 1%BSA/PBS and fixed with 75% cold ethanol for at least 1hr. PI (50 µg/ml) was then used to stain the cells, and the DNA content of these stained cells was measured by a FACScan cytometer equipped with Cell Quest software (BD Biosciences; Franklin Lakes, NJ).
2.7. Cell health assay
A549 cells cultured in 96-well plate were stained with Hoechst 33342 (5 µg/ml), PI (2.5 µg/ml) and YO-PRO-1 (0.1 µM; Invitrogen; Carlsbad, CA) at 4 °C for 30 min and analyzed by ImageXpress system (Molecular Devices, Sunnyvale, CA). PI-single positive and YO-PRO-1-single positive cells are recognized as necrotic and early apoptotic cells, respectively, whereas PI/ YO-PRO-1-double positive cells are referred to as late apoptotic cells [29]. The percentages of viable cells and dying/dead cells, which consist of necrotic, early and late apoptotic cells, were analyzed.
2.8. siRNA transfection
A549 cells were transiently transfected with oligonucleotide p38 siRNA (Cat. No. 6564; Cell Signaling, Beverly, MA) using oligofectamine transfection reagent (Cat. No. 12252011; Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. 48 hr following initial transfection, A549 cells were subjected to western blotting. For the SRB cell viability assay, cells underwent the same two rounds of transfection. However, immediately after the second transfection, cells were treated with EF24 for 48hr where indicated.
2.9. In vitro p38 kinase assay
The p38 recombinant kinase (Cat. No. PH00125; Invitrogen, Carlsbad, CA) was preincubated with test compound for 30 min. Test compounds are SB203580 (0.078–40 µM) with or without EF24 (0.078–40 µM). p38 was then incubated with a mixture of [γ-32P] ATP (0.8 µCi; NEG502A250UC; Perkin Elmer, Boston, MA) and the exogenous substrate MBP (0.4 mg/ml; Fisher Scientific; Pittsburgh, PA) at 30°C for 15 minutes. The reactions were stopped by spotting 20 µl of the reaction mixture onto individual pre-cut strips of phosphocellulose P81 paper (Cat. No. 3698915; Whatman, U.K.). The phosphocellulose paper was then washed three timesx for 15 min each with phosphoric acid (0.5%) to remove unincorporated ATP. Incorporated [γ-32P] ATP in MBP was measured using liquid scintillation counting.
3. Results
3.1. EF24 induces the activation of three MAPK pathways
EF24 is more potent than the parent compound curcumin in inhibiting A549 cell viability, consistent with the previous report (Fig 1) [7]. In order to understand the mechanism that contributes to this potent anti-cancer activity of EF24, we analyzed pathways that may be modulated upon treatment with this agent. Previously, we demonstrated that EF24 down-regulates TNFα-induced NF-κB activation by negatively regulating the activity of the upstream kinase of IκB, IKK. Cross talk is known to exist between the NF-κB pathway and other important mediators in regulating cell survival and proliferation including the three-tiered MAPK signaling pathways. To determine whether EF24 modulates any of the three well studied MAPKs, we monitored the activation states of ERK, JNK, and p38 in response to EF24 or curcumin. Interestingly, we found that EF24 induces the activation of each of the MAPKs in a dose-dependent manner revealed by their upregulated phosphorylation (Fig. 1C). ERK, as well as p38, were also activated by curcumin, however at higher concentrations than EF24 (20 µM vs 0.4 µM and 50 µM vs 0.8 µM). At the time and doses used, no phosphorylation of JNK was detected with curcumin treatment while activation of JNK was detected after treatment of cells with EF24 (5 µM). The lack of induction of JNK by curcumin is consistent with published data that curcumin inhibits, instead of activates, the signal transduction pathways leading to JNK activation [30–31].
3.2. Inhibition of ERK and JNK activation does not dramatically affect EF24-induced cytotoxicity
Since ERK participates in a mitogenic pathway that promotes cell survival and proliferation and EF24 induces activation of ERK, we determined whether this pathway is important for the potency of EF24, and if inhibition of ERK would further augment the loss of cell viability of EF24-treated cells. A low dose of EF24 (0.4 µM) was chosen for this experiment since it only induces a low level of growth inhibition (15–25%) of cells and detectable MAPK activation after 48 h of treatment. The chemical inhibitor U0126 was used to selectively inhibit ERK by targeting the upstream kinase MEK. The concentrations of U0126 used in this study were determined to inhibit EF24-induced ERK phosphorylation (data not shown). When the viability of cells with either agent alone was compared to those with the combination of EF24 and U0126, no significant change in cell viability with inhibition of ERK was observed (Fig. S2A). It is noted the addition of 5 µM of U0126 along with EF24 reduced protein levels of ERK and p38. It is possible that this combination inhibits a stabilization pathway for ERK, the mechanism of which requires further investigation.
Similarly, we determined if JNK inhibition would affect EF24-induced cytotoxicity. Several recent reports implicate JNK in the regulation of the apoptotic response in which activated JNK mediates the induction of cell death [32]. We hypothesized that inhibition of JNK would attenuate EF24-induced cell death. A549 cells were treated with a combination of EF24 and a JNK inhibitor, SP600125, for 48 h. JNK inhibition did not attenuate the loss of cell viability by EF24, instead caused a slightly additive drop in cell viability (Fig. S2C). This effect by the JNK inhibitor to negatively affect cell viability may be due to a prosurvival role of JNK in A549 cells or due to the nonspecific effect of the compound, SP600125. This compound has been shown to inhibit other cellular kinases [33].
3.3. p38 inhibitors enhance EF24-induced cytotoxicity
We then evaluated the effect of p38 inhibition on EF24-induced cytotoxicity. The same low dose of EF24 (0.4 µM) was used as in the previous experiments with MEK and JNK. We combined EF24 with increasing concentrations of a pyridinyl imidazole p38 inhibitor, SB203580 or SB202190. These p38 inhibitors alone did not significantly affect the cell viability at the test concentrations. When SB203580 was combined with EF24, the percentage of growth inhibition was significantly greater than the effect of each compound alone (Fig. 2A). The combination of SB202190 (12.5 µM) and EF24 also had a dramatic effect on cell viability (Fig. 2B), showing an approximate 30% reduction in cell viability below the additive effects of the combination.
Figure 2. Inhibition of p38 enhances EF24-induced growth inhibition.
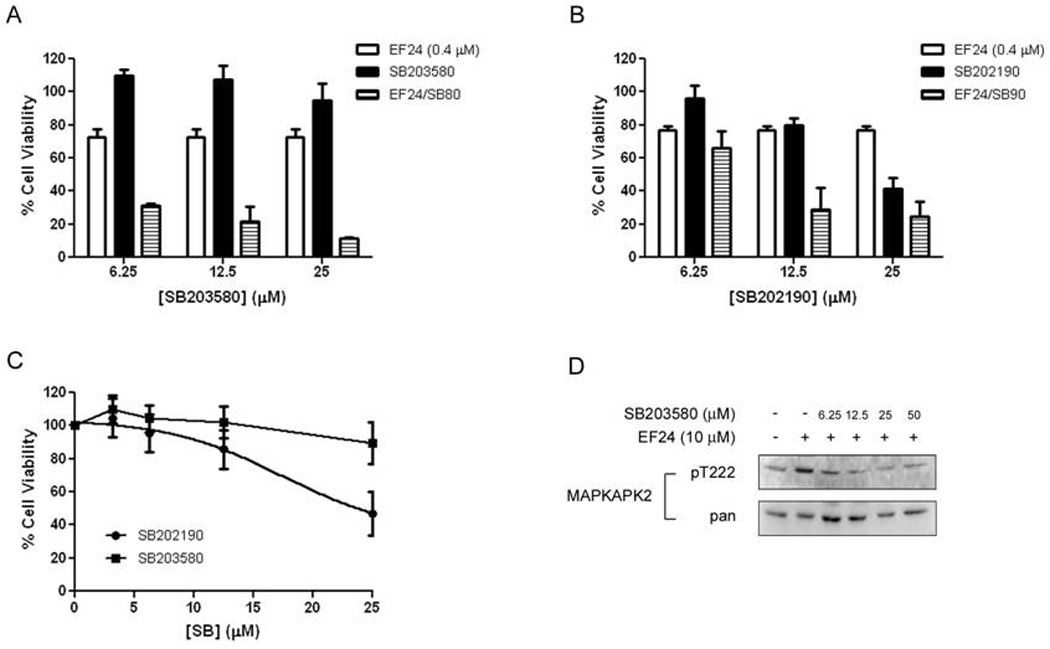
(A) Cells were treated with SB203580 or SB202190 for 48 hr. Cell viability was assessed using the SRB assay as the percentage of vehicle-treated (0.5% DMSO) control. (B, C) Combination treatment of A549 with EF24 (0.4µM) and SB203580 (B) or EF24 and SB202190 (C). The error bars in the experiments represent the standard deviation analyzed from three repeats. (D) A549 cells were pretreated with SB203580 for 1h before treatment with EF24 (10 µM) for 30 min. Cells were subjected to western blotting to access phosphorylation and protein expression of MAPKAPK-2 as a p38 substrate..
To determine whether the combination effect could be due to the cytotoxicity of the p38 inhibitors, the dose-dependent effect of both pyridinyl imidazoles on A549 cell growth was compared. SB203580 was found to have little effect on cell survival of A549 cells with the highest concentration tested (25 µM) only leading to a 10% loss in cell viability and an IC50 of approximately 50 µM (Fig. 2C). From the same analysis, the IC50 of SB202190 was determined to be approximately 25 µM.
It is important to note that caution should be taken when small molecule inhibitors are used in experiments. SB202190 and SB203580 have similar mechanisms in inhibition of p38 as ATP mimetics, though have shown differing effect on A549 cell viability. It has been well documented that SB202910 is a more promiscuous kinase inhibitor with a range of other targets [33], which leads credence as to why SB203580 is used more consistently in related experiments in the literature as a specific inhibitor of p38. This may be also evident in the fact that SB202190 and SB203580 have different IC50 values in their inhibition of A549 cell viability (approximately 25 µM and 50 µM, respectively), with SB202190 being more cytotoxic. The synergistic effect shown with the combination of EF24 and SB203850 may truly more adequately reflect the combinational activity of p38 inhibition and EF24 treatment. For this reason, SB203580 was employed for the remainder of the experiments.
The inhibition of p38 activity by SB203580 was also demonstrated by monitoring the phosphorylation of MAPKAPK-2, an immediate downstream effector of p38 (Fig. 2D). Again, inhibition of p38 by this compound significantly enhanced the EF24 effect. Since SB203580 has the least effect on A549 viability alone while still inhibiting p38 activity, this compound was used for the rest of the experiments.
3.4. The induction of apoptosis by EF24 is potentiated by p38 inhibition
EF24 has already been demonstrated to induce apoptosis in a various panel of cancer cells (5, 6). The enhanced cytotoxicity by the combination of EF24 with a p38 inhibitor is likely due to upregulated apoptosis function. We used a flow cytometry based assay to monitor the level of sub-G1 fraction of cells as an indication of apoptosis and examined whether the combination of EF24 and SB203580 caused an increased apoptotic response (Fig. 3A & 3B). Cells treated with EF24 (0.4 µM), or SB203580 (12.5µM), alone for 48 hr showed no significant difference in the percentage of apoptotic cells from that of the vehicle-control. However, when these two agents are combined, a synergistic accumulation of cells in the sub-G1 fraction was observed. A dramatic increase in sub-G1 cells were not observed between the curcumin and curcumin plus SB203580 treatment groups (Fig. 3A & 3B). Then, a cell imaging based high content analysis (HCA) approach and biochemical markers of apoptosis were used to validate the above results. The HCA based double-staining approach was able to differentiate apoptotic from necrotic cells. Indeed, this imaging based assay showed that the cells treated with the combination primarily undergo apoptosis (Fig 3C). In support of these observations, the combination treatment of A549 cells with a p38 inhibitor also increased EF24-induced cleavage of PARP, a well established substrate of effector caspases, which is another hallmark of apoptotic cell death (Fig. 3D). The enhanced PARP cleavage by the combination effect of EF24 and SB203580 suggests a caspases-mediated cell death process. To validate this model, we determined whether inhibiting global caspase activity could attenuate the loss of cell viability induced by this combination. To do so, we pretreated A549 cells with the pan caspase inhibitor zVAD-fmk for 1h before a 48 hr treatment of cells with the combination of EF24 and SB203580. The pretreatment of caspase inhibitor significantly blocked the cell death by EF24 and SB203580 in support of a caspase-mediated pathway (Fig. 3E). However, this caspase inhibitor only showed about 50% protection, suggesting an additional cell death mechanism is involved, the nature of which requires further investigations. In these experiments, the same p38 inhibitor, SB203580, failed to enhance the curcumin effect, which is consistent with the observation that p38 was not induced by curcumin at the concentration used.
Figure 3. Combination of EF24 with SB203580 synergistically induces apoptosis of lung cancer cells.
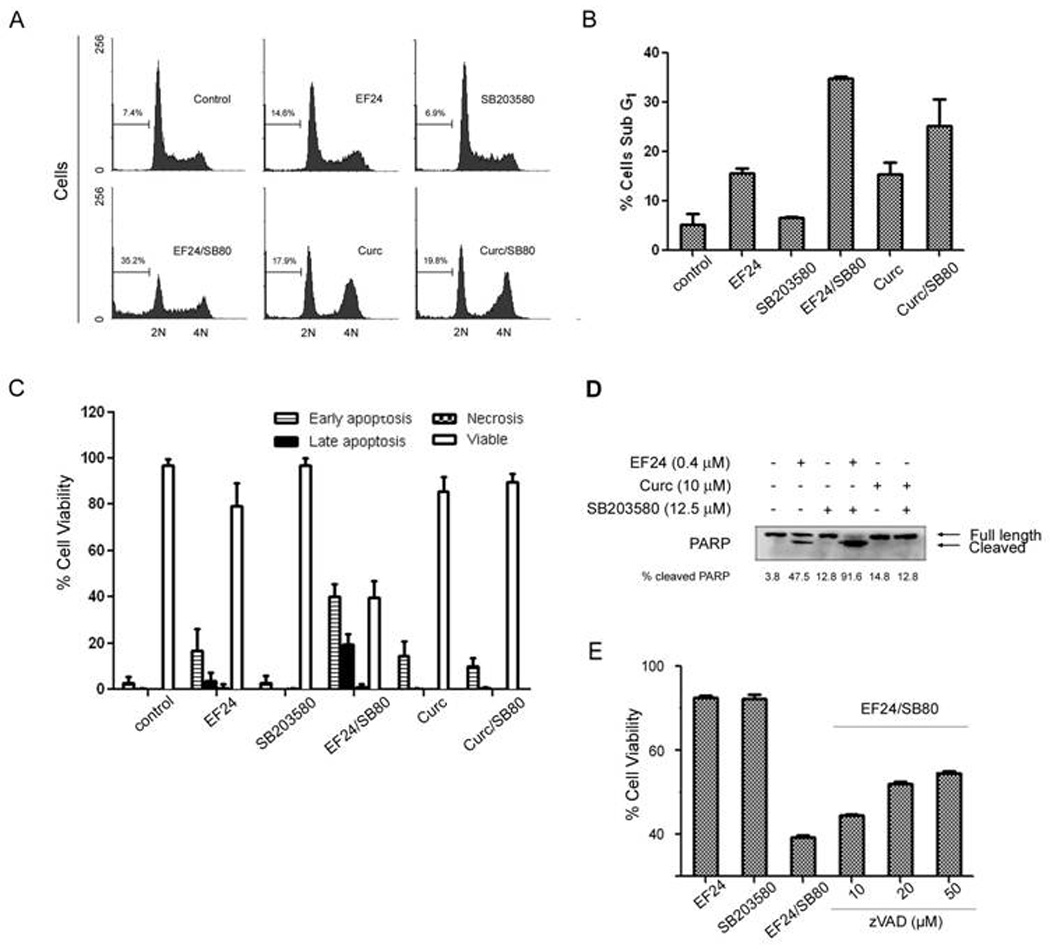
(A) Cell cycle analysis of combined treatment of EF24 (0.4 µM) and SB203580 (12.5 µM). A549 cells were exposed to the indicated compound for 48 hr. Flow cytometry was performed to define the cell cycle distribution based on nuclear content of cells as described in Materials and Methods. The data represent the cell cycle distribution for a representative experiment. (B) Summary of results with sub-G1 cells from three independent experiments. (C) Results of a cell health assay. A549 cells were plated in a 96 well plate at a density of 5000 cells/well. The following day, the cells were treated with the indicated concentrations of EF24, SB203580, Curcumin, or the combinations of EF24 or curcumin with SB203580 for 48 hr. The cellular stains DAPI, Yo-Pro-1, and PI were added to each well. Yo-Pro-1 positive cells are deemed early apoptotic, PI-positive cells are necrotic, and Yo-Pro-1 and PI-positive cells are late apoptotic [29]. Cells that excluded both stains but had intact nuclear staining were deemed viable. A second concentration of EF24 (0.6 µM) served as a control to ensure the assay could identify an increase in apoptotic cells. (D) Western blot analysis of A549 lysates treated with EF24 (0.4 µM), SB203580 (12.5µM), curcumin (10 µM) or the combination of two compounds after 48 hr. The amount of full length and cleaved PARP was revealed by Western blotting using an anti-PARP antibody. (E) Caspase inhibitor effect. A549 cells were pretreated with the caspase inhibitor zVAD-fmk at indicated concentrations for 1 hr before treatment with the combination of EF24 (0.4 µM) and SB203580 (12.5 µM).
3.5. Silencing of p38 sensitizes A549 cells to EF24-induced death
It is possible that the small molecule p38 inhibitors may induce some off-target effect which may be responsible for the enhanced EF24 activity. To test this possibility, we used a genetic approach to specifically knock down p38. A549 cells were treated with siRNA targeting p38, which resulted in a significant knockdown of p38. The relative specificity of the p38 siRNA was examined. Indeed, no change in other kinases was observed as seen for ERK expression levels (Fig. 4A & 4B). Then, these p38-knockdown cells were used to test the effect of reduced p38 on cell sensitivity to EF24 (0.4 µM) (Fig. 4C). Cell viability was analyzed 48 hr after treatment of cells with test agents. The effect of p38 knockdown was compared to a mock transfected control with a scramble siRNA. As shown in Fig. 3C, p38 silencing enhanced cell sensitivity to EF24, showing significant loss of cell viability similar to that of the combination of EF24 and SB203580. These results suggest that upregulated p38 upon EF24 treatment may provide a feedback loop in support of lung cancer cell survival. Disabling this p38-mediated negative feedback mechanism reduces the resistance to EF24, leading potentially to an enhanced therapeutic efficacy.
Figure 4. Knockdown of p38 sensitizes A549 cells to EF24-induced cytotoxicity.
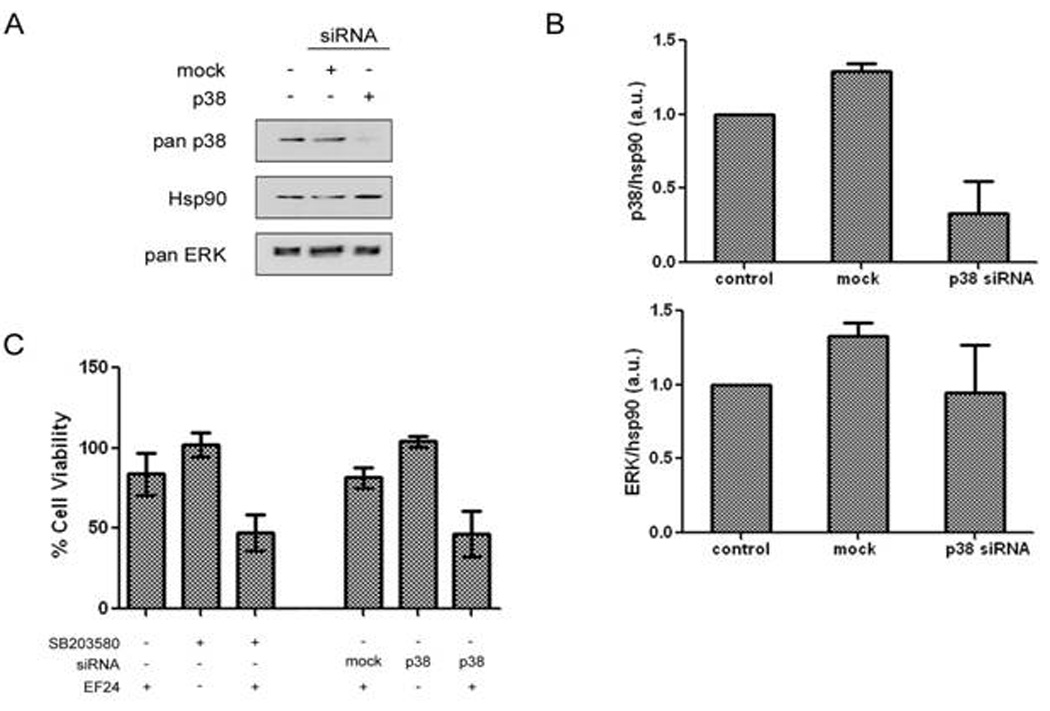
(A) A549 cells were transiently transfected with p38 siRNA as outlined in Materials and Methods. p38 as well as total Hsp90 and ERK protein was assessed by western blotting and quantified (B). (C) A549 cells were transiently transfected with mock or p38 siRNA and were incubated with or without 0.4 µM of EF24 for 48 hr where indicated. Cell growth was assessed by SRB assay. Results were compared to mock transfected control.
3.6. Combination of EF24 and SB203580 synergistically inhibits colony formation of lung cancer cells
Inhibition of p38 appears to reduce the cell resistance to EF24. To evaluate its potential therapeutic implications, we examined the effect of p38 inhibition on the efficacy of EF24 in suppressing transformation activity of A549 cells in a clonogenic assay. The IC50 for inhibition of colony formation of A549 cells was determined to be approximately 150 nM for EF24 and 10 µM for SB203580 from previous experiment (data not shown). For the combination treatment for this experiment, 100 nM of EF24 was used. To achieve the EF24:SB203580 ratio as used in cell viability assays (1:32.5 as in Fig. 2A), 5 µM of SB203580 was selected in this experiment. As shown in Fig. 5A & B, the combination of EF24 and SB203580 drastically reduced the number of colonies formed by A549 cells by approximately 50% as well as the overall size of the colonies. Thus, inhibiting p38 offers a promising strategy to enhance the EF24 anti-tumorigenic activity against lung cancer.
Figure 5. EF24 and SB203580 show a synergistic effect on the inhibition of A549 colony formation.
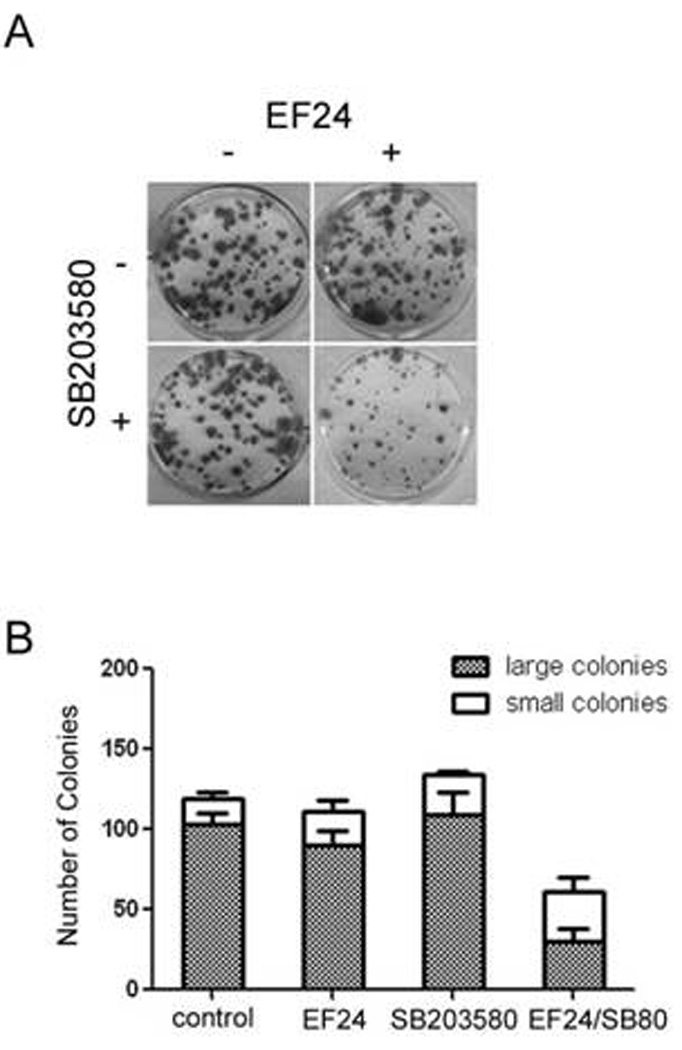
(A) A549 cells were first plated at a density of 450 cells/well and then treated every 3 days with the indicated concentration of test compounds. On day 10, the cells were fixed and stained with SRB. Colonies formed were counted using Image J. The effect of compound treatment on colony formation was compared to the vehicle-treated (0.5% DMSO) well. (B) Summary of data from two experiments as in A with both colony number and size included.
4. Discussion
Lung cancer is one of the leading causes of death worldwide, most people being diagnosed with advanced or metastatic non-small cell lung cancer (NSCLC). Currently, treatment options for this particular cancer are limited. In the present study, we used the A549 lung adenocarcinoma cell line, a cell model for NSCLC, to demonstrate that EF24 exhibits higher cytotoxicity than the lead compound curcumin. Interestingly, our work also reveals a negative feedback loop mediated by p38 that may restrict the efficacy of EF24. By disabling the EF24-induced upregulated p38, it may be possible to enhance the EF24 effect. Indeed, EF24 produces a dramatic synergistic growth inhibition of A549 cells and induction of apoptosis when combined with pyridinyl imidazole p38 inhibitors (Fig. 3). This study not only provides us with a potential combination that may be useful in a clinical setting, but also furnishes several important observations concerning the mechanism of action of EF24 against A549 cells.
The ERK, JNK, and p38 signaling pathways were activated in response to EF24 treatment as revealed by enhanced phosphorylation of the specific threonine and tyrosine motifs in the activation loops of each kinase. Though ERK has a widely accepted role in mediating cellular survival and proliferation, inhibiting the activation of ERK failed to potentiate the effect of EF24 to inhibit A549 cell growth (Fig. S2). Furthermore, JNK is thought to act as a pro-apoptotic kinase, but our studies show that inhibition of JNK activity in combination with EF24 was slightly additive (Fig. S2). We expected JNK inhibition to attenuate the anti-cancer activity of EF24. One conclusion from this observation is that either activation of JNK is not imperative for EF24-mediated cell death, or EF24-induced JNK has a pro-survival function [34–35]. However, the non-specific nature of the chemical inhibitor SP600125 certainly complicates the interpretation of the data [36].
p38 has been implicated in survival pathways in part due to its connection with the NF-κB transcriptional activation. The involvement of p38 in the induction of COX-2 and the biosynthesis of cytokines such as TNFα and IL-6 supports the importance of p38 in inflammation [37–38]. We demonstrated that SB203580, the prototypical p38 inhibitor used in our studies, blocks p38 activity induced by EF24 as evidenced by the inhibition of MAPKAPK-2 phosphorylation. SB203580 did not significantly affect A549 cell growth, indicating that inhibition of p38 alone does not affect cell proliferation. Concurrent inhibition of p38 activity in combination with EF24 enhances A549 cell death and thereby suggests that p38 has a pro-survival function in A549. Nuclear staining in flow cytometry as well as an increase in cleaved PARP further verified the synergistic nature of this treatment combination in triggering cellular apoptosis.
In support of a potential role of the EF24 combination with a p38 inhibitor for the treatment of lung cancer, we have demonstrated a potent combination effect in the suppression of A549 cell clonogenic activity. In analyzing the data from the 10 day colony formation assay, there was a dramatic decrease in the number of colonies formed, which may be a result of the death of cells with colony-formation potential. However, there was also a difference in the size of the colonies formed with the combination treatment, possibly suggesting growth suppression of cells in these colonies as well as apoptosis. This conclusion was supported by the experimental results with a pan caspase inhibitor zVAD-fmk, which showed significant reversal of cell death induced by EF24. However, it is still possible that EF24 induces cell death in part through another potentially caspase-independent mechanism [39].
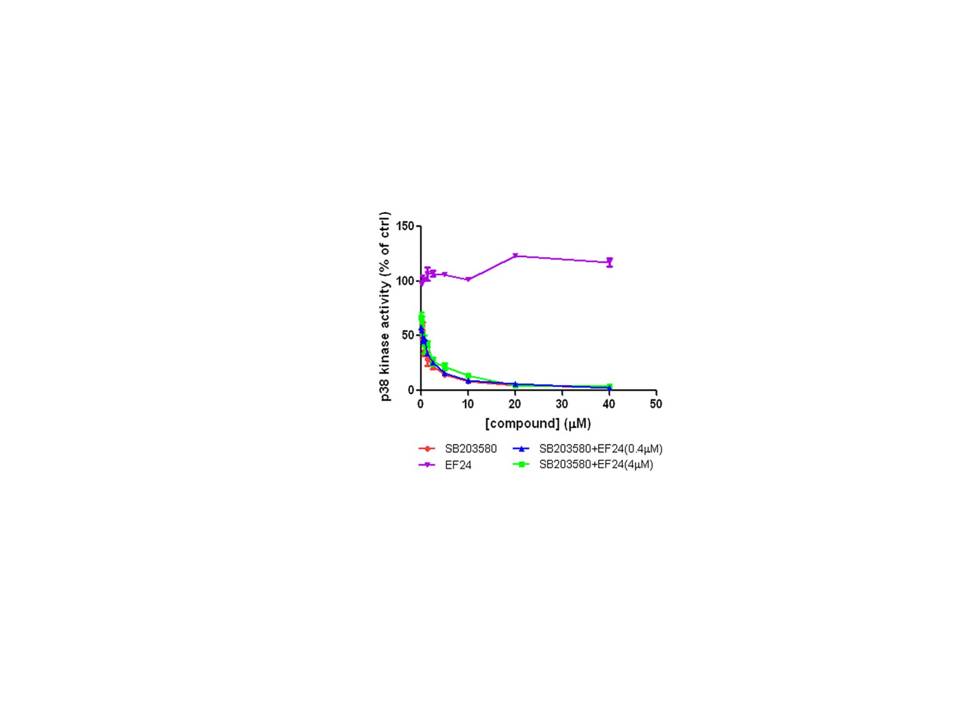
To begin to address the mechanism of the combination effect, we examined the possibility of a direct action of both EF24 and SB203580 on p38 and conducted a p38 in vitro kinase assay(Fig. 6). The enzymatic activity of p38 was examined in the presence of SB203580, EF24, or the combination of the two agents. As expected, the known p38 inhibitor SB203580 dramatically inhibited the activity of p38 with an IC50 of approximately 1 µM. However, the presence of EF24 alone (0.4 µM or 4 µM) in the reaction mixture did not inhibit the kinase activity of p38. Furthermore, no significant change in the IC50 of p38 inhibition was noted when EF24 (0.4 µM or 4 µM) was added to the reaction in the presence of SB203580. These studies rule out a direct action of EF24 on p38, in further support of the model that the p38 inhibition lowered the threshold of A549 lung cancer cells for the EF24 action. It is likely due to a disabled negative feedback loop mediated by the upregulated p38 upon EF24 treatment.
Figure 6. EF24 does not potentiate SB203580-mediated inhibition of p38 activity in vitro.
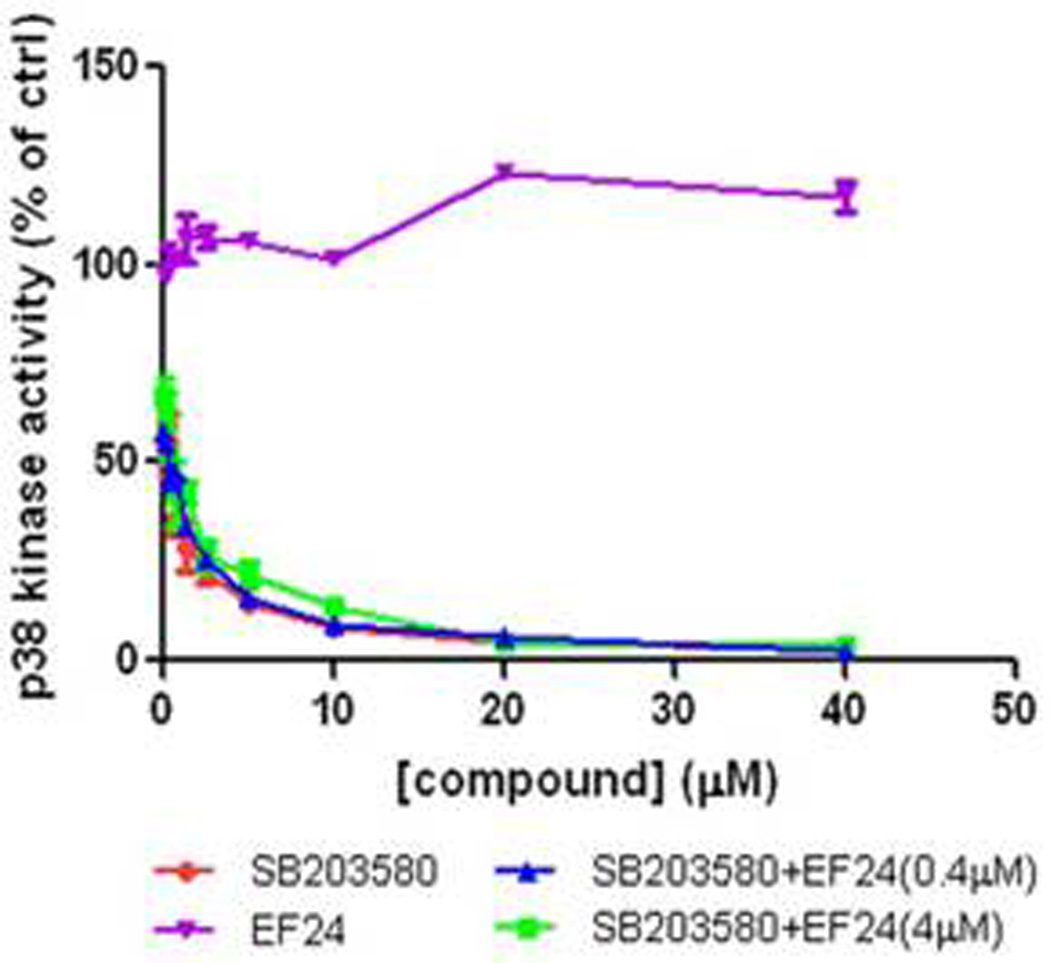
The ability of EF24, SB203580, and the combination of EF24 and SB203580 to attenuate p38 kinase activity was determined in vitro and graphed as the concentration of compound vs. % p38 activity. The compounds were incubated with the kinase for 30 min before the addition of the reaction mixture including the substrate MBP as described in Materials and Methods.
At the present time, p38 inhibitors are in clinical trials for a plethora of inflammatory diseases from arthritis to skin disorders such as psoriasis. Recently, these inhibitors have been found to be efficacious against tumors but often fail due to increased toxicity at the effective doses [40]. Development of combination treatments using these inhibitors could possibly decrease the dose needed to see a dramatic effect and therefore prevent side effects. We suggest that molecular-targeted agents like p38 inhibitors may serve as a companion agent to curcumin analogs with increased in vivo activity and bioavailability like EF24 in order to successfully treat lung cancer. Our work supports a combination strategy with EF24 and a specific p38 inhibitor for enhanced therapeutic efficacy in lung cancers.
Supplementary Material
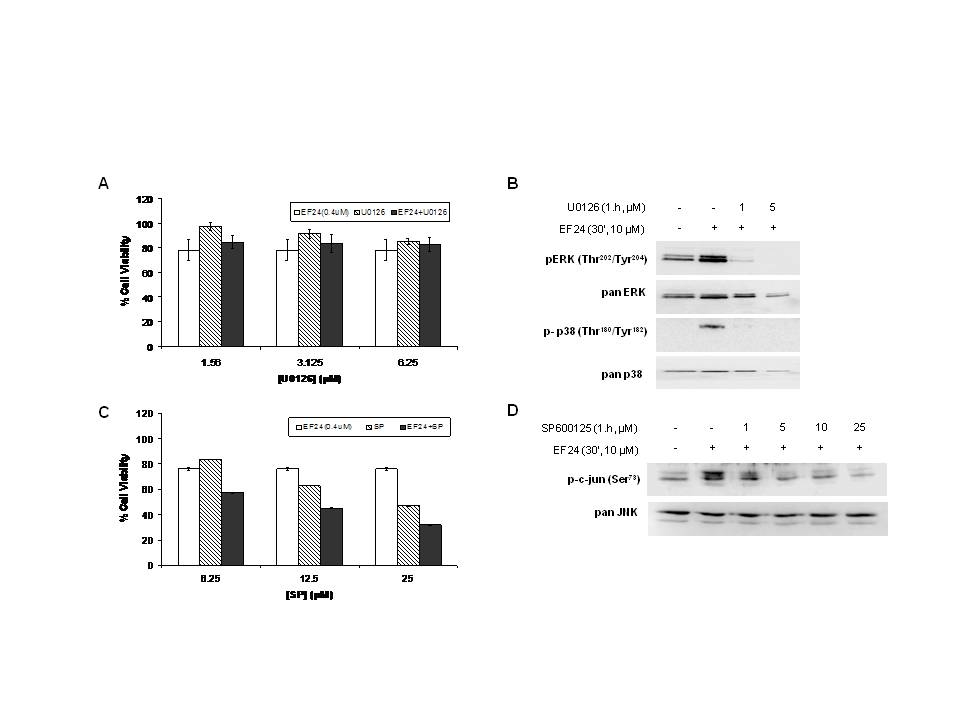
Cells were grown in 384-well plates and were treated with EF24 or curcumin as indicated for 48 or 72 hr. Cell viability was assessed by the SRB method and expressed as percentage of control (DMSO).
(A) Treatment of A549 with the combination of EF24 (0.4µM) and increasing concentrations of the MEK inhibitor U0126. Cell viability was then determined using the SRB assay method after 48h. The error bars in the experiments represent the standard deviation analyzed from three repeats. (B) A549 cells were pretreated with U0126 for 1h before treatment with EF24 (10 µM) for 30 min. Cells were subjected to western blotting to access phosphorylation and protein expression of ERK. (C) Treatment of A549 with the combination of EF24 (0.4µM) and increasing concentrations of the JNK inhibitor SP600125. Cell viability was then determined using the SRB assay method after 48h. The error bars in the experiments represent the standard deviation analyzed from three repeats. (D) A549 cells were pretreated with SP600125 for 1h before treatment with EF24 (10 µM) for 30 min. Cells were subjected to western blotting to access phosphorylation and protein expression of JNK.
{kind=link}
{kind=link}
Acknowledgements
We would like to thank members of the Fu lab for critiques and helpful discussions. This work was supported in part by grants from the National Institutes of Health (P01 CA116676 and SPORE P50 CA128613 to FRK and HF) and a fellowship from the American Association for the Advancement of Science/Packard Foundation (SLT). HF is a GRA distinguished investigator and FRK and HF are Georgia Cancer Coalition Distinguished Cancer Scholars.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aggarwal BB, Sung B. Pharmacological basis for the role of curcumin in chronic diseases: an age-old spice with modern targets. Trends Pharmacol Sci. 2009 Feb;30(2):85–94. doi: 10.1016/j.tips.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Gururaj AE, Beladkavadi M, Venkatesh DA, Marme D, Salimath BP. Molecular mechanisms of anti-angiogenic effect of curcumin. Biochem Biophys Res Commun. 2002 Oct 4;297(4):934–942. doi: 10.1016/s0006-291x(02)02306-9. [DOI] [PubMed] [Google Scholar]
- 3.Duvoix A, Balsius R, Delhalle S, et al. Chemopreventative and therapeutic effect of curcumin. Cancer Lett. 2005 Jun 8;223(2):181–190. doi: 10.1016/j.canlet.2004.09.041. [DOI] [PubMed] [Google Scholar]
- 4.Cheng AL, Hsu CH, Lin JK, et al. Phase I clinical trial of curcumin, a chemopreventative agent, in patients with high-risk or pre-malignant lesions. Anticancer Res. 2001;21:2895–2900. [PubMed] [Google Scholar]
- 5.Sharma RA, Euden SA, Platton SL, et al. Phase I clinical trial of oral curcumin: biomarkers of systemic activity and compliance. Clin Cancer Res. 2004 Oct 15;10(20):6847–6854. doi: 10.1158/1078-0432.CCR-04-0744. [DOI] [PubMed] [Google Scholar]
- 6.Adams BK, Ferstl EM, Davis MC, et al. Synthesis and biological evaluation of novel curcumin analogs as anti-cancer and anti-angiogenesis agents. Bioorg Med Chem. 2004 Jul 15;12(14):3871–3883. doi: 10.1016/j.bmc.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 7.Adams BK, Cai J, Armstrong J, et al. EF24, a novel synthetic curcumin analog, induces apoptosis in cancer cells via a redox-dependent mechanism. Anticancer Drugs. 2005 Mar;16(3):263–275. doi: 10.1097/00001813-200503000-00005. [DOI] [PubMed] [Google Scholar]
- 8.Singh S, Aggarwall BB. Activation of transcription factor NF-kappa B is suppressed by curcumin (diferuloylmethane) [corrected] J Biol Chem. 2001;270:24995–25000. doi: 10.1074/jbc.270.42.24995. [DOI] [PubMed] [Google Scholar]
- 9.Li L, Aggarwall BB, Shishodia S, Abbruzzese J, Kurkrock R. Nuclear factor-kappaB and IkappaB kinase are constitutively active in human pancreatic cells, and their down-regulation by curcumin (diferuloylmethane) is associated with the suppression of proliferation and the induction of apoptosis. Cancer. 2004 Nov 15;101(10):2351–2362. doi: 10.1002/cncr.20605. [DOI] [PubMed] [Google Scholar]
- 10.Vanden Berghe W, Plaisance S, Boone E, et al. p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-kappaB p65 transactivation mediated by tumor necrosis factor. J Biol Chem. 1998 Feb 6;273(6):3285–3290. doi: 10.1074/jbc.273.6.3285. [DOI] [PubMed] [Google Scholar]
- 11.Tuyt LM, Dokter WH, Birkenkamp K, et al. Extracellular-regulated kinase 1/2, Jun N-terminal kinase, and c-Jun are involved in NF-kappa B-dependent IL-6 expression in human monocytes. J Immunol. 1999 Apr 15;162(8):4893–4902. [PubMed] [Google Scholar]
- 12.Raingeaud J, Gupta S, Rogers JS, et al. Pro-inflammatory cytokines and environmental stress casue p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995 Mar 31;270(13):7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 13.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002 Dec 6;298(5600):1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 14.Madrid LV, Mayo MW, Reuther JY, Baldwin AS., Jr Akt stimulates the trasactivation potential of the RelA/p65 Subunit of NF-kappa B through utilization of the Ikappa B kinase and activation of the mitogen-activated protein kinase p38. J Biol Chem. 2001 Jun 1;276(22):18934–18940. doi: 10.1074/jbc.M101103200. [DOI] [PubMed] [Google Scholar]
- 15.Enslen H, Raingeaud J, Davis RJ. Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinases MKK3 and MKK6. J Biol Chem. 1998 Jan 16;273(3):1741–1748. doi: 10.1074/jbc.273.3.1741. [DOI] [PubMed] [Google Scholar]
- 16.Keller D, Zeng X, Li X, et al. The p38MAPK inhibitor SB203580 alleviates ultraviolet-induced phosphorylation at serine 389 but not serine 15 and activation of p53. Biochem Biophys Res Commun. 1999 Aug 2;261(2):464–471. doi: 10.1006/bbrc.1999.1023. [DOI] [PubMed] [Google Scholar]
- 17.Sanchez-Prieto R, Rojas JM, Taya Y, Gutkind JS. A role for the p38 mitogen-activated protein kinase pathway in the trascriptional activation of p53 on genotoxic stress by chemotherapeutic agents. Cancer Res. 2000 May 1;60(9):2464–2472. [PubMed] [Google Scholar]
- 18.Greenberg AK, Basu S, Hu J, et al. Selective p38 activation in human non-small cell lung cancer. Am J Respir Cell Mol Biol. 2002 May;26(5):558–564. doi: 10.1165/ajrcmb.26.5.4689. [DOI] [PubMed] [Google Scholar]
- 19.English JM, Cobb MH. Pharmacological inhibitors of MAPK pathways. Trends Pharmacol Sci. 2002;23(1):40–45. doi: 10.1016/s0165-6147(00)01865-4. [DOI] [PubMed] [Google Scholar]
- 20.Badger AM, Bradbeer JN, Votta B, et al. Pharmacological profile of SB 203580, a selective inhibitor of cytokine suppressive binding protein/p38 kinase, in animal models of arthritis, bone resorption, endotoxin shock and immune function. J Pharmacol Exp Ther. 1996 Dec;279(3):1453–1461. [PubMed] [Google Scholar]
- 21.Dominguez C, Powers DA, Tamayo N. p38 MAP kinase inhibitors: many are made, but few are chosen. Curr Opin Drug Discov Devel. 2005 Jul;8(4):421–430. [PubMed] [Google Scholar]
- 22.Young PR, McLaughlin MM, Kumar S, et al. Pyridinyl imidazole inhibitors of p38 mitogen-activated protein kinase bind in the ATP site. J Biol Chem. 1997;272:12116–12121. doi: 10.1074/jbc.272.18.12116. [DOI] [PubMed] [Google Scholar]
- 23.Wilson KP, McCaffrey PG, Hsiao K, et al. The structural basis for the specificity of pyridinylimidazole inhibitors of p38 MAP kinase. Chem Biol. 1997;4:423–431. doi: 10.1016/s1074-5521(97)90194-0. [DOI] [PubMed] [Google Scholar]
- 24.Bergstralh DT, Ting JP. Microtubule stabilizing agents: their molecular signaling consequences and the potential for enhancement by drug combination. Cancer Treat Rev. 2006 May;32(3):166–179. doi: 10.1016/j.ctrv.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 25.Dong QG, Sciabas GM, Fujioka S, et al. The function of multiple IkappB: NF-kappaB complexes in the resistance of cancer cells to Taxol-induced apoptosis. Oncogene. 2002 Sep 19;21(42):6510–6519. doi: 10.1038/sj.onc.1205848. [DOI] [PubMed] [Google Scholar]
- 26.Osaki S, Nakanishi Y, Pei XH, Ueno H, Hara N. Transfer of IkappaBalpha gene increase the sensitivity of paclitaxel mediated with caspase 3 activation in human lung cancer cell. J Exp Clin Cancer Res. 2003 Mar;22(1):69–75. [PubMed] [Google Scholar]
- 27.Mabuchi S, Ohmichi M, Nishio Y, et al. Inhibition of inhibitor of nuclear factor-kappaB phosphorylation increases the efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Clin Cancer Res. 2004 Nov 15;10(22):7645–7654. doi: 10.1158/1078-0432.CCR-04-0958. [DOI] [PubMed] [Google Scholar]
- 28.Liu GH, Wang SR, Wang B, Kong BH. Inhibition of nuclear factor-kappaB by an antioxidant enhances paclitaxel sensitivity in ovarian carcinoma cell line. Int J Gynecol Cancer. 2006 Sep-Oct;16(5):1777–1782. doi: 10.1111/j.1525-1438.2006.00652.x. [DOI] [PubMed] [Google Scholar]
- 29.Gawlitta D, Oomens GW, Baaijens FP, Bouten CV. Evaluation of a continuous quantification method of apoptosis and necrosis in tissue culture. Cytotechnology. 2004 Oct;46(2–3):139–150. doi: 10.1007/s10616-005-2551-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen YR, Tan TH. Inhibition of the c-Jun N-terminal kinase (JNK) signaling pathway by curcumin. Oncogene. 1998 Jul 16;17(2):173–178. doi: 10.1038/sj.onc.1201941. [DOI] [PubMed] [Google Scholar]
- 31.Squires MS, Hudson EA, Howells L, et al. Relevance of mitogen activated protein kinase (MAPK) and phosphotidylinositol-3-kinase/protein kinase B (PI3K/PKB) pathways to induction of apoposis by curcumin in breast cells. Biochem Pharmacol. 2003 Feb 1;65(3):361–376. doi: 10.1016/s0006-2952(02)01517-4. [DOI] [PubMed] [Google Scholar]
- 32.Chen YR, Tan TH. The c-Jun N-terminal kinase pathway and apoptotic signaling (review) Int J Oncol. 2000 Apr;16(4):651–662. doi: 10.3892/ijo.16.4.651. [DOI] [PubMed] [Google Scholar]
- 33.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000 Oct 1;351(1):95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bost F, McKay R, Bost M, et al. The Jun kinase 2 isoform is preferentially required for epidermal growth factor-induced transformation of human A459 lung carinoma cells. Mol Cell Biol. 1999 Mar;19(3):1938–1949. doi: 10.1128/mcb.19.3.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bost F, McKay R, Dean N, Mercola D. The JUN kinase/stress-activated protein kinase pathway is required for epidermal growth factor stimulation of growth of human A549 lung carcinoma cells. J Biol Chem. 1997 Dec 26;272(52):33422–33429. doi: 10.1074/jbc.272.52.33422. [DOI] [PubMed] [Google Scholar]
- 36.Bain J, McLauchlan H, Elliot M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003 Apr 1;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Craig R, Larkin A, Mingo AM, et al. p38 MAPK and NF-kappaB collaborate to incude interleukin-6 gene expression and release. Evidence for a cytoprotective autocrince signaling pathway in a cardiac myoctye model system. J Biol Chem. 2000 Aug 4;275(31):23814–23824. doi: 10.1074/jbc.M909695199. [DOI] [PubMed] [Google Scholar]
- 38.Singer CA, Baker KJ, McCaffrey A, et al. p38 MAPK and NF-kappaB mediate COX-2 expression in human airway myocytes. Am J Physiol Lung Cell Mol Physiol. 2003 Nov;285(5):L1087–L1098. doi: 10.1152/ajplung.00409.2002. [DOI] [PubMed] [Google Scholar]
- 39.Selvendiran K, Tong L, Vishwanath S, et al. EF24 induces G2/M arrest and apoptosis in cisplatin-resistant human ovarian cancer cells by increasing PTEN expression. J Biol Chem. 2007 Sep 28;282(39):28609–28618. doi: 10.1074/jbc.M703796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fabian MA, Biggs WH, 3rd, Treiber DK, et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005 Mar;23(3):329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cells were grown in 384-well plates and were treated with EF24 or curcumin as indicated for 48 or 72 hr. Cell viability was assessed by the SRB method and expressed as percentage of control (DMSO).
(A) Treatment of A549 with the combination of EF24 (0.4µM) and increasing concentrations of the MEK inhibitor U0126. Cell viability was then determined using the SRB assay method after 48h. The error bars in the experiments represent the standard deviation analyzed from three repeats. (B) A549 cells were pretreated with U0126 for 1h before treatment with EF24 (10 µM) for 30 min. Cells were subjected to western blotting to access phosphorylation and protein expression of ERK. (C) Treatment of A549 with the combination of EF24 (0.4µM) and increasing concentrations of the JNK inhibitor SP600125. Cell viability was then determined using the SRB assay method after 48h. The error bars in the experiments represent the standard deviation analyzed from three repeats. (D) A549 cells were pretreated with SP600125 for 1h before treatment with EF24 (10 µM) for 30 min. Cells were subjected to western blotting to access phosphorylation and protein expression of JNK.