

Abstract
Background
β-adrenergic stimulation is the main trigger for cardiac events in type-1 long QT syndrome (LQT1). We evaluated a possible association between ion channel response to β-adrenergic stimulation and clinical response to β-blocker therapy according to mutation location.
Methods and Results
The study sample comprised 860 patients with genetically-confirmed mutations in the KCNQ1 channel. Patients were categorized into carriers of missense mutations located in the cytoplasmic loops (C-loops), membrane spanning domain, C/N-terminus, and non-missense mutations. There were 27 aborted cardiac arrest [ACA] and 78 sudden cardiac death [SCD] events from birth through age 40 years. After multivariable adjustment for clinical factors, the presence of C-loop mutations was associated with the highest risk for ACA or SCD (hazard ratio [95% confidence interval] vs. non-missense mutations = 2.75 [1.29-5.86, P=0.009]). β-blocker therapy was associated with a significantly greater reduction in the risk of ACA or SCD among patients with C-loop mutations than among all other patients (hazard ratios = 0.12 [0.02-0.73, P=0.02] and 0.82 [0.31-2.13, P=0.68], respectively; P-for interaction = 0.04). Cellular expression studies showed that membrane spanning and C-loop mutations produced a similar decrease in current, but only C-loop mutations showed a pronounced reduction in channel activation in response to β-adrenergic stimulation.
Conclusions
Patients with C-loop missense mutations in the KCNQ1 channel exhibit a high-risk for life-threatening events and derive a pronounced benefit from treatment with β-blockers. Reduced channel activation following sympathetic activation can explain the increased clinical risk and response to therapy in patients with C-loop mutations.
Keywords: beta-blockers, ion channels, long QT syndrome, mutation
Long QT syndrome type-1 (LQT1) is the most common type of inherited long QT syndrome (LQTS), accounting for approximately 35 percent of all patients and more than 50 percent of genotyped patients.1 LQT1 arises from a decrease in repolarizing potassium current due to mutations in the KCNQ1 gene. Four KCNQ1 derived α-subunits assemble to form the IKs channel along with obligatory auxiliary subunits derived from KCNE1. Exercise is the main trigger for cardiac arrhythmic events in patients with LQT1.2 Activation of β1-adrenergic receptors (β1-AR) is the major signaling pathway contributing to increase in heart rate and cardiac output during exercise. β1-AR activation leads to activation of protein kinase A (PKA), which directly phosphorylates the KCNQ1 subunit, increasing IKs function.3-4 The increase in IKs is thought to suppress the premature beats and afterdepolarization induced by increased L-type Ca2+ currents during β-adrenergic stimulation.5 Accordingly, ß-blockers have been considered the first-line therapy in LQT1 patients without a history of aborted cardiac arrest (ACA). Data from several prior long QT syndrome (LQTS) studies 1, 6 demonstrate that despite the reduction in the risk of cardiac events with ß-blocker therapy among LQT1 patients, there is a considerable cardiac residual event rate among patients who are being treated with this mode of medical therapy (about 10 cardiac events per 100 person-years),6 suggesting that ß-blockers may be less effective in certain subgroups of LQT1 patients.
The KCNQ1 protein consists of 676 amino acid residues with an intracellular N-terminus region, 6 membrane-spanning segments with two connecting cytoplasmic loops (C-loops) and an intracellular C-terminus region.7 Prior genotype-phenotype studies have provided important information regarding the effect of location and coding type of the channel mutations on the phenotypic manifestations and clinical course of LQT1 patients. These studies have shown that missense mutations and mutations located at the transmembrane region (including the C-loops) were associated with greater risk for cardiac events.8 However, the mechanism related to the increased risk associated with transmembrane mutations has not been studied. C-loops, part of the transmembrane region, were suggested to affect adrenergic channel regulation by PKA.9 We therefore hypothesized that the previously reported findings regarding the risk associated with transmembrane mutations 8 is related to the effect of C-loop mutations within this region. Accordingly, the present study was carried out in a large cohort of subjects having a spectrum of KCNQ1 mutations from the International LQTS Registry, and was designed to: 1) investigate the clinical outcomes among KCNQ1 mutation carriers by further dividing the transmembrane region into membrane spanning and C-loop domains; 2) determine a possible differential response to ß-blocker therapy depending on mutation location and function related to PKA regulation; and 3) relate the clinical data to functional studies of changes in IKs function and β-AR regulation in mammalian cells.
Methods
Study sample
The study comprised 860 patients with genetically confirmed KCNQ1 mutations derived from 170 proband-identified families. The proband in each family had QTc prolongation not due to a known secondary cause. The subjects were drawn from the Rochester (n = 637), the Netherlands (n = 94), the Japanese (n = 82), the Danish (n = 43), and the Swedish (n=4) portions of the Multicenter Mutation Registry. All subjects or their guardians provided informed consent for the genetic and clinical studies. Patients with congenital deafness or patients with multiple LQTS associated mutations were excluded from the study.
Phenotype characterization
On enrollment, routine clinical and electrocardiographic information was obtained from birth to their enrolled age, and ongoing clinical information was obtained at yearly intervals thereafter. For each patient, data on personal and family histories, cardiac events, and therapy were systematically recorded at enrollment and at each visit or medical contact. Clinical data, recorded on prospectively designed forms, included patient and family histories and demographic, ECG, therapeutic, and cardiac event information. Data on β-blocker therapy included the starting date, and discontinuation date if appropriate. Information on the endpoint of ACA or SCD was also verified through requested medical records. Every effort was made to confirm an underlying life threatening arrhythmia when observed or documented by medical staff.
Genotype characterization
The KCNQ1 mutations were identified with the use of standard genetic tests performed in academic molecular-genetic laboratories. Genetic alterations of the amino acid sequence were characterized by location and by the specific type of mutation (missense, splice site, in-frame insertions/deletions, nonsense, stop codon, and frameshift).
We evaluated the risk associated with 4 main prespecified subgroups: 1) C- or N-terminus-missense; 2) membrane spanning-missense; 3) C-loops-missense; and 4) non-missense (i.e. splice sites, in-frame insertions, in-frame deletions, stop codons, and frameshift). The membrane spanning region of the KCNQ1-encoded channel was defined as the coding sequence involving amino acid residues between 124-170 (S1-S2), 196-241 (S3-S4), and 263-355 (S5-S6), with the C-loops region between residues 171-195 (S2-S3) and 242-262 (S4-S5) (Figure 1). The N-terminus region was defined before residue 124 and the C-terminus region after residue 355.
Figure 1.
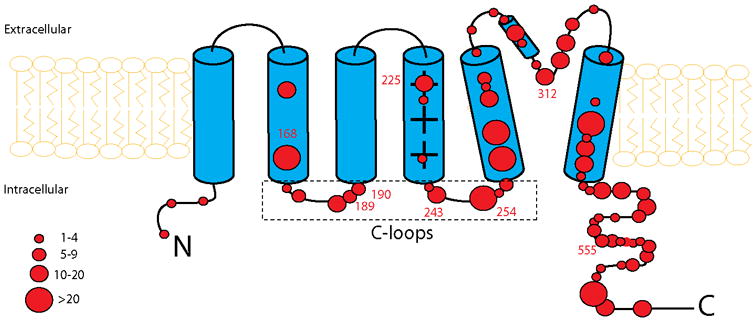
Frequency and location of mutations in the KCNQ1 potassium channel. Diagramatic location of 99 different mutations in the KCNQ1 potassium channel involving 860 subjects. The α subunit involves the N-terminus (N), 6 membrane-spanning segments, 2 cytoplasmic loops (S2-S3 and S4-S5) and the C-terminus portion (C). The size of the circles reflects the number of subjects with mutations at the respective locations.
To minimize survival bias, we did include patients who died before they were genotyped (n=64). They were assumed to have the mutation their first degree relatives had. All other patients were confirmed through genotyping.
Cellular Expression Studies
In order to study the mechanism underlying the risk for cardiac events in patients with missense C-loop mutations, we measured channel function and regulation for channels formed with wild-type subunits co-expressed with four mutant subunits present in C-loops (G189R, R190Q, R243C and V254M) and four mutant subunits present in the non-C-loop domains, three in the membrane spanning domain (T312I, G168R and S225L) and one in the C-terminus (R555C). The mutations chosen included the most common mutations in the LQT1 registry. Wild type-and mutant KCNQ1 subunits, together with KCNE1 subunits cDNA were transfected into HEK293T cells. 10 Mutant KCNQ1 cDNA was transfected in combination with WT-KCNQ1 to mimic the heterozygous nature of the disease (WT- KCNQ1:mutant KCNQ1:KCNE1=0.5:0.5:1).
Fluorescence conjugated and tagged constructs were used to evaluate the efficiency of the co-transfection of wild-type and mutant subunits 11 (supplementary data). 85-90% of HEK293T cells co-transfected with both wild-type and mutant subunits showed fluorescence of at least one subunit transfected and 85-95% of transfected cells expressed all the subunits transfected (supplementary figure 1S). All electrophysiology determinations were performed with the untagged subunit. Expression of wild-type and mutant subunits were confirmed by Western blot (supplementary figure 2S). Expression levels were not significantly decreased for the mutant subunits when compared to wild-type. We measured ion channel currents after channel depolarization to +20 mV for 4s from -80 mV holding potential, before and after application of forskolin, a protein kinase A activator (10 μmol/mL) using standard electrophysiological techniques and physiological solutions. Current was normalized for all voltages to cell capacitance and further normalization was performed between wild type and mutant. The normalization to wild type currents was accomplished using wild type cells currents transfected and measured in the same day as the currents measured from mutant channel.10 Pipettes used had resistances ranging from 2-6 MΩ. Series resistance compensation of >70% was used to compensate for voltage drops in the pipette. All experiments were performed at room temperature. Details of the molecular biology and electrophysiological methods are described in the materials and methods section of the Supplementary Appendix.
End point
The primary end point of the study was the occurrence of a first life-threatening cardiac event, comprising aborted cardiac arrest ([ACA] requiring external defibrillation as part of the resuscitation) or LQTS-related sudden cardiac death ([SCD] abrupt in onset without evident cause, if witnessed, or death that was not explained by any other cause if it occurred in a non-witnessed setting) from birth through age 40 years. Follow up after age 40 years was not included to minimize the influence of coronary disease on cardiac events. The consistency of the results among patients who received an implantable cardioverter defibrillator (ICD) during follow-up was evaluated in a secondary analysis that included the occurrence of a first appropriate ICD shock in the composite ACA or SCD end point.
Statistical analysis
Characteristics of the 4 subgroups of patients categorized by mutation location and type were compared with the one way ANOVA test or Chi square and Fisher exact tests, as appropriate. The probability of a first life-threatening cardiac event by the mutation-location and type subgroup was graphically displayed according to the method of Kaplan and Meier, with comparison of instantaneous risk by the log-rank test. The Cox proportional-hazards survivorship model was used to evaluate the independent contribution of clinical and genetic factors to the first occurrence of a life-threatening cardiac event from birth through age 40 years. The Cox regression models, stratified by decade of birth year and allowing for time-dependent covariates, were fitted to estimate the adjusted hazard ratio function of age. Therefore, to fulfill the assumption of proportional hazards for sex over the entire age range, a time-dependent covariate for sex (via an interaction with time) was incorporated, allowing for different hazard ratios by sex before and after age 13 years. This was justified due to known higher risk of cardiac events or life threatening cardiac events among males before adolescence and a similar risk or superior female risk after the onset of adolescence. 12-16 Patients who did not have an ECG for QTc measurement (n=127) were identified in the Cox models as “QTc missing”, and all Cox models were adjusted for this QTc missing parameter. The influence of time-dependent ß-blocker therapy (the age at which ß-blocker therapy was initiated) on outcome in the subgroups of patients with and without C-loop-missense mutations was determined by adding “time-dependent ß-blocker ”-by-“mutation category” interaction term to the multivariable Cox model. We have adjusted for the effect of potential lack of independence between subjects using the robust sandwich estimator for family membership.17-18 This robust sandwich covariance estimator is used with correlated data. Correlations among data points in the Cox model lead to underestimation of the standard error used in significance testing, whereas the robust estimator uses an inflated variance estimate, taking family membership or other clustering connection into account. All significant predictors of life threatening event risk remained significant using or not using this robust measure of variance. It should be noted that there is seldom more than a single observed outcome (ACA/SCD) per family (only 7% of families had more than 1 event), thus the standard model-based SE's, CI's, and p-values and likelihood ratio tests are valid.16
We have carried out the following additional secondary analyses: 1) Including the biophysical function of the mutations (categorized as dominant-negative, haploinsufficiency, and unknown) as a covariate in the model, 2) excluding the large subgroup of patients with V254M mutations, and 3) including appropriate ICD shocks in the composite end point. Also, to assess whether fuller adjustment for family membership was important, regression models which included shared frailty terms (i.e. random effects) for family were fit.
The statistical software used for the analyses was SAS version 9.20 (SAS Institute Inc, Cary, NC). For the fitting of models with frailty terms, the software used was Splus 7.0.0 for Sun SPARC. For electrophysiology and biochemistry experiments One-way ANOVA followed by Tukey Post Hoc test was applied for the assessment of statistical significance for multiple group comparison by using SPSS Statistics (IBM). Unpaired Student t-test was used for two group comparison. A 2-sided 0.05 significance level was used for hypothesis testing.
Results
Study sample
The spectrum of mutations as categorized by location and type and their respective number of carriers are presented in Table 1S-A (Supplementary Appendix). The location and frequency of missense mutations is presented diagrammatically in Figure 1. Of the total 99 different KCNQ1 mutations identified, 77 were missense mutations and 22 non-missense mutations. Missense mutations were further categorized according to their location: there were 28 different mutations in C-terminus or N-terminus regions (26 in C-terminus), 34 mutations in membrane spanning regions, and 15 mutations in the C-loop regions (8 in S2-S3 loop and 7 in S4-S5 loop). The clinical characteristics of patients in the 4 mutation location/type subgroups are presented in Table 1. Of the 860 study subjects, 20 percent had C/N terminal-missense mutations, 44 percent had membrane spanning-missense mutations, 15 percent had C-loop-missense mutations, and 22 percent had non-missense mutations. Patients with C-loop-missense mutations exhibited the longest QTc interval at enrollment, were treated with β-blockers more frequently during follow-up, and had a higher frequency of cardiac events of any type, including syncope, ACA, and LQTS death, as compared with the other mutation subgroups. The clinical characteristics of probands only are presented in Table 1S-B (Supplementary Appendix).
Table 1. Demographic and clinical characteristics.
| Parameter | Missense | Non- missense | ||
|---|---|---|---|---|
|
| ||||
| C/N-Term | membrane spanning | C-loops | ||
| Patients, n (%) | 172 (20.0) | 376 (43.7) | 125 (14.5) | 187 (21.7) |
| Female, n (%) | 94 (54.7) | 221 (58.8) | 70 (56.0) | 119 (63.6) |
| Age at enrollment, years, median (interquartile range) | 21 (9-41) | 25 (11-41) | 19 (5-35) | 21 (11-39) |
| QTc at enrollment, msec, mean±SD* | 467±63 | 480±51 | 503±58 | 470±41 |
| QTc at enrollment ≥ 500 msec, n (%) | 39/154 (25.3) | 94/312 (30.1) | 47/102 (46.1) | 41/165 (24.8) |
| Therapy during follow-up | ||||
| ß-blockers, n (%) | 64 (37.0) | 167 (44.4) | 63 (50.4) | 74 (39.8) |
| Pacemaker, n (%) | 2 (1.2) | 7 (1.9) | 1 (0.8) | 4 (2.2) |
| Defibrillator, n (%) | 6 (3.5) | 25 (6.6) | 10 (8.0) | 13 (7.0) |
| Sympathectomy, n (%) | 0 (0.0) | 1 (0.3) | 1 (0.8) | 1 (0.5) |
| Cardiac event during follow-up | ||||
| Syncope, n (%) | 52 (30.1) | 122 (32.4) | 69 (55.2) | 46 (24.7) |
| Aborted cardiac arrest, n (%) | 6 (3.5) | 10 (2.7) | 8 (6.4) | 3 (1.6) |
| Sudden cardiac death, n (%) | 18 (10.4) | 29 (7.7) | 24 (19.2) | 13 (7.0) |
| Any cardiac event, n (%) | 63 (36.4) | 144 (38.3) | 84 (67.2) | 55 (29.6) |
One hundred twenty seven LQT1 mutation carriers did not have an ECG for QTc measurement, of them 58 (46%) died suddenly at a young age without a documented ECG.
Clinical outcome of patients according to mutation location and type
There were 105 first life threatening cardiac events (27 first ACA events and 78 first LQTS-related SCD events) among the 860 study patients. Patients were enrolled to the registry between years 1978 - 2007 with follow up through 2008; the last reported life threatening cardiac event occurred in 2005. Figure 2 presents the cumulative probabilities of first life threatening cardiac events in the 4 subgroups. There was a significantly higher event rate in the C-loop-missense subgroup as compared with the other 3 subgroups (p log rank < 0.001). Thus, at age 40 years, the rate of life threatening cardiac events was 33 percent in patients with C-loop-missense mutations as compared with ≤ 16 percent in patients with other mutations.
Figure 2.
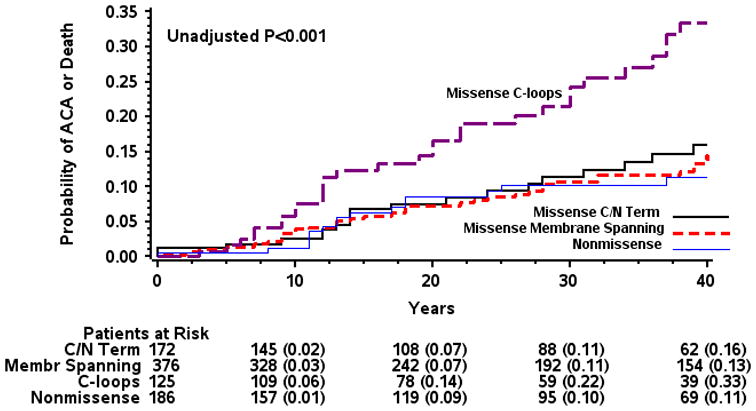
Kaplan-Meier estimates of cumulative probability of life threatening cardiac events by mutation location and type. ACA= aborted cardiac arrest. LQTS= long QT syndrome. The number in parentheses reflects the cumulative event rate at that point in time.
The findings from the multivariable analysis for the end point of a first life threatening cardiac event are shown in Table 2. Notably, the adjusted hazard ratio for C-loop-missense vs. non-missense mutation was 2.75 (P=0.009), and there was no statistically significant difference in the risk among the other mutation location/type subgroups.
Table 2.
Multivariable analysis: Risk factors for aborted cardiac arrest or sudden cardiac death.
| Hazard ratio | 95% CI | P Value | ||
|---|---|---|---|---|
|
|
||||
| Gender/age | ||||
| Males (vs. females) age < 13 yrs | 1.93 | 1.08-3.45 | 0.03 | |
| Males (vs. females) age 13 to 40 yrs | 1.13 | 0.67-1.91 | 0.65 | |
| QTc ≥ 500 msec (vs. QTc<500) | 3.55 | 1.83-6.89 | <0.001 | |
| Mutation type and location | ||||
| Cytoplasmic loops (missense) vs. non-missense | 2.75 | 1.29-5.86 | 0.009 | |
| C/N Terminus (missense) vs. non-missense | 1.47 | 0.64-3.39 | 0.37 | |
| Membrane spanning (missense) vs. non-missense | 0.85 | 0.41-1.78 | 0.67 | |
The models are adjusted for sex X age, corrected QT category (including missing QT), mutation type and location category, and time-dependent ß-blocker treatment.
There were 127 LQT1 mutation carriers without available ECG data; 58 (46%) of them died suddenly at a young age without a documented ECG. The hazard ratio (95% CI) for missing QTc vs. available QTc = 10.49 (95% CI 6.61-16.66), p<0.001.
Secondary confirmatory analyses (Table 2S, supplementary appendix) showed that patients with C-loop-missense mutations had an adjusted hazard ratio of 2.74 (95 percent confidence interval 1.68 to 4.46 [P<0.001]) for life threatening events as compared with patients with other mutations. The results were consistent when the biophysical function of the mutations was added as a covariate to the multivariable model. In order to show that our results do not depend on the C-loop V254M mutation, which is the most common mutation in the C-loop subgroup (Table 1S), accounting for 50% of C-loop patients, we have carried out an additional separate analysis excluding patients who carried this mutation. Results were consistent, with patients with C-loop mutations having a greater risk for life threatening events, demonstrating that our findings were independent of this mutation. The results were also consistent after inclusion of appropriate ICD shocks in the composite end point (adjusted hazard ratio for C-loop-missense mutations vs. non-missense mutations of 2.64 [95 percent confidence interval 1.64 to 4.23; P<0.001]), and after stratifying patients by enrolling center. To assess whether fuller adjustment for family membership was important, regression models which included frailty terms (i.e. random effects) for family were fit in the multivariable Cox models. Models with Gamma and Gaussian frailty terms were fit and the C-loop term had a consistent effect size with the original models while remaining statistically significant. Further, in both these models the frailty terms were non-significant. The consistency of the results provides further support for the higher risk associated with C-loop mutations.
ß-blocker therapy
In the current study the effect of β-blocker therapy on the risk of life-threatening events among the different mutation subgroups was assessed as a time dependent covariate (i.e. β-blockers were given to patients at different time-points during follow-up, and this information was taken into account in the multivariable models). Multivariable analysis showed a significant differential effect of ß-blocker therapy on the outcome of patients with C-loop-missense mutations as compared with those who had other mutations (Table 3). ß-blocker therapy was associated with a significant 88% reduction (P=0.02) in the risk of life threatening events among patients with C-loop-missense mutations, whereas the benefit of ß-blocker therapy was significantly attenuated among patients with other mutations in the KCNQ1 channel (adjusted hazard ratio of 0.82 [P=0.68]; P-value for treatment-by-mutation-location/type interaction = 0.04).
Table 3. Multivariable analysis: response to ß-blocker therapy.
| β -blocker vs. no β -blocker therapy | Hazard ratio | 95% CI | P Value |
|---|---|---|---|
| All LQT1 patients | 0.49 | 0.19-1.23 | 0.13 |
| LQT1 patients with C-loop-missense mutations* | 0.12 | 0.02-0.73 | 0.02 |
| LQT1 patients with other mutations (non-[C-loop-missense] mutations* | 0.82 | 0.31-2.13 | 0.68 |
The models are adjusted for sex X age, corrected QT category (including missing QT), mutation type and location category, and time-dependent ß-blocker treatment.
p for interaction for mutation location-by-β blocker treatment =0.04
Consistent with those findings, the rate of ACA or SCD (Figure 3) was lowest among patients with C-loop-missense mutations who were treated with β-blockers and highest among patients with C-loop-missense mutations who were not treated with β-blockers (0.17 vs. 1.11 per 100 patient-years, respectively), whereas patients with other mutations in the KCNQ1 channel exhibited intermediate and similar rates of life-threatening events with- and without β-blocker therapy (0.36 and 0.38 per 100 patient-years, respectively [Figure 3]).
Figure 3.
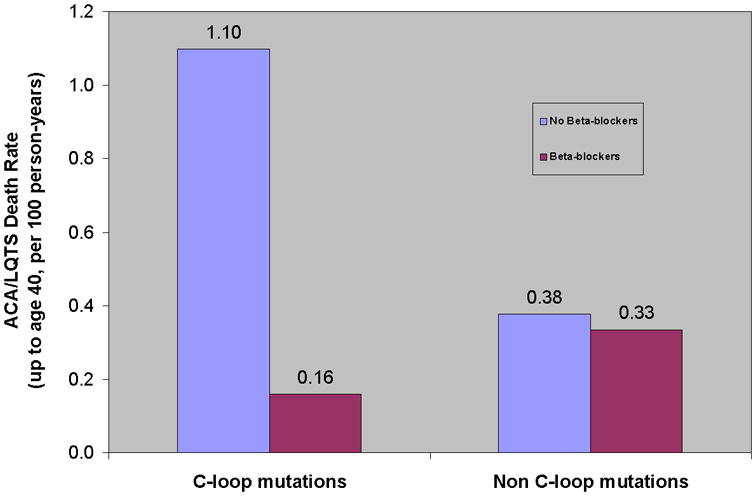
Risk for life threatening cardiac events by mutation location and ß-blocker treatment. 63 of the 125 (50%) subjects with C-loop missense mutations were treated with beta-blockers during a mean follow-up of 26.2 years; 305 of the 735 (42%) subjects with non-C-loop-missense mutations were treated by beta-blockers during a mean follow-up of 27.5 years. Event rates per 100 person-years were calculated by dividing the number of events during the period of β-blocker therapy or the absence of β-blocker therapy by person-years, and multiplying the results by 100. ACA= aborted cardiac death. LQTS= long QT syndrome.
In addition, we have repeated analysis for only patients who were treated by β-blockers (at any point in time) and found consistent results, that there is differential response to ß-blocker therapy depending on mutation location. ß -blocker vs. no ß -blocker therapy in patients with C-loop mutations HR = 0.15 (95% CI 0.01-1.73, p=0.13) and ß -blocker vs. no ß -blocker therapy in patients with non C-loop mutations HR = 1.89 (95% CI 0.54-6.63, p=0.32) with P-value for treatment-by-mutation-location/type interaction = 0.028.
Cellular Expression Studies
In order to understand the mechanism underlying the increase in risk associated with C-loop mutations we measured channel basal function and regulation in eight mutant channels associated with LQT1, three in the membrane spanning domains (T312I, G168R and S225L), four located in C-loops (G189R, R190Q, R243C and V254M) and one in the C-terminus (R555C) (Figure 4A). Wild-type and mutant subunits were co-expressed for all experiments. Basal channel current was decreased for all mutations studied when compared to wild type subunits (Figure 4B). In addition, because activation by PKA is thought to be particularly important for IKs function and to underlie arrhythmogenesis in LQT1,4, 19-20 we measured the effect of the PKA activator forskolin. All C-loop mutations tested showed dramatically impaired response to forskolin, whereas the other mutations showed a strong activation by forskolin, as did the wild type KCNQ1 channel (Figure 4C and D).
Figure 4.
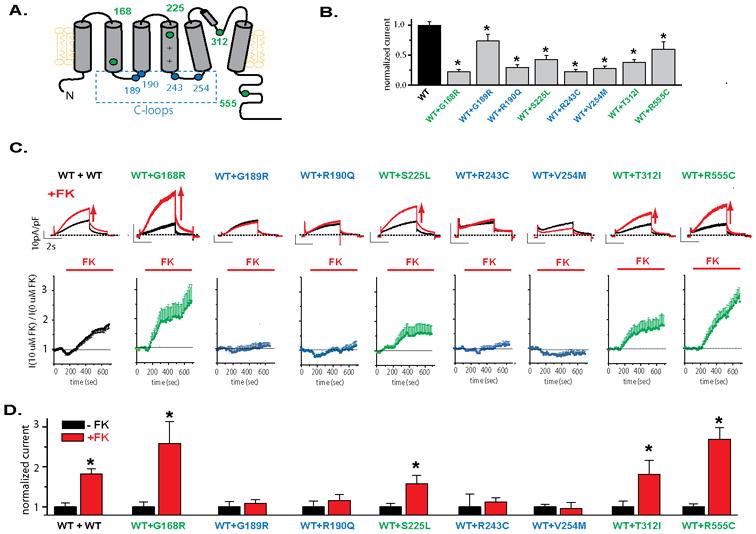
Regulation of LQT1 mutant channels by PKA. A. Schematic representation of location of the mutations used in the study. B: Effect of each of the mutations studied in basal non stimulated cell currents. Average current measured for cells expressing WT and mutant subunits measured at +40 mV after 3sec depolarization. KCNQ1 and KCNE1 subunit were expressed at a ratio 0.5 KCNQ1WT: 0.5 KCNQ1mut : 1 KCNE1 or 0.5 KCNQ1WT : 0.5vector : 1 KCNE1 for wild-type haploinsuficient channels, *p<0.05 compared to WT. C: Top panel: typical ion channel current measured before and after 10 min application of the PKA activator forskolin (FK, 10 μM) for wild-type (WT) and WT and mutant subunits co-expressed. Scale bars in each panel are 10pA/pF and 2sec. Scale bars are the same for all constructs. Bottom panel: time course of current regulation by forskolin measured at +20 mV after 3sec depolarization for channels formed by either WT or mutant co-expressed with WT subunits, as indicated. Current was normalized to current in the absence of forskolin application. KCNQ1 and KCNE1 subunits were expressed at a ratio 0.5 KCNQ1WT : 0.5 KCNQ1mut : 1 KCNE1 or 1 KCNQ1WT : 1 KCNE1 for wild-type channels. Currents were activated by 4sec depolarizing steps to +20 mV from a -80mV holding potential. These were followed by a step to −20mV. D: Summary data for experiments done as in C, *p<0.05 compared to the current before stimulation (black bar) in each group.
Discussion
The present analysis among 860 LQT1 patients with a wide range of mutations in the KCNQ1 channel provides several important implications regarding risk assessment and management in this study sample: (1) patients with missense mutations located in the C-loops exhibit the highest risk for life threatening cardiac events, independently of clinical and electrocardiographic variables; (2) β-blocker therapy is associated with a pronounced reduction in the risk of ACA or SCD among carriers of missense mutations in the C-loop, whereas the benefit of this mode of medical therapy is significantly attenuated in LQT1 patients with other mutations; (3) expression studies of C-loop mutations suggest that an impaired regulation by PKA is the mechanism underlying the increased risk for cardiac events independent of patient QTc, and may explain the pronounced response to medical therapy with β-blockers among patients with C-loop mutation carriers.
We have recently shown that patients with mutations located in the transmembrane region have a significantly higher rate of cardiac events than those with mutations located in the C-terminus.8 In addition, mutations in the transmembrane domain were suggested to be associated with a greater prolongation of the QTc during exercise.21 The present findings confirm previous work, indicating mutations in the transmembrane region are associated with higher risk, but suggest that within the transmembrane region there are distinct functional domains, with the C-loops (but not in the membrane spanning domain) being associated with increased risk for life threatening cardiac events compared with other mutations in the KCNQ1 channel. The S2-S3 and S4-S5 C-loops have been previously suggested to have an important functional role in modifying the function of voltage gated potassium channels.22 In particular for IKs, the S4-S5 loop has been suggested to mediate a functional interaction with the auxiliary KCNE1 subunits.23 Most recently, LQT1 mutations in C-loops, when expressed in the absence of wild-type subunits, were suggested to affect adrenergic channel regulation.9 Our results showed that even when expressed in the presence of wild-type subunits, C-loop mutations can dramatically affect channel regulation. Consistent with our results, induced pluripotent stem cells differentiated into cardiomyocytes from a patient carrying R190Q were recently shown to lack adrenergic regulation of their IKs current.24 Also consistent with our results, for haploinsuficient mutations, not tested here, a simple lack of mutant subunit expression is expected to maintain normal adrenergic regulation, contributing to the milder phenotype of these mutations.8 It is conceivable that a decrease in channel regulation, as observed for the C-loop mutations, will lead to an increase in the burden of the mutation during adrenergic stimulus. The increase in cardiac risk associated with C-loop mutations is independent of traditional clinical variables; this can be explained by a blunted PKA-mediated activation, because QTc is generally measured at rest. Thus, our results suggest that exercise may exacerbate the QTc prolongation for C-loop mutants. It has been recently suggested that the mutation KNCQ1(A341V) also caused an impairment in beta-adrenergic activation.25 This mutation is located at the end of the S6 domain, a region suggested to interact with the S4-S5 loop.26 It is possible that other mutations causing similar functional impairment as the c-loop mutations may also carry the increased cardiac risk and beta-blocker efficacy.
Current guidelines recommend empiric therapy with β-blockers in all LQTS patients.27 The present study shows for the first time, a mutation-specific response to β-blocker therapy in type-1 long QT syndrome, demonstrating that β-blockers were associated with a significantly greater reduction in the risk of life-threatening cardiac events among patients with mutations located in the C-loops as compared with all other mutations. It is conceivable that during β-adrenergic stimulation patients with mutations located in the C-loops have an unopposed increase in inward Ca2+ currents and prolongation of repolarization due to blunted PKA-mediated activation of IKs.5 β-blockers may decrease these unopposed inward Ca2+ currents, shorten repolarization and reduce the risk of ventricular arrhythmias,28 whereas patients with other mutations do not exhibit such an effect.
Study limitations
Upon enrollment in the registry clinical history was obtained, thus follow-up data in the current study comprised historical data from birth to enrollment and prospective information collected at yearly intervals after enrollment.
The International LQTS Registry records therapies that are prescribed at the discretion of the treating physicians to enrolled subjects, and therefore β-blocker administration was not randomized. However, since the patient's physician would have been blind to whether the patient had a C-loop mutation the interaction of this with β-blocker therapy is still compelling. Prior studies from the International LQTS Registry have shown that β-blocker therapy is associated with a significant reduction in the risk of cardiac events in LQTS patients. However, the present study is the first to assess the benefit of β-blocker therapy for the reduction in the risk of ACA or SCD among LQT1 patients. We have shown that β-blocker therapy is associated with a significant 88% (p=0.02) reduction in the risk of life-threatening cardiac events among LQT1 carriers of the higher risk C-loop mutations. Risk reduction associated with β-blocker therapy in the total study sample, and among carriers of the low-risk non-C-loop mutations did not reach statistical significance. The lack of a significant β-blocker effect may be due to sample size limitation and a more limited number of events among carriers of lower risk mutations. Thus, lower-risk patients should still be treated with β-blocker therapy according to guidelines 27 since the cumulative probability of ACA or SCD from birth through age 40 years among patients with non-C-loop-missense mutations was still considerable (between 11 and 16 percent). These limitations also suggest that further studies, in independent population, are needed before extrapolating them to clinical practice.
The present results, derived from LQTS families enrolled in the Registry, may be confounded by familial factors such as ethnicity. To minimize bias we: 1) adjusted for family membership in the multivariable models; and 2) carried out a secondary analysis, in which additional adjustment was made for proband status. These analyses yielded similar results, further supporting the consistency of our findings. Of the 127 LQT1 mutation carriers without available ECG data, 58 (46%) died suddenly at a young age without a documented ECG. To minimize this bias related to exclusion of higher risk patients, all multivariable analyses included adjustment for a QTc missing category in addition to the QTc>500 ms category.
Channel current and response to Forskolin were analyzed for 8 mutations out of 99 mutation types observed in this study, but the robust findings in these expression studies do strongly support our suggested mechanism. Experimental data was performed at room temperature, and results may be different at physiological temperature.
Conclusions
We used a combination of clinical analysis and cellular electrophysiology experiments to investigate the molecular determinants and mechanism underlying the clinical outcomes of a large cohort of subjects having a spectrum of KCNQ1 mutations categorized by their code type and location. Patients with KCNQ1 missense mutations located in the cytoplasmic loops had a significantly greater risk for life threatening cardiac events and gained grater benefit when treated with ß-blockers as compared with patients having other KCNQ1 missense or non-missense mutations independently of clinical risk factors. We suggest that a combination of decrease in basal function and altered adrenergic regulation of the IKs channel underlies the increased cardiac risk in this subgroup of patients. Our results highlight the importance of understanding the molecular determinants and mechanisms underlying arrhythmogenesis to identify cardiac risk factors for LQT1 patients.
Supplementary Material
Acknowledgments
This research was carried out while Dr. Alon Barsheshet was a Mirowski-Moss Career Development Awardee at the University of Rochester Medical Center, Rochester, NY. The authors thank Ms. Jaime Sorenson, Ms. Nobiru Suzuki and Ms. Mehreen Butt for their technical assistance.
Funding Sources: This work was supported by research grants HL-33843 and HL-51618 from the National Institutes of Health, Bethesda, Md and by a research grant from BioReference Labs to the Heart Research Follow-Up Program in support of the LQTS Registry.
Footnotes
Conflict of Interest Disclosures: Dr. Ackerman has a consulting relationship and license agreement/royalty arrangement with Transgenomic. Dr. Kaufman receives research support from Cambridge Heart. Dr. O-Uchi is the recipient of American Heart Association (AHA) Postdoctoral Fellowship grant (09POST2310079), a Foreign Study Grant Award of Kanae Foundation (Tokyo, Japan) and Irisawa Memorial Promotion Award for Young Physiologists (The Physiological Society of Japan). Dr. Kanters is a recipient of the Danish Council for Strategic Research grant. Dr. Shimizu is a recipient of research grant from the Ministry of Health, Labour and Welfare, Japan. Dr. Moss is a recipient of NIH grants and receives research support from BioReference Labs. Dr. Wilde has a consulting relationship with Transgenomic.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Goldenberg I, Moss AJ. Long QT syndrome. J Am Coll Cardiol. 2008;51:2291–2300. doi: 10.1016/j.jacc.2008.02.068. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Denjoy I, Guicheney P, Breithardt G, Keating MT, Towbin JA, Beggs AH, Brink P, Wilde AA, Toivonen L, Zareba W, Robinson JL, Timothy KW, Corfield V, Wattanasirichaigoon D, Corbett C, Haverkamp W, Schulze-Bahr E, Lehmann MH, Schwartz K, Coumel P, Bloise R. Genotype-phenotype correlation in the long-QT syndrome: Gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 3.Walsh KB, Kass RS. Regulation of a heart potassium channel by protein kinase A and C. Science. 1988;242:67–69. doi: 10.1126/science.2845575. [DOI] [PubMed] [Google Scholar]
- 4.Marx SO, Kurokawa J, Reiken S, Motoike H, D'Armiento J, Marks AR, Kass RS. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science. 2002;295:496–499. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- 5.Shimizu W, Antzelevitch C. Differential effects of beta-adrenergic agonists and antagonists in LQT1, LQT2 and LQT3 models of the long QT syndrome. J Am Coll Cardiol. 2000;35:778–786. doi: 10.1016/s0735-1097(99)00582-3. [DOI] [PubMed] [Google Scholar]
- 6.Goldenberg I, Bradley J, Moss A, McNitt S, Polonsky S, Robinson JL, Andrews M, Zareba W. Beta-blocker efficacy in high-risk patients with the congenital long-QT syndrome types 1 and 2: Implications for patient management. J Cardiovasc Electrophysiol. 2010;21:893–901. doi: 10.1111/j.1540-8167.2010.01737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jespersen T, Grunnet M, Olesen SP. The KCNQ1 potassium channel: From gene to physiological function. Physiology. 2005;20:408–416. doi: 10.1152/physiol.00031.2005. [DOI] [PubMed] [Google Scholar]
- 8.Moss AJ, Shimizu W, Wilde AA, Towbin JA, Zareba W, Robinson JL, Qi M, Vincent GM, Ackerman MJ, Kaufman ES, Hofman N, Seth R, Kamakura S, Miyamoto Y, Goldenberg I, Andrews ML, McNitt S. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation. 2007;115:2481–2489. doi: 10.1161/CIRCULATIONAHA.106.665406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matavel A, Medei E, Lopes CM. PKA and PKC partially rescue long QT type 1 phenotype by restoring channel-PIP2 interactions. Channels. 2010;4:3–11. doi: 10.4161/chan.4.1.10227. [DOI] [PubMed] [Google Scholar]
- 10.Jons C, J OU, Moss AJ, Reumann M, Rice JJ, Goldenberg I, Zareba W, Wilde AA, Shimizu W, Kanters JK, McNitt S, Hofman N, Robinson JL, Lopes CM. Use of mutant-specific ion channel characteristics for risk stratification of long QT syndrome patients. Sci Transl Med. 2011;3:76ra28. doi: 10.1126/scitranslmed.3001551. [DOI] [PubMed] [Google Scholar]
- 11.Jhun BS, J OU, Wang W, Ha CH, Zhao J, Kim JY, Wong C, Dirksen RT, Lopes CM, Jin ZG. Adrenergic signaling controls RGK-dependent trafficking of cardiac voltage-gated L-type Ca2+ channels through PKD1. Circ Res. 2012;110:59–70. doi: 10.1161/CIRCRESAHA.111.254672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hobbs JB, Peterson DR, Moss AJ, McNitt S, Zareba W, Goldenberg I, Qi M, Robinson JL, Sauer AJ, Ackerman MJ, Benhorin J, Kaufman ES, Locati EH, Napolitano C, Priori SG, Towbin JA, Vincent GM, Zhang L. Risk of aborted cardiac arrest or sudden cardiac death during adolescence in the long-QT syndrome. JAMA. 2006;296:1249–1254. doi: 10.1001/jama.296.10.1249. [DOI] [PubMed] [Google Scholar]
- 13.Goldenberg I, Moss AJ, Peterson DR, McNitt S, Zareba W, Andrews ML, Robinson JL, Locati EH, Ackerman MJ, Benhorin J, Kaufman ES, Napolitano C, Priori SG, Qi M, Schwartz PJ, Towbin JA, Vincent GM, Zhang L. Risk factors for aborted cardiac arrest and sudden cardiac death in children with the congenital long-QT syndrome. Circulation. 2008;117:2184–2191. doi: 10.1161/CIRCULATIONAHA.107.701243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Locati EH, Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Lehmann MH, Towbin JA, Priori SG, Napolitano C, Robinson JL, Andrews M, Timothy K, Hall WJ. Age- and sex-related differences in clinical manifestations in patients with congenital long-QT syndrome: Findings from the international LQTS registry. Circulation. 1998;97:2237–2244. doi: 10.1161/01.cir.97.22.2237. [DOI] [PubMed] [Google Scholar]
- 15.Zareba W, Moss AJ, Sheu G, Kaufman ES, Priori S, Vincent GM, Towbin JA, Benhorin J, Schwartz PJ, Napolitano C, Hall WJ, Keating MT, Qi M, Robinson JL, Andrews ML. Location of mutation in the KCNQ1 and phenotypic presentation of long QT syndrome. J Cardiovasc Electrophysiol. 2003;14:1149–1153. doi: 10.1046/j.1540-8167.2003.03177.x. [DOI] [PubMed] [Google Scholar]
- 16.Sauer AJ, Moss AJ, McNitt S, Peterson DR, Zareba W, Robinson JL, Qi M, Goldenberg I, Hobbs JB, Ackerman MJ, Benhorin J, Hall WJ, Kaufman ES, Locati EH, Napolitano C, Priori SG, Schwartz PJ, Towbin JA, Vincent GM, Zhang L. Long QT syndrome in adults. J Am Coll Cardiol. 2007;49:329–337. doi: 10.1016/j.jacc.2006.08.057. [DOI] [PubMed] [Google Scholar]
- 17.Lin DY, Wei LJ. The robust inference for the proportional hazards model. J Am Stat Assoc. 1989;84:1074–1078. [Google Scholar]
- 18.Therneau TM, Grambsch PM. Modeling survival data: Extending the cox model. New York, NY: Springer-Verlag; 2000. [Google Scholar]
- 19.Terrenoire C, Clancy CE, Cormier JW, Sampson KJ, Kass RS. Autonomic control of cardiac action potentials: Role of potassium channel kinetics in response to sympathetic stimulation. Circ Res. 2005;96:e25–34. doi: 10.1161/01.RES.0000160555.58046.9a. [DOI] [PubMed] [Google Scholar]
- 20.Potet F, Scott JD, Mohammad-Panah R, Escande D, Baro I. Akap proteins anchor CAMP-dependent protein kinase to KVLQT1/ISK channel complex. Am J Physiol Heart Circ Physiol. 2001;280:H2038–2045. doi: 10.1152/ajpheart.2001.280.5.H2038. [DOI] [PubMed] [Google Scholar]
- 21.Shimizu W, Horie M, Ohno S, Takenaka K, Yamaguchi M, Shimizu M, Washizuka T, Aizawa Y, Nakamura K, Ohe T, Aiba T, Miyamoto Y, Yoshimasa Y, Towbin JA, Priori SG, Kamakura S. Mutation site-specific differences in arrhythmic risk and sensitivity to sympathetic stimulation in the LQT1 form of congenital long QT syndrome: Multicenter study in japan. J Am Coll Cardiol. 2004;44:117–125. doi: 10.1016/j.jacc.2004.03.043. [DOI] [PubMed] [Google Scholar]
- 22.Isacoff EY, Jan YN, Jan LY. Putative receptor for the cytoplasmic inactivation gate in the shaker K+ channel. Nature. 1991;353:86–90. doi: 10.1038/353086a0. [DOI] [PubMed] [Google Scholar]
- 23.Franqueza L, Lin M, Shen J, Splawski I, Keating MT, Sanguinetti MC. Long QT syndrome-associated mutations in the S4-S5 linker of KVLQT1 potassium channels modify gating and interaction with mink subunits. J Biol Chem. 1999;274:21063–21070. doi: 10.1074/jbc.274.30.21063. [DOI] [PubMed] [Google Scholar]
- 24.Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flugel L, Dorn T, Goedel A, Hohnke C, Hofmann F, Seyfarth M, Sinnecker D, Schomig A, Laugwitz KL. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010;363:1397–1409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- 25.Spatjens R, Heijman J, Lentink V, Volders PGA. Phosphomimetic KCNQ1 substitution S27D rescues loss of cAMP-dependent IKs upregulation by the long-QT1 mutation A341V. Heart Rhythm. 2011;8:S–180. [Google Scholar]
- 26.Choveau FS, Rodriguez N, Ali FA, Labro AJ, Rose T, Dahimene S, Boudin H, Le Henaff C, Escande D, Snyders DJ, Charpentier F, Merot J, Baro I, Loussouarn G. KCNQ1 channels voltage dependence through a voltage-dependent binding of the S4-S5 linker to the pore domain. J Biol Chem. 2011;286:707–716. doi: 10.1074/jbc.M110.146324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, Gregoratos G, Klein G, Moss AJ, Myerburg RJ, Priori SG, Quinones MA, Roden DM, Silka MJ, Tracy C, Smith SC, Jr, Jacobs AK, Adams CD, Antman EM, Anderson JL, Hunt SA, Halperin JL, Nishimura R, Ornato JP, Page RL, Riegel B, Blanc JJ, Budaj A, Dean V, Deckers JW, Despres C, Dickstein K, Lekakis J, McGregor K, Metra M, Morais J, Osterspey A, Tamargo JL, Zamorano JL. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: A report of the american college of cardiology/american heart association task force and the european society of cardiology committee for practice guidelines (writing committee to develop guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death): Developed in collaboration with the european heart rhythm association and the heart rhythm society. Circulation. 2006;114:e385–484. doi: 10.1161/CIRCULATIONAHA.106.178233. [DOI] [PubMed] [Google Scholar]
- 28.Huffaker R, Lamp ST, Weiss JN, Kogan B. Intracellular calcium cycling, early afterdepolarizations, and reentry in simulated long QT syndrome. Heart Rhythm. 2004;1:441–448. doi: 10.1016/j.hrthm.2004.06.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.