

Abstract
The membrane tyrosine kinase receptors, AXL and MET, are implicated in GnRH neuron migration and/or survival. We hypothesized that the receptors with their ligands, GAS6 and HGF, respectively may cross-talk in GnRH neuronal function. In NLT GnRH neuronal cells, MET co-immunoprecipitated with AXL, although HGF or GAS6 did not transphosphorylate AXL or MET, respectively. Co-expression of a kinase dead AXL blocked HGF activation of MET and indirectly AKT and p38MAPK. Silencing of AXL decreased HGF’s ability to phosphorylate MET and activate AXL’s downstream effectors, p38MAPK and AKT. HGF/MET signaling modulated neuron migration dependent and independent of AXL co-expression and p38MAPK. Conversely, AXL’s control of GnRH neuronal survival was dependent on HGF/MET signaling. Together, these data support that the importance of membrane tyrosine kinase receptor crosstalk to regulate neuronal cell-specific developmental functions.
Keywords: GnRH neurons, AXL, MET
1. Introduction
Hypothalamic Gonadotropin releasing hormone (GnRH) neurons are the primary regulators of the reproductive axis. They release the decapeptide hormone, GnRH, in an episodic fashion to control pituitary gonadotropin production and normal reproductive functions (Wierman et al., 2011). During embryogenesis, GnRH neurons originate near the olfactory placode and migrate across cribriform plate into the basal forebrain (Schwanzel-Fukuda and Pfaff, 1989). Failure in GnRH neuronal migration or loss of these neurons can result in delayed or absent pubertal maturation in mice and men (Wierman et al., 2004; Wierman et al., 2011; Wray, 2010).
Hepatocyte Growth Factor/Scatter Factor (HGF/SF) is a multifunctional cytokine secreted as pro-HGF which is activated by serine proteases, tissue and urokinase type plasminogen activators (tPA/uPA) (Bottaro et al., 1991; Mars et al., 1993; Naldini et al., 1991). HGF activates its hetrodimeric tyrosine kinase receptor MET which plays a pivotal role in epithelial-mesenchyme-transition, embryonic development, organogenesis, development of neuronal precursors, regulation of olfactory axon outgrowth, epithelial cell growth, morphogenesis and differentiation, tissue regeneration and wound healing (Comoglio, 2001; Comoglio et al., 2008; Furge et al., 2000; Gentile A, 2008). Recent studies have shown complex interactions between HGF/MET with other membrane receptor tyrosine kinases (RTKs) such as Semaphorin-4D/Plexin B1(Soong et al., 2012) and SDF1/CXCR4 (Tu et al., 2009), suggesting the importance of crosstalk between membrane receptors of various types (Giacobini et al., 2007; Giacobini et al., 2008).
HGF and MET are widely expressed in the developing and adult brain. Prior work suggests an HGF gradient can induce chemotaxis and act as a guiding signal in migrating GnRH neurons in immortalized cells and in vivo (Giacobini et al., 2007; Jung et al., 1994). HGF and MET null mice are embryonically lethal (Bladt et al., 1995; Schmidt et al., 1995; Uehara et al., 1995), whereas both tPA and uPA knockouts are sub-fertile with a decreased number of GnRH neurons (Giacobini et al., 2007). Whether the reduced GnRH neuronal numbers are due to a lack of migration or decreased survival of GnRH neurons was not determined.
The dispersed, low numbers of GnRH neurons in vivo have led the field to use immortalized NLT GnRH neuronal cell line as a model of early neuronal development as the cells express low levels of GnRH with an intrinsic migratory phenotype (Allen et al., 1999; Giacobini et al., 2007; Radovick et al., 1991). Our prior studies on the TAM (TYRO3, AXL and MER) RTK family showed that AXL and TYRO3, but not MER, were expressed in NLT GnRH neuronal cells and that Axl/Tyro3 null mice have delayed sexual maturation and estrus cycle defects due to defects in GnRH neuronal cell migration and survival (Allen et al., 1999; Pierce et al., 2008; Pierce et al., 2011). In addition to AXL and TYRO3, MET is expressed at higher levels in the migratory NLT compared to the post migratory GT1–7 GnRH neuronal cell lines (Allen et al., 1999; Giacobini et al., 2002; Giacobini et al., 2008; Pierce et al., 2008). Since both AXL and MET are activated by heparan sulfate proteoglycans tethered ligands, GAS6 and HGF respectively, and both display directional chemotaxis in GnRH neurons, we postulated that their pathways may interact in GnRH neuronal cells.
We previously showed that GAS6/AXL signaling protects from growth factor withdrawal induced apoptosis via ERK-MAPK and the PI3 kinase (PI3K) pathway to AKT (Allen et al., 1999). In addition, GAS6/AXL activation induces GnRH neuronal migration via the p38MAPK pathway(Allen et al., 2002b). HGF/MET has been shown to signal via multiple MAPK pathways for growth and invasion in human gastric carcinoma cells (Wang et al., 2012), through ERK, PI3K and p38MAPK for cortical neuron and oral squamous cell migration (Brusevold et al., 2012; Segarra J, 2006), through PI3K for survival and migration in lung epithelial cell lines (Graziani et al., 1991), via STAT3 for morphogenesis (Boccaccio et al., 1998), invasion in skin tumor cells (Syed et al., 2011) and cell survival and motility in gastric cancer cells (Okamoto et al., 2011). The downstream components of MET signaling in GnRH neuronal cells had not been evaluated. Thus, we asked whether crosstalk between the AXL and MET and/or their ligands to intracellular signaling pathways impacted GnRH neuronal cell migration or survival.
2. Material and Methods
2.1. Cell Culture and Transfection
NLT (Mellon PL, 1990) and GT1–7 (Radovick et al., 1991) GnRH neuronal cells were grown in DMEM (HyClone, Logan, UT) supplemented with 10% FBS (Atlanta Biologicals, Lawrenceville, GA), penicillin (100 U/ml), and streptomycin (100μg/ml) in humidified 5% CO2 incubator at 37°C. Transfections on NLT cells were performed using Lipofectamine Plus reagents as per the manufacturer’s instructions(Invitrogen, CA).
2.2. Reagents
Human recombinant HGF (H9661) and DMSO were purchased from Sigma-Aldrich (St. Louis, MO). Full length recombinant GAS6 was provided by B Varnum at Amgen (Thousand Oaks CA). Pure MET tyrosine kinase inhibitor (PF-04217903) was a kind gift from Ross Camidge (University of Colorado, Denver). LY294002, SB203580 and PD98059 were purchased from Calbiochem (San Diego, CA). AXL (M-20), MET (sc-8057) and HGF (sc-13087) antibodies, normal mouse, rabbit and goat IgG and HRP-linked anti-goat antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal TYRO3 (MAB579) antibody and anti-CASPASE 3 antibody were purchased from R&D Systems (Minneapolis, MN). Horseradish peroxidase (HRP)-conjugated secondary antibodies (Goat anti-rabbit IgG and Goat anti-mouse IgG) were purchased from Bio-rad (Hercules, CA). Antibodies specific to AKT, phospho-AKT(Ser 473), ERK, phospho-ERK(Thr 202/204), p38MAPK, phospho-p38MAPK(Thr180/Tyr 182), phospho-MET(Tyr 1234/1235), phospho-AXL(Tyr 702), STAT3, phospho-STAT3(Tyr 705) and PARP were purchased from Cell Signaling Technology (Beverly, MA). β-Tubulin and phospho-STAT3 (Ser 727) were purchased from Abcam (Cambridge, MA) and GAPDH antibodies was purchased from Millipore (Billerica, MA).
2.3. Plasmids
pRK5, PRK5-AXL WT plasmid containing the mouse Axl cDNA (wild type) and PRK5-AXL KD plasmid containing the truncated tyrosine kinase receptor Axl was provided by Paola Bellosta (New York University)(Bellosta et al., 1995). For domain analysis, AXL mutant plasmids encoding deletion in first immunoglobulin domain (pcDNA3 Δ AXL-IG), Fibronectin III domain I (pcDNA3 Δ AXL-FNI), Fibronectin III domain II (pcDNA3 Δ AXL-FNII), Fibronectin III domain I and II (pcDNA3 Δ Axl-FNI/II) and intracellular domains (pcDNA3 Δ AXL-ICD) were generously provided by Shuang-En Chuang (National Institute for Cancer Research, Taiwan) (Lay et al., 2007). Sequences for all plasmids were confirmed by DNA sequencing (UCD, Cancer Center Sequencing Core Facility).
2.4. DNA Microarray Analysis
Total RNA was isolated from 3 different batches of NLT and GT1–7 cells and DNA microarrays and bioinformatics analysis of the data were performed as previously described (Pierce et al., 2008).
2.5. Semi-quantitative qPCR
Total RNA was isolated from NLT or GT1–7 cells using RNeasy kit according to the manufacturer’s protocol (Qiagen, Valencia, CA). 2μg RNA was reverse transcribed using RT-PCR system from Biorad (Hercules, CA) in PTC-200 thermal cycler (MJ Research, Waltham, MA). qPCR was performed in a Applied Biosystems real time PCR system using Power SYBR Green PCR master mix (Applied Biosystems, Foster city, CA). Primer sequences for amplifying Met were Fwd 5 ′ -TCTGGGAGCTCATGACGAGA-3 ′ and Rev 3 ′ - CTTCGTACAAGGCGTCTGGA -5 ′, Hgf were: Fwd 5 ′ - GCTTGGCATCCACGATGTTC-3 ′ and Rev 3 ′ - AGGATTGCAGGTCGAGCAAG-5 ′. The quantity of Met and Hgf mRNA were compared to that of housekeeping control glyceraldehyde-3-phosphatedehydrogenase (Gapdh) in the qPCR reactions using following primers Fwd 5 ′ -CGACCCCTTCATTGACCTCA-3 ′ and 3 ′ -GCCACGACTCATACAGCACC −5 ′. Amplification reactions were set up of 25μl containing 5μM of each primer and SYBR Green qPCR master mix with following program, consisting of initial denaturation for 10min at 95°C, followed by 40 cycles of 95°C for 15s, primer annealing and acquisition at 60°C for 1min. The cycle threshold values (Ct) obtained for Met and Hgf genes were normalized against Gapdh to calculate Δ Ct values from triplicate experiments.
2.6. Immunoblot Analysis
NLT and GT1–7 cells were harvested in 1xRIPA buffer (150mM NaCl, 1% NP40, 0.5% deoxycholate, 0.1% SDS, 59mM Tris, pH8.0) and 0.5 mM PMSF, 1x protease inhibitor (Sigma-Aldrich, St. Louis, MO), 20mM Na3VO4 and 25mM NaF (Fisher, Pittsburgh, PA) were freshly added to the buffer. Protein lysates were quantified using BCA assay (Pierce, Rockford, IL) and equivalent amount of protein were resolved on 7.5% SDS/PAGE gels. The PVDF membranes were blocked 3% BSA in TBS-T buffer (20mM Tris-Cl, pH7.6, 137mM NaCl and 0.1% Tween-20) for 1hr at room temperature. Immunoblot analyses and stripping to reprobe with pan antibodies were performed as described (Nielsen-Preiss et al., 2007). Densitometry analysis was performed with the Bio-Rad Fluor-S multi-imager and NIH Image J software.
2.7. Immunoprecipitation
Immunoprecipitation (IP) experiments were performed as previously described (Pierce et al., 2008). Immunoprecipitated lysates were then loaded on 7.5% SDS gel and membranes were subsequently used for immunoblotting experiments. IP MET was probed for MET, AXL, TYRO3, MER and EGFR. IP of IgG mouse were probed with AXL TYRO3, MER and EGFR and immunoprecipitations were repeated in three independent experiments. Data were quantified using Image J software.
2.8. Small Interfering RNA (siRNA) for AXL
Scrambled siRNAs and four individual siRNAs targeting Axl were designed and synthesized using siRNA Target Finder and Silencer siRNA Construction Kit (Ambion, Austin, TX). siRNA sequences and transfection was performed as described (Pierce et al., 2008). Twenty-four hrs after transfection, cells were placed in media without serum for 8 hrs and used for signaling, migration or apoptosis experiments.
2.9. Scratch Assay
NLT cells were grown in DMEM with 10% FBS in 6 well plates up to 90% confluency. Cells were transiently transfected with scrambled or AXL-specific siRNAs for 24 hrs. Cells were subsequently incubated in serum free DMEM for 16–18 hrs and washed with PBS. Cells were treated with 25μm SB203580 or 10μm LY294002 or DMSO for 2 hrs, in DMEM with 2% FBS followed by incubation in presence of PBS or HGF (50 ng/ml) for 48h. A scratch was made using a sterile 200μl micropipette tip and three different scratch areas in marked fields per well were photographed at 0 hr using a phase-contrast inverted microscope and Moticam 2000 camera and Moticam image acquisition software. Cells were re-photographed 24hrs later, the area of the scratch was measured and the number of cells migrated into the scratch for each well was counted using Image J software. Migration of neuronal cells per well was calculated from three fields in triplicate experiments and data was normalized to scratch area from each well.
2.10. Apoptosis Assay
NLT cells were transiently transfected with scrambled (control) and AXL-specific siRNAs. After starving for 6 hrs NLT neuronal cells were counted, plated on coverslips and treated with 10μm LY294002 or 30μm PD98059or mock inhibited with DMSO for 2 hrs followed by incubation in presence of PBS or HGF (50 ng/ml) for 48hrs. Hoechst staining was then performed as described earlier (Allen et al., 1999). One thousand cells were counted from eight randomly chosen fields using a fluorescent microscope (Axiovert 200 Zeiss microscope, Carl Zeiss Germany). Duplicate coverslips in three separate experiments were evaluated. The rate of apoptosis was expressed as a percentage of total counted cells.
2.11. Statistical Analysis
All data are expressed as mean ± sem for each group. Statistical differences between two groups was analyzed using Student’s t test and among multiple groups using one way ANOVA with Bonferronis post-hoc test (for silencing experiment) and Tukeys multiple comparisions post-hoc test (for scratch and apoptosis assay), P values < 0.05 were considered statistically significant.
3. Results
3.1. Differential expression of Met, Axl and Tyro3 in GnRH neuronal cells
DNA microarrays were performed in triplicate in NLT and GT1–7 neuronal cells as previously reported (Pierce et al., 2008). 9881 transcripts were found to be differentially regulated (p<0.005) and 1827 and 901 transcripts were expressed in only GT and NLT cells respectively (using Gene Spring software). Data were analyzed for transcript levels of Met and its ligand Hgf and TAM family receptor (Axl) and its ligand Gas6. Among the TAM family members, the Axl transcript was more abundant in the NLT cells (log2 transformed 11.4) as compared to the GT1–7 (log2 transformed 8.2) GnRH neuronal cells, whereas transcript levels of Tyro3 and Gas6 were similar between the two neuronal cell lines as previously reported (Pierce et al., 2008). Both Met and Hgf transcripts were higher in NLT cells (log2 transformed 9.8 and 8.1 respectively) compared to GT1–7 neuronal cells (log2 transformed 3.7 and 3.8 respectively). Differential transcript levels were confirmed by measuring mRNAs using a SYBR green semiquantitative qPCR normalized to Gapdh as an internal control. Both Met and Hgf were higher in NLT cells (Δ Ct 5.2 ±0.8 and 4.7±0.6 respectively) as compared to GT1–7 neuronal cells (Δ Ct 9.1 ±0.6 and 11.4±0.6 respectively). Immunoblotting of neuronal lysates demonstrated that HGF protein levels were not different (NS) (Fig. 1A). Since immunobloting of whole cell lysates failed to detect the low levels of MET protein, immunoprecipitation (IP) was used to confirm higher levels of MET in NLT as compared to GT1–7 neuronal cells (Fig. 1B). The glycosylated MET precursor band and β-subunit of MET receptor were detected at 170 and 145 kDa respectively. AXL protein was detected only in NLT cells, TYRO3 levels were equivalent in both cell lines and no GAS6 protein was detected in either cell lines, as previously reported (Pierce et al., 2008).
Figure 1. HGF/MET expression and signaling in GnRH Neuronal Cells.
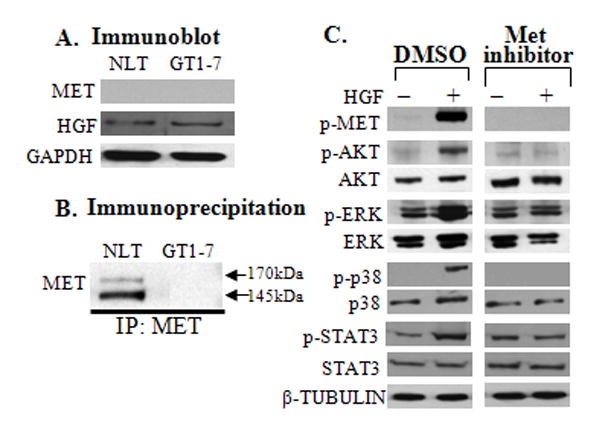
A: Immunoblots depicting protein levels of MET and HGF expressed in NLT and GT1–7 GnRH neuronal cells with GAPDH (loading control), n=3. B: Immunoprecipitation of MET in NLT and GT1–7 GnRH neuronal cells. C: HGF/MET interaction is required for activation of MET and it’s downstream signaling pathways. Inhibition of MET decreased levels of activated p-MET, p-AKT, p-ERK, p-p38MAPK and p-STAT3 after HGF stimulation, β-TUBULIN as loading control, n=3.
3.2. HGF phosphorylates MET to signal via AKT, ERK, STAT3 and p38MAPK in GnRH neuronal cells
We previously showed that GAS6/AXL phosphorylates ERK, PI3K and p38MAPK pathways in GnRH neuronal cells (Allen et al., 1999; Allen et al., 2002a). To define the downstream signaling pathways activated by HGF/MET in GnRH neurons, NLT cells were serum starved for 6–8 hrs and treated with a selective MET tyrosine kinase inhibitor (PF-04217903, 500μM) or DMSO. After 16–18 hrs, cells were treated with or without HGF (50 ng/ml for 15 min) and harvested. DMSO was used as vehicle control. Inhibition of MET decreased HGF phosphorylation of MET (0.4 versus 45-fold in control, P=0.01), AKT (0.6 versus 14.7-fold in control, P=0.0001), ERK (0.9 versus 3.0 -fold in control, P=0.0008), p38MAPK (0.6 versus 35-fold in control, P=0.01) and STAT3 (2.4 versus 4.7-fold in control, P=0.004). Interestingly, HGF failed to phosphorylate STAT3 at the Tyr705 residue, but instead phosphorylated STAT3 at Ser727 residue in NLT GnRH neuronal cells. These data suggest that HGF through MET phosphorylates multiple downstream signaling pathway components in NLT GnRH neuronal cells, including ERK, AKT, p38MAPK and STAT-3 (Fig. 1C).
3.3. AXL directly interacts with MET in NLT GnRH neuronal cells
We asked whether there is a direct interaction between MET and AXL, and examined if their respective ligands (HGF and GAS6) can phosphorylate each other’s receptor in GnRH neuronal cells (Fig. 2A). An anti-MET antibody was used to immunoprecipitate MET protein and lysates were probed with anti-MET or anti-AXL antibodies. To demonstrate the basal levels of MET and AXL in NLT cells, lysates (50 μg) were used as an input control. AXL protein (120 and 140 kDa) was detected in MET immuoprecipitated NLT lysates (0.6 -fold as compared to input lysate, n=3), indicating a direct but weak interaction of MET with AXL (Fig. 2B). IP of control mouse IgG probed with AXL showed no band. Additional experiments showed that MET can also interact with TYRO3 (120 and 140 kDa) but not with the other TAM family member, MER or with a control tyrosine kinase receptor, EGFR (170 kDa) (Supplementary Fig. S1). These data indicate that MET and AXL receptors interact in GnRH neuronal cells.
Figure 2. Crosstalk of AXL and MET in NLT GnRH neurons.
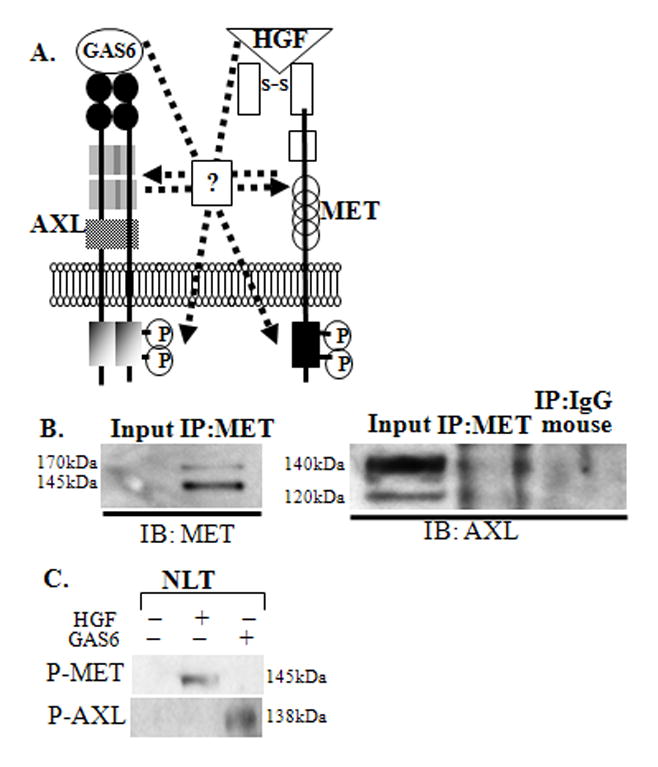
A: Schematic illustrating the possible crosstalk of GAS6/AXL and HGF/MET in migrating GnRH neurons. B: MET and AXL directly interact in GnRH neuronal cells. NLT lysate (input control), lysates immunoprecipitated with anti-MET and mouse IgG (negative control) were immunoblotted with anti-MET and anti-AXL, n=3. C: HGF and Gas6 do not activate AXL and MET. NLT lysates treated with HGF or GAS6 were immunoblotted with anti-phospho-MET or anti-phosho-AXL, n=3.
3.4. HGF and GAS6 do not phosphorylate each other’s receptors
Next we explored whether GAS6, the AXL ligand, can phosphorylate MET, or conversely HGF can phosphorylate AXL (Fig 2A). NLT cells were starved for 6–8 hrs followed by treatment with PBS, HGF (50 ng/ml for 15 min), or GAS6 (400 ng/ml for 10 min, (Allen et al., 1999; Allen et al., 2002b) (Fig. 2C). Immunoblots of NLT lysates stimulated with HGF and probed with phosphor- MET revealed that MET was phosphorylated only after HGF stimulation, with no effect after GAS6 (Fig. 2C). Similarly, NLT lysates probed with P-AXL demonstrated that GAS6 phosphorylated AXL, but HGF had no effect (Fig. 2C). Thus HGF and GAS6 failed to trans-phosphorylate the opposite receptor, and the ligands are receptor specific in GnRH neuronal cells.
3.5. AXL modulation of HGF /MET signaling is dependent on the intracellular domain
To elucidate which domains of AXL interact with MET to modulate HGF/MET signaling, AXL mutants (Fig. 3A) were expressed and then treated in the presence or absence of HGF in GnRH neuronal cells. Over expression of WT AXL induced MET phosphorylation by 1.6 -fold. As compared to vector control, AXL mutants did not alter HGF phosphorylation of MET with deletion of the AXL immunoglobulin (ΔAXL-IgG, 1.4 -fold), the fibronectin I and II (ΔAXL -FNI/II, 0.9 -fold), only fibronectin I (ΔAXL-FNI, 0.8 -fold) or only the fibronectin II (ΔAXL-FNII, 0.7 -fold) domains (see Fig. 3B). AXL mutant in which the intracellular domain which contains the kinase domain was deleted (ΔAXL-ICD mutant) was less well expressed in NLT neuronal cells. However, despite a lower level of expression, the ΔAXL-ICD mutant resulted in a loss of MET phosphorylation (0.06 -fold as compared to vector and 0.03 -fold as compared to wild type AXL, P=0.04) (Fig. 3B). Thus, the intracellular domain of AXL, including the tyrosine kinase domain, is important for AXL modulation of HGF/MET signaling. Interestingly, deletion of only the AXL fibronectin II domain as compared to overexpressed wild type AXL (0.4-fold) did alter HGF phosphorylation of MET. To confirm the importance of the FNII domain of AXL in AXL/MET interaction, coimmunoprecipitation experiment was performed. Lysates of cells expressing empty vector and ΔAXL-FNII were immunoprecipitated with a MET antibody. Immunoblots were then probed with MET and revealed that FN2 domain of AXL may be involved in bringing the AXL and MET receptors into close proximity (Fig. 3C)
Figure 3. Mutation in the kinase domain of AXL prevents HGF activation of MET, AKT and p38MAPK.
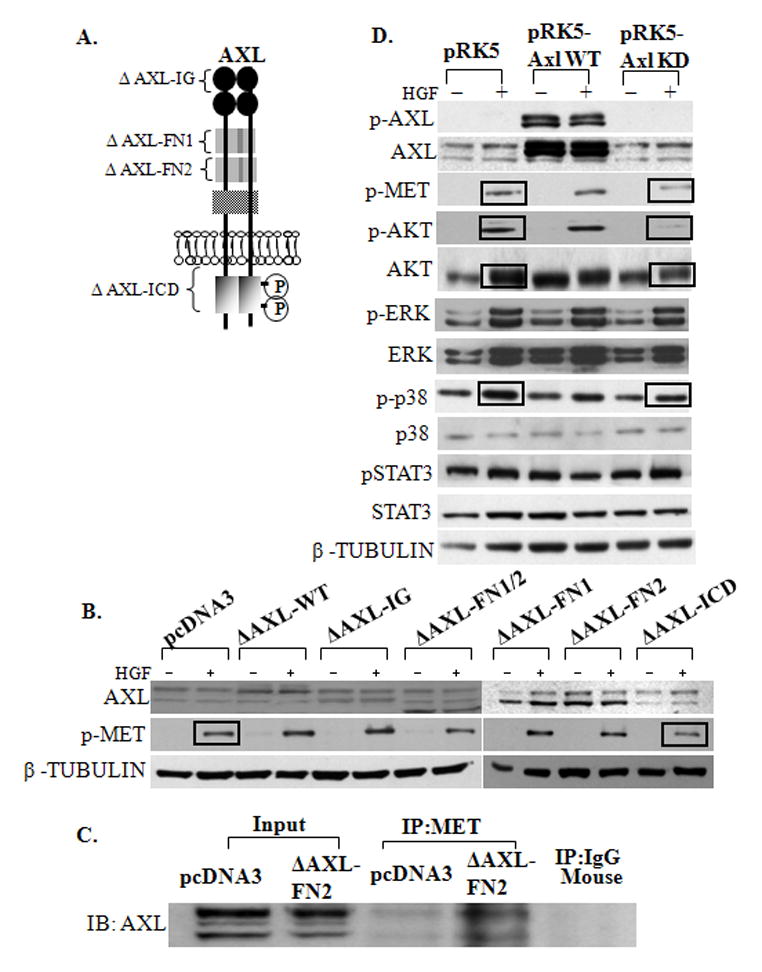
A: Schematic illustrating the different domain deletion mutants of AXL. B: Intracytoplasmic domain of AXL can modulate HGF induced MET activation. NLT cells were transfected with pcDNA3 or Axl domain deleted mutants (ΔpcDNA3 AXL-Ig, ΔpcDNA3 AXL-FN1, ΔpcDNA3 AXL-FN2, ΔpcDNA3 AXL-FN1/2 and ΔpcDNA3 Axl-ICD) (see methods for details). Cells were immunoblotted with AXL, p-MET and β-TUBULIN (loading control), n=3. Intracellular domain deleted AXL mutant resulted in a loss of HGF induced MET activation. C: AXL-FN2 domain is involved in protein-protein interaction of AXL and MET. Overexpressed NLT lysates (input control) were immunoprecipitated with anti-MET and Mouse IgG (negative control) and were immunoblotted with anti-AXL. D: Tyrosine kinase domain of AXL can alter HGF/ MET signaling and activation of AKT and p38MAPK pathway. NLT cells were transfected with control vectors (pRK5) or wild type-AXL (pRK5-WT AXL) or tyrosine kinase domain deleted AXL mutant (pRK5-KD AXL) (see methods for details). Cells were harvested for immunoblot, and probed with antibodies for AXL, p-AXL, p-MET, p-AKT, AKT, p-ERK, ERK, p-p38MAPK, p38MAPK, p-STAT3, STAT3 and β-TUBULIN (loading control), n=3. Kinase dead AXL mutant decreased HGF induced MET, AKT and p38MAPK activation.
3.6. AXL/MET interaction occurs via AKT and p38MAPK pathways
To confirm the functional relevance of the AXL tyrosine kinase phosphorylation on downstream signaling of HGF, wild type AXL (pRK5-AXL-WT) and the AXL mutant with mutated kinase domain (pRK5-AXL-KD) were overexpressed and then cells incubated in the absence or presence of HGF. HGF stimulation increased the levels of phosphorylated MET and AKT, but not ERK, p38MAPK or STAT3 when wild type AXL was overexpressed (9.4 -fold) (see Fig. 3D). In contrast, expression of pRK5-AXL-KD resulted in a diminished MET phosphorylation (1.1-fold as compared to 46 -fold in vector, P=0.05), decreased phospho-AKT (1.6-fold as compared to 28-fold in vector, P=0.03) and phospho-p38MAPK (0.2-fold as compared to 2.0-fold in vector, P=0.02) in response to HGF. Importantly, ERK phosphorylation (1.1-fold as compared to 1.4-fold in vector) and STAT3 phosphorylation (1.1-fold as compared to 1.0-fold in vector) was unchanged (Fig. 3D). Together, our data suggest that the AXL kinase domain is required for efficient HGF-induced MET signaling to the PI3K/AKT and p38MAPK, but not the ERK MAPK and STAT3 pathways in the GnRH neuronal cells.
3.7. Levels of AXL protein in GnRH neuronal cells modulates downstream HGF/MET signaling
We then investigated if altering the endogenous levels of AXL modulated HGF-induced MET signaling. Silencing of endogenous AXL (50–80% by siRNA, Fig. 4A) resulted in a decreased ability of HGF to phosphorylate MET (19.1 to 9.9-fold, P<0.001), AKT (39.7 to 22-fold, P<0.05), and p38MAPK (21.8-fold to 8.1-fold, P<0.05), respectively as compared to the scrambled control (Fig. 4B). In contrast, there were no effects of silencing AXL on HGF activation of ERK phosphorylation (2.0-fold versus 2.1-fold in scrambled controls, NS) and STAT3 phosphorylation (1.6-fold versus 1.9-fold in scrambled controls, NS), suggesting cell specific downstream effector pathways dependent on AXL/MET interaction in GnRH neurons (Fig. 4A and 4B). Additionally, since TYRO3 another TAM family member can directly interact with MET in the neuronal cells, we silenced endogenous TYRO3 (47%). Loss of TYRO3 in the neuronal cells had no effect on HGF induced MET activation (data not shown), confirming AXL-specific interaction with MET.
Figure 4. Silencing of AXL alters HGF/MET signaling in NLT GnRH neurons.
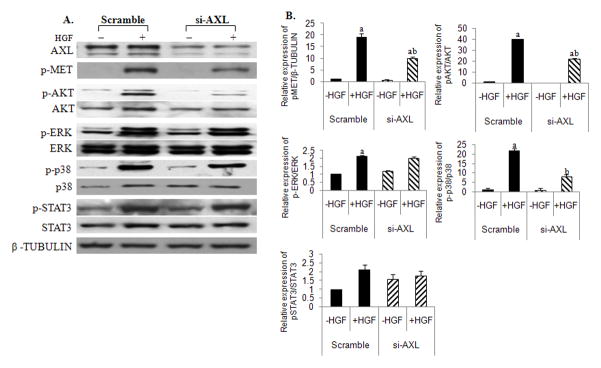
A: Silencing AXL decreased HGF stimulated activation of MET, AKT and p38MAPK. NLT cells were transfected with control scrambled siRNA or siRNA specific for AXL (see methods for details). Cells were immunoblotted with AXL, p-MET, p-AKT, AKT, p-ERK, ERK, p-p38MAPK, p38MAPK, p-STAT3, STAT3 and β-TUBULIN (loading control), n=3. B: Bar graphs depicting effect of HGF on the mean relative expression of pMET, pAKT, pERK, p38MAPK and p-STAT3 to that of β-TUBULIN, AKT, ERK, p38MAPK and STAT3 respectively in scrambled and AXL inhibited NLT GnRH neuronal cells. Letters represent significant difference from each other (a, difference from HGF untreated cells and b, difference from HGF treated scramble controls) p<0.05, ANOVA with post hoc Bonferronis multiple comparisons test, n=3.
3.8. HGF/MET activation of GnRH neuronal cell migration is dependent on AXL
We previously showed that GAS6/AXL signaling through p38MAPK mediates GnRH neuronal migration (Allen et al., 2002a). To test the hypothesis that AXL levels modulate the effects of HGF/MET promotion of GnRH neuron migration, scratch assays were performed in NLT GnRH neuronal cells with and without silencing of AXL. Cells were incubated with SB203580 (25μM), a p38MAPK inhibitor and LY294002 (10μM), a PI3Kinase inhibitor in the presence or absence of DMSO and HGF (50 ng/ml) for 24 hrs (see Fig. 5A). Silencing of AXL (85% by siRNA as compared to scramble control) expression was confirmed using immunoblot (inset of Fig. 5B). NLT cells that migrated after 24hrs were counted and data normalized with their respective scratch area (see Fig. 5B). HGF activated migration as compared to the DMSO treated controls (1.8± 0.1 vs. 1.1±0.1 respectively, P<0.01) in NLT neuronal cells transfected with scrambled oligomers. However, HGF failed to induce migration in NLT neurons where AXL expression was silenced (1.1±0.07 vs. 1.8±0.1, as compared to HGF treated scramble controls, NS) indicating that AXL co-expression was required for HGF induction of GnRH migration. Treatment with the p38MAPK inhibitor, SB203580 significantly reduced the basal migration in both DMSO treated scramble controls (1.1±0.1 to 0.4±0.02, P<0.01) and DMSO treated siAXL group (0.9±0.01 to 0.2±0.1, P<0.001), suggesting that basal neuronal migration is critically dependent on p38MAPK. Addition of HGF increased rates of migration back to basal levels in the presence of the p38MAPK inhibitor in scrambled controls (0.9±0.02 vs. 0.4±0.4, as compared to SB203580 treated scrambled controls, P<0.05). Inhibition of AXL blocked this effect, suggesting that HGF effects on migration are independent of AXL’s activation of p38MAPK.
Figure 5. Interaction of AXL and MET influence migration in NLT GnRH neuronal cells.

A: Representative wound-healing images of functional effects of silencing AXL on HGF induced NLT GnRH neuronal migration (see methods for details). B: Bar graph depicting effect of HGF on migration of NLT neurons in presence of p38MAPK inhibitor (SB203580) and PI3K inhibitor (LY294002) in scramble and AXL inhibited (as shown in inset) neuronal cells. Results are normalized with scratch area and expressed as mean ± SEM. Letters represent significant difference from each other (a and b, difference from DMSO and HGF scrambled control groups, c and d, difference from DMSO and HGF siAXL groups and e, difference from SB203580+HGF treated scrambled control group) p<0.05; ANOVA with post hoc Tukeys multiple comparisons test, n=3.
In contrast, treatment with the PI3K inhibitor, LY294002, had no significant effects on HGF-induced GnRH migration in either scrambled controls (1.0±0.07 as compared to DMSO treated scrambled group, NS) or AXL inhibited neuronal cells (1.0±0.2, as compared to DMSO treated siAXL group and also as compared to LY294002 and HGF treated scrambled groups, NS). Additionally, treatment with PD98059 (10μM), a MEK inhibitor that blocks ERKMAPK signaling had no effect on basal migration of NLT cells (data not shown)implicating that GnRH neuronal migration is not dependent on ERK pathway. Immunoblots confirmed that samples treated with SB203580 or LY294002 showed significant reduction in the phospho-p38MAPK and phospho-AKT levels (supplementary Fig. S2). Together these data indicate that HGF/MET activation of GnRH neuronal migration is dependent on the presence of AXL, yet independent of AXL’s actions via the p38MAPK pathway. The neuronal migration pathway that mediates these effects remains to be elucidated.
3.9. HGF/MET modulates the effects of AXL on GnRH neuronal cell survival
Similarly, we asked whether HGF/MET conferred effects on GnRH neuronal cell survival in response to growth factor withdrawal and whether these are dependent on AXL coexpression and its downstream effectors (see Fig. 6). NLT cells were transfected with scrambled control or si-AXL for 24hrs. Cells were pre-treated with LY294002 or PD98059 or DMSO for 2hrs followed by incubation in the presence or absence of PBS or HGF in serum-free medium for 48hrs. Silencing of AXL (85% by siRNA as compared to scramble control) expression was confirmed using immunoblot (inset of Fig. 6). Apoptotic neuronal cells were measured by counting the number of cells with condensed or fragmented chromatin using Hoechst staining (Fig. 6). Inhibition of AXL expression increased apoptotic cells in baseline conditions (2.5-fold, 3.4±0.2 in scrambled group to 8.8±1.5 in si-AXL group, P<0.05) confirming its importance in survival even in the absence of its ligand, GAS6. Somewhat to our surprise, HGF did not affect rates of cell death after growth factor withdrawal in baseline conditions (0.7-fold, 3.4±0.2 in DMSO treated to 2.6±0.3 in HGF treated scrambled controls, NS). However, HGF rescued the cells from programmed cell death when AXL expression was inhibited (2.3-fold, 8.8±1.5 in si-AXL with DMSO to 3.8±0.6 in si-AXL with HGF, P<0.05). These data suggest that HGF/MET signaling modulates AXL’s ability to promote neuronal cell survival.
Figure 6. AXL and MET interaction influence survival in NLT GnRH neuronal cells.
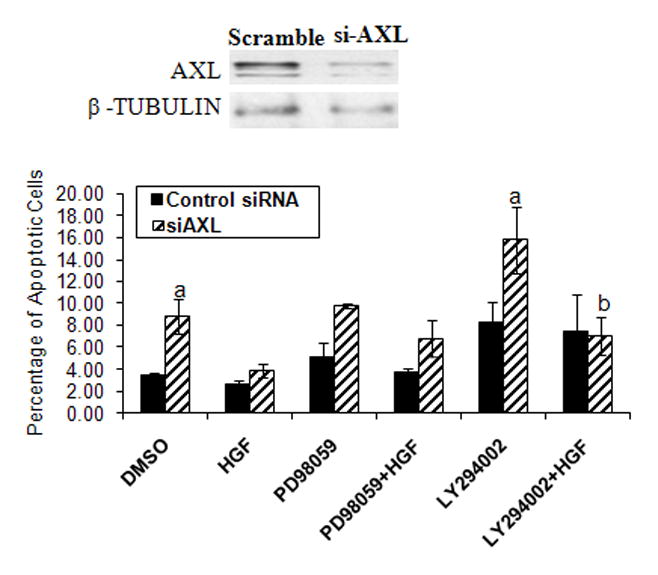
Functional effects of silencing AXL (as shown in inset) on HGF induced on rates of apoptosis in response to serum deprivation in a PI3K dependent pathway as assessed by Hoechst staining (see methods for details). Letters represent significant difference from each other (a, difference from DMSO treated scrambled control group and b, difference from LY294002 treated siAXL group) p<0.05; ANOVA with post hoc Tukeys multiple comparisons test, n=3.
In cells where AXL was silenced, inhibition of the PI3Kpathway with LY294002 or ERK pathway with the MEK inhibitor PD98059 resulted in increased apoptotic cells, 1.9-fold (8.2 ±1.8 in scramble to 15.8±3 in si-AXL, NS) and 1.9-fold (5.0±1.2 in scramble to 9.7±0.2 in si-AXL, NS) respectively, confirming these effector pathways are downstream of AXL as previously observed (Allen et al., 1999; Allen et al., 2002a). In cells where AXL was silenced, addition of HGF rescued cells treated with the PI3K inhibitor (2.2–fold, 15.8±3 in si-AXL with LY294002 to 7.0±1.7 in si-AXL with LY294002 and HGF, P<0.05). In contrast, HGF did not significantly alter the rates of apoptosis in cells incubated with the MEK inhibitor (9.7±0.2 in si-AXL with PD98059 to 6.7±1.6 in si-AXL with PD98059 and HGF, NS). Immunoblots confirmed that samples treated with LY294002 or PD98059 showed significant reduction in the phospho-AKT and phospho-ERK levels (supplementary Fig. S2). Together, these data suggest that HGF signaling to MET modulates AXL’s influence on survival in response to growth factor withdrawal via the PI3K, but not the ERK pathway.
4. Discussion
The present study identifies the importance of AXL co-expression in modulating HGF/MET and conversely HGF/MET to modulate AXL function in a model of early migrating GnRH neurons. Co-immunoprecipitation of MET with AXL supports a direct interaction between these two tyrosine kinase receptors in GnRH neuronal cells. We also identify that HGF/MET directly or indirectly activates multiple pathways including ERK, PI3K/AKT and p38MAPK in GnRH neuronal cells. Although the ligands, GAS6and HGF do not transactivate the opposite receptor, AXL or MET respectively, the presence of AXL modulates HGF/MET signaling and conversely the presence of MET modulates AXL effects. HGF/MET mediates neuronal migration dependent on interaction with AXL but independent of AXL’s actions via the p38MAPK or AKT pathways. Conversely, HGF/MET is not critical for GnRH neuronal survival in the presence of AXL, but rescues GnRH neuronal cells from growth factor withdrawal induced apoptosis when AXL is silenced. Despite being a robust effector pathway downstream of MET, the ERK MAPK pathway does not mediate MET effects on migration or survival in GnRH neuronal cells. We further show that coexpression of a kinase dead AXL disrupted AXL’S ability to modulate HGF/MET signaling, suggesting the importance of the AXL kinase domain to mediate the crosstalk between the 2 RTKs.
Prior work has shown that HGF/MET interacts with other membrane receptors such as MSP/Ron to promote tumorigenesis (Jo et al., 2000). In gastric cancer cells, HGF/MET can form hetrodimeric complexes with Ron in which MSP (the ligand for Ron) and HGF specific activation resulted in transphosphorylation of each other’s receptors(Follenzi et al., 2000). In contrast, in our system, GAS6 and HGF failed to transphosphorylate MET and AXL, respectively suggesting that in GnRH neuronal cells, activation of both AXL and MET receptors are ligand specific processes.
In glioma cells, crosstalk between SDF-1 alpha/CXCR4 and HGF/MET is mediated by NF-Kappaβ and leads to migration (Esencay M, 2012). Khoury and coworkers have reported that the physiological signals downstream of HGF/MET can synergize with ErbB2/Neu receptors to further enhance the malignant phenotype in kidney cells (Khoury et al., 2005). We asked if AXL and MET crosstalk affects their respective downstream signaling in the early migrating GnRH neurons. Earlier studies from our lab have shown that GAS6/AXL drives migration via Rac-p38MAPK pathway and cell survival via PI3K/AKT and ERK pathways in GnRH neurons (Allen et al., 1999; Allen et al., 2002a). Activation of the HGF/MET downstream signaling has been implicated in regulating a wide number of pathways including AKT (survival), FAK (invasion), ERK (proliferation) and STAT3 (morphogenesis) in cancer cells (as reviewed in (Birchmeier C, 2003). Addition of a selective MET inhibitor leads to a significant reduction in the expression of AKT, ERK, p38MAPK and STAT3 in NLT neurons. Altering the levels of AXL influenced the bi-directional cross-talk between AXL and MET. Expression of a kinase dead AXL mutant reduced HGF induced MET phosphorylation of AKT and p38MAPKwith no effect on ERK or STAT3, confirming the cell specific pathways downstream of the interaction between AXL and MET in GnRH neurons. Similarly, either deletion of the intracytoplasmic domain or specifically mutating the tyrosine kinase domain of AXL resulted in a significant reduction in HGF- induced activation of MET. These data suggest that the kinase domain of AXL is the major locus of receptor interaction. Inability to observe complete inhibition of phospho-MET could be due to the inadequate silencing of AXL or subtle roles played by other AXL domains such as fibronectin II domain that may be involved in bringing the receptors into close proximity.
HGF has previously been shown to induce a chemotactic response for migration of GN11 cells (similar to NLT cells) in a Boyden chamber assay (Giacobini et al., 2007). Our data revealed that silencing of AXL decreases HGF induced migration, suggesting that AXL/MET interaction is required for optimal GnRH neuronal migration in response to HGF. Whereas AXL induced migration is dependent on p38MAPK, the effects of HGF appear to be independent of p38MAPK or PI3K/AKT (Fig 7). HGF is known to exert a protective effect in the hippocampal CA1 after transient forebrain ischemia in an ERK-dependent pathway (Niimura et al., 2006). In GnRH neuronal cells, HGF had no effects on survival in response to growth factor withdrawal, but partially rescued apoptosis when AXL or the PI3K/AKT pathway was inhibited. Together, these data confirm the bi-directional crosstalk between the two receptors and their downstream effector pathways in GnRH neuronal cells.
Figure 7.
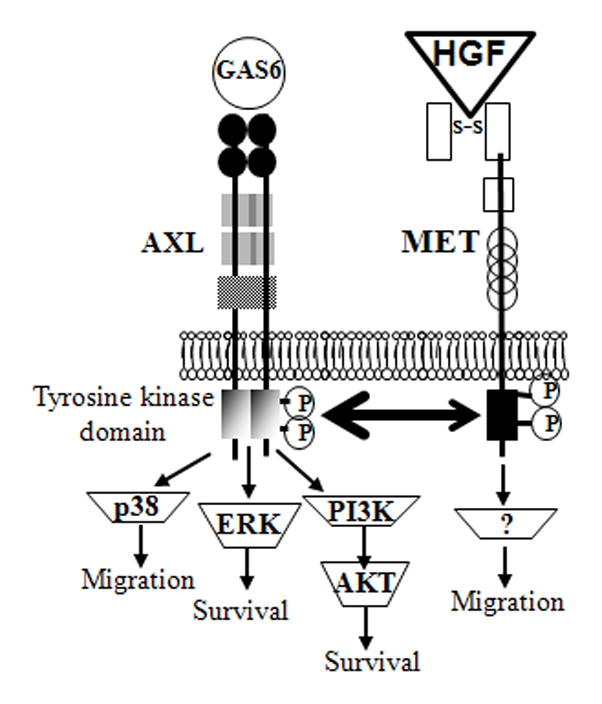
Schematic illustration of the crosstalk of GAS6/AXL and HGF/MET and their putative effects on various downstream signaling molecules in early migrating GnRH neurons.
5. Conclusion
In conclusion, our studies have demonstrated novel pathways for MET signaling in GnRH neuronal cells. Importantly, AXL, but not TYRO3 or MER members of the TAM family communicates with MET. HGF/MET signaling activates migration independent of AXL via an as yet unknown pathway in addition to acting via AXL to trigger p38MAPK induced neuronal migration. HGF/MET has no effects on its own to modulate survival but rescues cells where AXL is deficient via the PI3K/AKT pathway. These data add to the growing observations that membrane receptor tyrosine kinases may crosstalk during development to mediate their cell specific actions and suggest that mutations in these pathway components may be candidates for study as underlying defects in lack of pubertal development and disorders of sexual maturation in humans.
Highlights.
Multiple membrane receptor tyrosine kinases can influence early migrating GnRH neurons.
AXL coexpression is required for HGF/MET signaling via a p38MAPK and AKT specific pathways.
HGF/MET promotes GnRH neuronal migration via AXL dependent and independent pathways and modulates AXL promotion of neuronal survival.
Tyrosine kinase receptor crosstalk is important for optimal GnRH neuronal development and function.
Acknowledgments
The authors would like to thank Melinda Schaller for conducting early studies on this project and thank Maozhen Tian and Katja Kiseljak-Vassiliades for their review of the manuscript. We would also like to thank Amanda Flockton, Kurt Stenmark and Maria Frid (UCD) for their generous help during scratch assay experiments.
This work was supported by National Institute of Health grant HD31191 to MEW.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen MP, Zeng C, Schneider K, Xiong X, Meintzer MK, Bellosta P, Basilico C, Varnum B, Heidenreich KA, Wierman ME. Growth arrest-specific gene 6 (Gas6)/adhesion related kinase (Ark) signaling promotes gonadotropin-releasing hormone neuronal survival via extracellular signal-regulated kinase (ERK) and Akt. Mol Endocrinol. 1999;13:191–201. doi: 10.1210/mend.13.2.0230. [DOI] [PubMed] [Google Scholar]
- Allen MP, Xu M, Linseman DA, Pawlowski JE, Bokoch GM, Heidenreich KA, Wierman ME. Adhesion-related kinase repression of gonadotropin-releasing hormone gene expression requires Rac activation of the extracellular signal-regulated kinase pathway. J Biol Chem. 2002a;277:38133–38140. doi: 10.1074/jbc.M200826200. [DOI] [PubMed] [Google Scholar]
- Allen MP, Linseman DA, Udo H, Xu M, Schaack JB, Varnum B, Kandel ER, Heidenreich KA, Wierman ME. Novel mechanism for gonadotropin-releasing hormone neuronal migration involving Gas6/Ark signaling to p38 mitogen-activated protein kinase. Mol Cell Biol. 2002b;22:599–613. doi: 10.1128/MCB.22.2.599-613.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellosta P, Costa M, Lin DA, Basilico C. The receptor tyrosine kinase ARK mediates cell aggregation by homophilic binding. Mol Cell Biol. 1995;15:614–625. doi: 10.1128/mcb.15.2.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchmeier C, BW, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- Bladt F, Riethmacher D, Isenmann S, Aguzzi A, Birchmeier C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature. 1995;376:768–771. doi: 10.1038/376768a0. [DOI] [PubMed] [Google Scholar]
- Boccaccio C, Ando M, Tamagnone L, Bardelli A, Michieli P, Battistini C, Comoglio PM. Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature. 1998;391:285–288. doi: 10.1038/34657. [DOI] [PubMed] [Google Scholar]
- Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, Aaronson SA. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251:802–804. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- Brusevold IJ, Aasrum M, Bryne M, Christoffersen T. Migration induced by epidermal and hepatocyte growth factors in oral squamous carcinoma cells in vitro: role of MEK/ERK, p38 and PI-3 kinase/Akt. J Oral Pathol Med. 2012 doi: 10.1111/j.1600-0714.2012.01139.x. [DOI] [PubMed] [Google Scholar]
- Comoglio PM. Pathway specificity for Met signalling. Nat Cell Biol. 2001;3:161–162. doi: 10.1038/35083116. [DOI] [PubMed] [Google Scholar]
- Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7:504–516. doi: 10.1038/nrd2530. [DOI] [PubMed] [Google Scholar]
- Esencay M, NE, Zagzag D. HGF upregulates CXCR4 expression in gliomas via NF-kappaB: implications for glioma cell migration. J Neurooncol. 2012;99:33–40. doi: 10.1007/s11060-010-0111-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follenzi A, Bakovic S, Gual P, Stella MC, Longati P, Comoglio PM. Cross-talk between the proto-oncogenes Met and Ron. Oncogene. 2000;19:3041–3049. doi: 10.1038/sj.onc.1203620. [DOI] [PubMed] [Google Scholar]
- Furge KA, Zhang YW, Vande Woude GF. Met receptor tyrosine kinase: enhanced signaling through adapter proteins. Oncogene. 2000;19:5582–5589. doi: 10.1038/sj.onc.1203859. [DOI] [PubMed] [Google Scholar]
- Gentile A, TL, Comoglio PM. The Met tyrosine kinase receptor in development and cancer. Cancer Metastasis Rev. 2008;27:85–94. doi: 10.1007/s10555-007-9107-6. [DOI] [PubMed] [Google Scholar]
- Giacobini P, Giampietro C, Fioretto M, Maggi R, Cariboni A, Perroteau I, Fasolo A. Hepatocyte growth factor/scatter factor facilitates migration of GN-11 immortalized LHRH neurons. Endocrinology. 2002;143:3306–3315. doi: 10.1210/en.2002-220146. [DOI] [PubMed] [Google Scholar]
- Giacobini P, Messina A, Wray S, Giampietro C, Crepaldi T, Carmeliet P, Fasolo A. Hepatocyte growth factor acts as a motogen and guidance signal for gonadotropin hormone-releasing hormone-1 neuronal migration. J Neurosci. 2007;27:431–445. doi: 10.1523/JNEUROSCI.4979-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacobini P, Messina A, Morello F, Ferraris N, Corso S, Penachioni J, Giordano S, Tamagnone L, Fasolo A. Semaphorin 4D regulates gonadotropin hormone-releasing hormone-1 neuronal migration through PlexinB1-Met complex. J Cell Biol. 2008;183:555–566. doi: 10.1083/jcb.200806160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziani A, Gramaglia D, Cantley LC, Comoglio PM. The tyrosine-phosphorylated hepatocyte growth factor/scatter factor receptor associates with phosphatidylinositol 3-kinase. J Biol Chem. 1991;266:22087–22090. [PubMed] [Google Scholar]
- Jo M, Stolz DB, Esplen JE, Dorko K, Michalopoulos GK, Strom SC. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J Biol Chem. 2000;275:8806–8811. doi: 10.1074/jbc.275.12.8806. [DOI] [PubMed] [Google Scholar]
- Jung W, Castren E, Odenthal M, Vande Woude GF, Ishii T, Dienes HP, Lindholm D, Schirmacher P. Expression and functional interaction of hepatocyte growth factor-scatter factor and its receptor c-met in mammalian brain. J Cell Biol. 1994;126:485–494. doi: 10.1083/jcb.126.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury H, Naujokas MA, Zuo D, Sangwan V, Frigault MM, Petkiewicz S, Dankort DL, Muller WJ, Park M. HGF converts ErbB2/Neu epithelial morphogenesis to cell invasion. Mol Biol Cell. 2005;16:550–561. doi: 10.1091/mbc.E04-07-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lay JD, Hong CC, Huang JS, Yang YY, Pao CY, Liu CH, Lai YP, Lai GM, Cheng AL, Su IJ, Chuang SE. Sulfasalazine suppresses drug resistance and invasiveness of lung adenocarcinoma cells expressing AXL. Cancer Res. 2007;67:3878–3887. doi: 10.1158/0008-5472.CAN-06-3191. [DOI] [PubMed] [Google Scholar]
- Mars WM, Zarnegar R, Michalopoulos GK. Activation of hepatocyte growth factor by the plasminogen activators uPA and tPA. Am J Pathol. 1993;143:949–958. [PMC free article] [PubMed] [Google Scholar]
- Mellon PL, WJ, Goldsmith PC, Padula CA, Roberts JL, Weiner RI. Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron. 1990;5:1–10. doi: 10.1016/0896-6273(90)90028-e. [DOI] [PubMed] [Google Scholar]
- Naldini L, Vigna E, Narsimhan RP, Gaudino G, Zarnegar R, Michalopoulos GK, Comoglio PM. Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto-oncogene c-MET. Oncogene. 1991;6:501–504. [PubMed] [Google Scholar]
- Nielsen-Preiss SM, Allen MP, Xu M, Linseman DA, Pawlowski JE, Bouchard RJ, Varnum BC, Heidenreich KA, Wierman ME. Adhesion-related kinase induction of migration requires phosphatidylinositol-3-kinase and ras stimulation of rac activity in immortalized gonadotropin-releasing hormone neuronal cells. Endocrinology. 2007;148:2806–2814. doi: 10.1210/en.2007-0039. [DOI] [PubMed] [Google Scholar]
- Niimura M, Takagi N, Takagi K, Funakoshi H, Nakamura T, Takeo S. Effects of hepatocyte growth factor on phosphorylation of extracellular signal-regulated kinase and hippocampal cell death in rats with transient forebrain ischemia. Eur J Pharmacol. 2006;535:114–124. doi: 10.1016/j.ejphar.2006.01.037. [DOI] [PubMed] [Google Scholar]
- Okamoto W, Okamoto I, Arao T, Yanagihara K, Nishio K, Nakagawa K. Differential roles of STAT3 depending on the mechanism of STAT3 activation in gastric cancer cells. Br J Cancer. 2011;105:407–412. doi: 10.1038/bjc.2011.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce A, Bliesner B, Xu M, Nielsen-Preiss S, Lemke G, Tobet S, Wierman ME. Axl and Tyro3 modulate female reproduction by influencing gonadotropin-releasing hormone neuron survival and migration. Mol Endocrinol. 2008;22:2481–2495. doi: 10.1210/me.2008-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce A, Xu M, Bliesner B, Liu Z, Richards J, Tobet S, Wierman ME. Hypothalamic but not pituitary or ovarian defects underlie the reproductive abnormalities in Axl/Tyro3 null mice. Mol Cell Endocrinol. 2011;339:151–158. doi: 10.1016/j.mce.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radovick S, Wray S, Lee E, Nicols DK, Nakayama Y, Weintraub BD, Westphal H, Cutler GB, Jr, Wondisford FE. Migratory arrest of gonadotropin-releasing hormone neurons in transgenic mice. Proc Natl Acad Sci U S A. 1991;88:3402–3406. doi: 10.1073/pnas.88.8.3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C, Bladt F, Goedecke S, Brinkmann V, Zschiesche W, Sharpe M, Gherardi E, Birchmeier C. Scatter factor/hepatocyte growth factor is essential for liver development. Nature. 1995;373:699–702. doi: 10.1038/373699a0. [DOI] [PubMed] [Google Scholar]
- Schwanzel-Fukuda M, Pfaff DW. Origin of luteinizing hormone-releasing hormone neurons. Nature. 1989;338:161–164. doi: 10.1038/338161a0. [DOI] [PubMed] [Google Scholar]
- Segarra J, BL, Drenth T, Maina F, Lamballe F. Combined signaling through ERK, PI3K/AKT, and RAC1/p38 is required for met-triggered cortical neuron migration. J Biol Chem. 2006;281:4771–4780. doi: 10.1074/jbc.M508298200. [DOI] [PubMed] [Google Scholar]
- Soong J, Chen Y, Shustef EM, Scott GA. Sema4D, the ligand for Plexin B1, suppresses c-Met activation and migration and promotes melanocyte survival and growth. J Invest Dermatol. 2012;132:1230–1238. doi: 10.1038/jid.2011.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed ZA, Yin W, Hughes K, Gill JN, Shi R, Clifford JL. HGF/c-met/Stat3 signaling during skin tumor cell invasion: indications for a positive feedback loop. BMC Cancer. 2011;11:180. doi: 10.1186/1471-2407-11-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu H, Zhou Z, Liang Q, Li Z, Li D, Qing J, Wang H, Zhang L. CXCR4 and SDF-1 production are stimulated by hepatocyte growth factor and promote glioma cell invasion. Onkologie. 2009;32:331–336. doi: 10.1159/000216352. [DOI] [PubMed] [Google Scholar]
- Uehara Y, Minowa O, Mori C, Shiota K, Kuno J, Noda T, Kitamura N. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature. 1995;373:702–705. doi: 10.1038/373702a0. [DOI] [PubMed] [Google Scholar]
- Wang J, Gui Z, Deng L, Sun M, Guo R, Zhang W, Shen L. c-Met upregulates aquaporin 3 expression in human gastric carcinoma cells via the ERK signalling pathway. Cancer Lett. 2012;319:109–117. doi: 10.1016/j.canlet.2011.12.040. [DOI] [PubMed] [Google Scholar]
- Wierman ME, Pawlowski JE, Allen MP, Xu M, Linseman DA, Nielsen-Preiss S. Molecular mechanisms of gonadotropin-releasing hormone neuronal migration. Trends Endocrinol Metab. 2004;15:96–102. doi: 10.1016/j.tem.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Wierman ME, Kiseljak-Vassiliades K, Tobet S. Gonadotropin-releasing hormone (GnRH) neuron migration: initiation, maintenance and cessation as critical steps to ensure normal reproductive function. Front Neuroendocrinol. 2011;32:43–52. doi: 10.1016/j.yfrne.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray S. From nose to brain: development of gonadotrophin-releasing hormone-1 neurones. J Neuroendocrinol. 2010;22:743–753. doi: 10.1111/j.1365-2826.2010.02034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]