

Abstract
The cardinal symptom of migraine is headache pain. In this paper we review the neurobiology of this pain as it is currently understood. In recent years, we discovered that the network of neurons that sense pain signals from the dura changes rapidly during the course of a single migraine attack and that the treatment of an attack is a moving target. We found that if the pain is not stopped within 10–20 minutes after it starts, the first set of neurons in the network, those located in the trigeminal ganglion, undergo molecular changes that make them hypersensitive to the changing pressure inside the head, which explains why migraine headache throbs and is worsened by bending over and sneezing. We found that if the pain is not stopped within 60–120 minutes, the second group of neurons in the network, those located in the spinal trigeminal nucleus, undergoes molecular changes that convert them from being dependent on sensory signals they receive from the dura by the first set of neurons, into an independent state in which they themselves become the pain generator of the headache. When this happens, patients notice that brushing their hair, taking a shower, touching their periorbital skin, shaving, wearing earrings, etc become painful, a condition called cutaneous allodynia. Based on this scenario, we showed recently that the success rate of rendering migraine patients pain-free increased dramatically if medication was given before the establishment of cutaneous allodynia and central sensitization. The molecular shift from activity-dependent to activity-independent central sensitization together with our recent conclusion that triptans have the ability to disrupt communications between peripheral and central trigeminovascular neurons (rather than inhibiting directly peripheral or central neurons) explain their clinical effects. Both our clinical and pre-clinical findings of the last five years point to possible short- and long-term advantages in using an early-treatment approach in the treatment of acute migraine attacks.
Keywords: Migraine, headache, pain, sensitization
1. Sensitization
1.1. Peripheral sensitization
Peripheral sensitization is thought to be a major contributor to hypersensitivity in many painful syndromes including migraine headaches [2,51,91]. It generally refers to a state where primary afferent nociceptive neurons exhibit increased responsiveness to external mechanical or thermal stimuli at the original site of inflammation or injury. Such changes can be manifested as a novel response to previously ineffective stimulus intensities, indicating decreased activation thresholds [20, 52,61,81,104], and increased response magnitude to suprathreshold stimuli either with or without a noticeable change in threshold [2,16,30,92] In addition to marked changes in their stimulus response properties, peripheral sensitization can also be manifested as an increased level of ongoing discharge (i.e. spontaneous activity) in the absence of externally applied stimuli.
Among the common symptoms of peripheral sensitization during migraine are the throbbing of the headache and its aggravation during routine physical activities that increase intracranial pressure such as coughing and bending over [7,74]. Such intracranial hypersensitivity involves the sensitization of nociceptors that innervate the meninges [91]. Accordingly, fluctuations in intracranial pressure [19] associated with normal vascular pulsation (4–10 mmHg), as well as those associated with bending over or coughing (4–25 mmHg), effectively activate meningeal nociceptors during migraine, when they are sensitized, but not in the absence of migraine, when they are not sensitized.
1.2. Inflammatory mediators of peripheral sensitization
A large number of chemical mediators produced at the site of tissue injury and inflammation can promote the excitation and sensitization of nociceptors. Mediators such as bradykinin, histamine, serotonin (5-HT), and prostaglandin E2, (PGE2) have been shown to produce both excitation and mechanical sensitization of somatic [89] and meningeal nociceptors [52,91]. Other inflammatory mediators known to promote peripheral sensitization are cytokines, most notably interleukins 1, 6 and 8 (IL-1, IL-6, IL-8) and tumor necrosis factor-alpha (TNF-alpha) [67,79]. These mediators are believed to promote nociceptor sensitization through the endogenous release of eicosanoids and sympathetic amines [79]. Additional inflammatory mediators proposed to promote peripheral sensitization include protons, proteases and nitric oxide. Increased levels of protons (acidic pH) found in inflamed tissues, produce not only activation and sensitization of meningeal nociceptors [91], but also enhance the effects of other inflammatory mediators [89]. Inflammatory proteases, especially tryptase and trypsins, activate protease-activated receptors (PARs) on nociceptors, most notably PAR-2 [31]. Nitric oxide has been shown to produce local inflammation within the meninges [76], sensitize meningeal nociceptors [53], and induce headache or migraine in patients [68].
1.3. Cellular mechanisms of peripheral sensitization
Most sensitizing agents activate receptors that are coupled to second-messenger cascades which, in turn, modulate voltage-gated ion channels. Other potential targets for the actions of sensitizing agents on nociceptors may also include direct action on sensory transduction elements. Mechanical and thermal sensitivity can be modulated independently in individual nociceptors, suggesting the existence of separate, possibly multiple, transduction mechanisms [5,71]. For example, increased thermal skin sensitivity can be mediated by the transient receptor potential ion channel 1 (TRPV1), a transducer of noxious heat [72,96] when its threshold is lowered by bradykinin, PAR-2 agonist, and protons [18, 78,93]. The mechanism underlying increased mechanical sensitivity remains largely unknown as transducers of noxious mechanical stimuli are yet to be identified.
Peripheral sensitization can also be promoted by changes in the properties of voltage-gated ion channels, such as the TTX-resistant sodium channel (TTX-R). Inflammatory agents such as PGE2 and 5-HT are thought to sensitize sensory neurons by modulating TTX-R sodium currents [28] through activation of the cAMP-PKA second messenger cascade [29]. Such action may be involved in sensitization of mechanosensitive meningeal nociceptors, as they express the TTX-R channels [90] and are sensitized by the cAMP-PKA cascade [52]. The cAMP-PKA cascade is also likely to be involved in mechanical sensitization through the suppression of the sustained (delayed rectifier) outward K+ current that is thought to modulate the firing threshold [24,25] and the enhancement of Ih, the hyperpolarization-activated cation current that is thought to facilitate repetitive firing [37]. Another second messenger cascade that may promote mechanical sensitization is the cGMP-PKG cascade that is activated by nitric oxide. The sensitizing action of nitric oxide on meningeal nociceptors may involve the activation of this cascade probably through the facilitation of Ca+2-activated potassium (BK) channels [48].
The proximate factors that cause local release of sensitizing chemicals during migraine remain unknown. One presumed factor is cortical spreading depression –a slowly propagating wave of neural inhibition and excitation associated with extracellular release of excitatory agents such as potassium and glutamate. Bolay et al. [8] have shown that cortical spreading depression activates the trigeminovascular system. One of the potential consequences of sensory fiber activation is the release of neuropeptides such as substance P (SP) and calcitonin gene–related peptide (CGRP) from the peripheral terminals of meningeal nociceptors which, in turn, promotes vasodilatation and plasma extravasation [15,50,85]. CGRP and SP are thought to sensitize nociceptors indirectly by inducing the release of sensitizing inflammatory mediators such as histamine, 5HT, BK, TNF-alpha and nitric oxide from other immune cells, especially mast cells, in a process known as mast cell degranulation [77,94,112]. The trigeminovascular system also innervates the inner ear, and auditory brainstem [17,86,99–103,114]. These pathways may play a role in the vertigo, hearing loss, tinnitus, and aural fullness experienced by some migraineurs during their attacks, and which can mimic Meniere’s disease.
1.4. Central sensitization
Central sensitization in somatosensory pain pathways was first discovered in the rat spinal cord, where it was shown to play a role in post-injury pain hypersensitivity [108], and later documented in several animal models and in humans [33,49,57,75,87,97]. Central sensitization refers to a condition where nociceptive neurons in the dorsal horn of the spinal cord exhibit increased excitability, increased synaptic strength, and enlargement of their receptive fields beyond the original site of inflammation or injury [64,109,110]. Central sensitization is triggered by sensory input arriving from sensitized nociceptors that supply the affected site. Once initiated, central sensitization may remain dependent on incoming input (i.e., activity-dependent) or become self-sufficient altogether (i.e., activity independent). Sensitized dorsal horn nociceptors become responsive to innocuous (i.e., previously sub-threshold) sensory signals that arrive from areas outside the affected site, resulting in expansion of their receptive fields. Clinically, central sensitization is manifested as decreased pain threshold and exaggerated pain response that is referred outside the original pain site.
Among the common symptoms of central sensitization during migraine is the phenomenon of allodynia, where patients become irritated by mundane mechanical and thermal stimulation of the scalp and facial skin [6,14,22,27,41–43,54,55,83,95,103]. This hypersensitivity is manifested in response to activities such as combing, shaving, breathing cold air, wearing eyeglasses, contact lenses, earrings, or necklaces. Such allodynia involves the sensitization of nociceptive trigeminovascular neurons of the medullary dorsal horn that receive converging sensory input from the dura and skin [13]. Accordingly, innocuous skin stimuli evoke dramatic activity in central trigeminovascular neurons during migraine, when they are sensitized, but produce little or no response in the absence of migraine, when they are not sensitized.
There is good evidence that central sensitization can also be produced in trigeminovascular neurons in the deep laminae of the medullary dorsal horn, and that these sensitized neurons may play a role in the pathogenesis of migraine headache [9]. Topical application of inflammatory agents on the exposed rat dura, which activates the trigeminovascular pathway for many hours [13,23,82], induces long-lasting sensitization in medullary dorsal horn neurons that receive convergent intracranial input from the dura and extracranial input from the periorbital skin. This neuronal sensitization is manifested as increased responsiveness to mechanical stimulation of the dura increased responsiveness to mechanical and thermal stimulation of the skin decreased response thresholds and increased response magnitude to dural and skin stimulation, and expansion of dural and cutaneous receptive fields [13].
The history of migraine literature is dotted with accounts of increased skin sensitivity during migraine. In 1873, Edward Living [55] quoted Tissot in his seminal book “on Megrim” as saying: “so painful is this hyperasthesia in a certain stage of the seizure with some people that, he (the patient) could not bear anything to touch his head”. In 1953, Harold Wolff [107] found that in cranial tissue, deep-pain thresholds were high during headache-free periods and low during the headache, that the zone of low-threshold expanded to include areas on the non-painful side of the head, that the deep-pain threshold begun to decrease several hours after the onset of headache, and that commonly, this hypersensitivity outlasted the headache for hours and even days. In 1960, James Lance [83] found that about 2/3 (317/500) of migraine patients experienced scalp tenderness during migraine.
Assuming that migraine in humans is associated with sensitization of medullary dorsal horn neurons as observed in the rat, it is reasonable to predict that it should also be associated with allodynia in the periorbital skin. Using quantitative sensory testing technique, it was shown recently that 79% of the patients developed mechanical and/or thermal allodynia on the facial skin ipsilateral to the migraine pain 1–2 hours after onset of the attack [10]. By 4 hours, allodynia frequently extended outside the referred pain area to the skin over the contralateral head and both arms [10]. A standardized questionnaire can now be used to reliably identify patients as allodynic or non-allodynic [3,40].
1.5. Cellular mechanisms of central sensitization
Central sensitization can be divided into two distinct phases, the initiation phase and the maintenance phase, each mediated by different mechanisms.
Initiation of sensitization in the spinal cord depends on the input from nociceptive neurons of the dorsal horn that receive from signals C-fiber nociceptors that contain the excitatory amino acid glutamate and neuropeptides such as substance P and CGRP [106]. Once activated from the periphery (leading to action potential invasion of the central terminals), calcium inflow into the central terminals of C-fiber nociceptors causes them to release a number of neuropeptides in the superficial layers of the medullary dorsal horn. Consequently, activation of C-fibers elicits slow synaptic potentials [66,88], leading to cumulative depolarization of dorsal horn neurons. C-fiber input can also contribute to the progression and establishment of sustained depolarization in dorsal horn neurons by recruiting L-type calcium plateau currents [65].
Maintenance of sensitization in spinal cord neurons can be activity-dependent or activity-independent [45]. Activity-dependent central sensitization is the consequence of neurotransmitter (glutamate) and neuromodulator (substance P, brain-derived neurotrophic factor, ephrin-B ligand) induced activation of multiple intra-cellular signaling pathways in dorsal horn neurons by virtue of activation of ligand-gated ion channels (NM-DA, AMPA/Kainate), G-protein coupled metabotropic receptors (NK1, mGluR) and tyrosine kinase receptors (TrkB, EphR). Enhanced neuronal excitability in this form of central sensitization involves phosphorylation of intracellular (PKA, PKC) and extracellular (ERK) kinases and enhanced production of cyclooxygenase in the spinal cord. PKA and PKC activation leads to the phosphorylation of ionotropic glutamate receptors (NMDA and AMPA), which increases synaptic efficacy by altering channel open-time and promoting the trafficking of receptors to the synaptic membrane [58, 111]. ERK phosphorylation increases A-type K+ current via Kv4.2 channels regulation [32]. Increased cyclooxygenase level in the dorsal horn activates EP receptors to facilitate transmitter release from nociceptor central terminals [98], produce a direct depolarization of dorsal horn neurons [4], and reduce glycine receptor activity [1]. Activity-dependent central sensitization is displayed by many cells in both the superficial and deep laminae of the dorsal horn, but its contribution to pain sensitivity appears to be mediated by lamina I neurons, particularly those expressing the NK1 receptor [36,60].
Activity-independent central sensitization develops slowly over several hours and lasts for prolonged periods. This form of sensitization, like the activity dependent form, is initiated by intense activity in nociceptors [44], which induces activation of NMDA, mGlu, NK1 and trkB receptors in the central neurons. This, in turn, activates PKA, PKC and ERK as described above, but it also increases production of the cytokine interleukins-1β (IL-1βpgn endothelial cells and spinal microglia [21]. Central sensitization shifts from activity-dependent mode to activity-independent mode upon widespread increase in expression of transcription factor genes such as CRE-binding protein [46,47], immediate early genes such as c-fos and COX-2 [34, 80], and late-response genes encoding prodynorphine, NK1 and trkB [33,42,56,60]. Interestingly, these genes share cAMP-response element (CRE) sites in their promoter region [44,84].
1.6. Implications for migraine therapy
Migraine patients with and without allodynia exhibit different responses to abortive migraine treatments. Treatment of acute or chronic pain is generally more complicated to treat with triptans in the presence of allodynia. Patients who do not exhibit allodynia during migraine are highly responsive to triptans; they are typically rendered pain-free within 2 hours of treatment [12]. Patients whose migraine headache is accompanied by cutaneous allodynia become increasingly resistant to triptan therapy with the progression of the attack [12]. These patients are highly likely to be rendered pain-free by triptan treatment early in the attack, when they have not yet exhibited signs of allodynia, or even in the early presence of allodynia. The same patients, however, would fail to respond to triptans if treatment were delayed until they have fully developed allodynia over a period of several hours [12]. These observations can be explained by the effects of triptans on central trigeminovascular neurons that mediate allodynia during migraine.
In the rat, sensitization in central trigeminovascular neurons induced by topical application of “inflammatory soup” (histamine/bradykinin/prostaglandin E2 in Hepes buffer) to the dura is blocked by co-administration of sumatriptan (i.e., early treatment paradigm). In contrast, sumatriptan intervention several hours after inflammatory soup application (i.e., late treatment paradigm) could not reverse the ongoing sensitization in the central neurons [11]. Therefore, central trigeminovascular neurons appear to be unequipped to respond to triptans directly. A similar experimental paradigm showed that early triptan treatment cannot prevent the induction of sensitization in meningeal nociceptors, and that late treatment has no effect at the ongoing state of peripheral sensitization [52]. Collectively, these data suggest that the action of triptans in the dorsal horn is mediated mainly by presynaptic inhibition of signal transmission between peripheral (first-order) and central (second-order) trigeminovascular neurons (Fig. 1). This conclusion is consistent with the selective presence of presynaptic 5HT1D receptors on central terminals of peripheral nociceptors in the dorsal horn [70]. Accordingly, termination of migraine headache and the associated allodynia using triptan treatment is possible as long as the excitability of the central neurons remains driven by incoming signals from the meninges, but not after they develop autonomous activity [11].
Fig. 1.
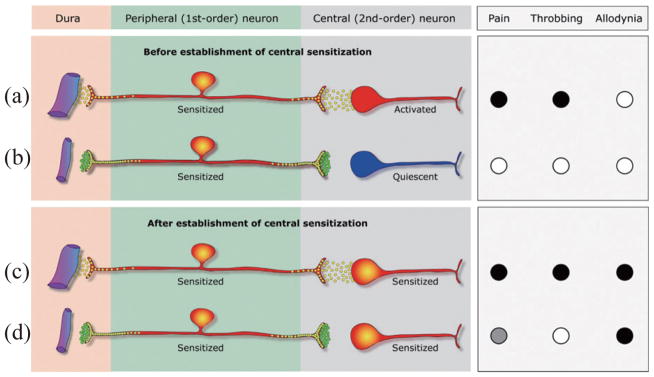
Proposed mechanism of action for 5HT1B/1D agonists during migraine. (a) Peripheral sensitization begins with release of neuropeptides (yellow circles) that promote local vasodilatation and plasma extravasation through their peripheral branch in the meninges, and activation of central trigeminovascular neurons through their central branch in the dorsal horn. Consequently, rhythmic pulsation of the meninges generate bursts of action potentials that activate the central trigeminovascular neuron (shown in red) and the pain (●) begins to throb (●). (b) Systemically-administered triptan molecules (green circles) bind to presynaptic 5HT1B/1D receptors on terminals of both the peripheral and central branches of the meningeal nociceptor; this blocks neuropeptide release from the peripheral terminal, but has no effect on the hyper-excitability of the meningeal nociceptor. However, blockade of neuropeptide release from the central terminal of meningeal nociceptor renders the central trigeminovascular neuron inactive (shown in blue), resulting in termination of pain (○) and throbbing (○). (c) After the establishment of central sensitization, the pain continues to throb (●) and the skin becomes allodynic (●). (d) At this stage, blockade of neuropeptide release from the central terminals of the meningeal nociceptor cannot reverse the hyper-excitability of the central trigeminovascular neuron because its activity no longer depends on input from the meningeal nociceptor. In the face of the autonomous activity of the central trigeminovascular neuron, this blockade of synaptic transmission provides partial pain relief (
 ), terminates the throbbing (○) and does not resolve the allodynia (●).
), terminates the throbbing (○) and does not resolve the allodynia (●).
Most patients testify that triptans are much more likely to render them pain-free when taken early rather than late, but routinely delay treatment until attacks are fully developed or the pain is severe. Justifying the delayed treatment are concerns about side effects, addiction, limits on supply imposed by prescribers, cost, and most commonly waiting to see if headache develops into a severe migraine attack [26]. For these patients, one way to terminate migraine with allodynia and fully developed central sensitization is parenteral administration of COX1/COX2 inhibitors [38]. Infusion of the COX1/COX2 inhibitor ketorolac in allodynic patients who already missed the critical period for triptan therapy terminated both the headache and the allodynia provided that the patient had no history of using opioids to treat her/his migraines. In the rat, infusion of COX-1/COX-2 inhibitors blocked sensitization in meningeal nociceptors and suppressed ongoing sensitization in spinal trigeminovascular neurons, suggesting that parenteral NSAID administration acts in the dorsal horn to inhibit the central neurons directly and reduce the synaptic input from the peripheral trigeminovascular neuron [38,39].
Though impractical as a routine migraine therapy, parenteral NSAID administration should be useful as a non-narcotic rescue therapy for migraine in the setting of the emergency department. Patients who use an opioid therapy over an extended period of time are at high risk of developing medication-overuse headache and low response to non-narcotic drugs. The rational for recommending against the use of opioids in allodynic migraine patients is based on evidence that opioids can facilitate sensitization in the dorsal horn [62, 69,73,105] through: (a) upregulation of NMDA receptors, (b) downregulation of glutamate transporters, (c) production of nitric oxide, (d) activation of spinal glia, and (e) increased extracellular kevel of prostaglandins.
2. Summary
We have presented a review of some of the current information regarding the neural pathways of pain symptoms in migraine. Peripheral senistization refers to a state where primary afferent nociceptive neurons exhibit increased responsiveness to external mechanical or thermal stimuli at the original site of inflammation or injury. Common symptoms of peripheral sensitization during migraine are the throbbing of the headache and its aggravation during routine physical activities that increase intracranial pressure such as coughing and bending over. These effects are mediated by bradykinin, histamine, serotonic, prostaglandin E2 and a number of cytokines and other inflammatory mediators. In contrast, central sensitization refers to a condition where nociceptive neurons in the dorsal horn of the spinal cord exhibit increased excitability, increased synaptic strength, and enlargement of their receptive fields beyond the original site of inflammation or injury. Allodynia is the archetypical manifestation of central sensitization. It is mediated by central trigeminovascular neurons. Central sensitization undergoes an initiation phase and a maintenance phase, each mediated by different neurons. Once initiated, maintenance of central sensitization can be activity-dependent or activity-independent. The activity-dependent form is the consequence of neurotransmitter and neuromodulator induced activation of multiple intracellular signalling pathways. Activity-independent sensitization develops slowly over several hours and lasts for prolonged periods. The pathways and time course of central sensitization have clinical relevance. Early in a migraine attack, triptans are highly effective abortives. However, in patients whose migraine is accompanied by allodynia, the patient becomes increasingly resistant to triptan therapy as the allodynia develops.
There are a host of other migraine symptoms that have not been studied as thoroughly as allodynia. In some cases, distortion or intensification of other sensory modalities, as seen in photophobia, phonophobia, osmophobia, and vestibular symptoms, may be mediated by the same pathways and could have similar response or resistance to therapy. For example, thalamic neurons may have dual innervation and be modulated by normally non-nociceptive modalities. In the case of vestibular symptoms in particular, the trigeminovascular pathways have been shown to affect inner ear bloodflow, which could conceivably create peripheral vestibular dysfunction as a downstream consequence of central sensitization. Both basic and clinical research will be needed to dissect these phenomena and lead to more appropriate and effective treatments.
Fig. 2.
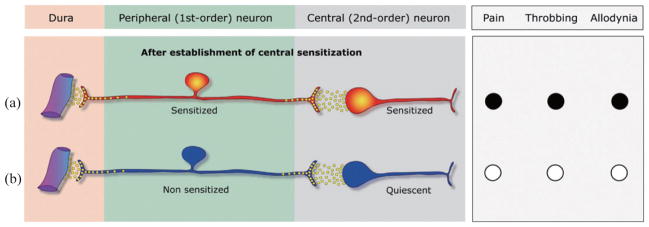
Proposed mechanism of action for COX1/COX2 inhibitors during migraine. (a) After the establishment of central sensitization, the pain throbs (●) and the skin is allodynic (●). (b) At this stage, COX1/COX2 inhibitors reverse the sensitization of both the peripheral and central trigeminovascular neurons, resulting in termination of pain, throbbing and allodynia (○).
Acknowledgments
Supported by NIH grants NS051484 and NS069847 from NINDS.
References
- 1.Ahmadi S, Lippross S, Neuhuber WL, Zeilhofer HU. PGE (2) selectively blocks inhibitory glycinergic neurotransmission onto rat superficial dorsal horn neurons. Nat Neurosci. 2002;5:34–40. doi: 10.1038/nn778. [DOI] [PubMed] [Google Scholar]
- 2.Andrew D, Greenspan JD. Mechanical and Heat Sensitization of Cutaneous Nociceptors After Peripheral Inflammation in The Rat. Journal of Neurophysiology. 1999;82:2649–2656. doi: 10.1152/jn.1999.82.5.2649. [DOI] [PubMed] [Google Scholar]
- 3.Ashkenazi A, Silberstein S, Jakubowski M, Burstein R. Improved identification of allodynic migraine patients using a questionnaire. Cephalalgia. 2007;27:325–329. doi: 10.1111/j.1468-2982.2007.01291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baba H, Kohno T, Moore KA, Woolf CJ. Direct activation of rat spinal dorsal horn neurons by prostaglandin E2. J Neurosci. 2001;21:1750–1756. doi: 10.1523/JNEUROSCI.21-05-01750.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belmonte C, Gallar J, Pozo MA, Rebollo I. Excitation by irritant chemical substances of sensory afferent units in the cat’s cornea. J Physiol (Lond) 1991;437:709–725. doi: 10.1113/jphysiol.1991.sp018621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blau JN. Adult migraine: the patient observed. In: Blau JN, editor. Migraine – Clinical, therapeutic, conceptual and research aspects. Chapman and Hall Ltd; Cambridge: 1987. pp. 3–30. [Google Scholar]
- 7.Blau JN, Dexter SL. The site of pain origin during migraine attacks. Cephalalgia. 1981;1:143–147. doi: 10.1046/j.1468-2982.1981.0103143.x. [DOI] [PubMed] [Google Scholar]
- 8.Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med. 2002;8:136–142. doi: 10.1038/nm0202-136. [DOI] [PubMed] [Google Scholar]
- 9.Burstein R. Deconstructing migraine headache into peripheral and central sensitization. Pain. 2001;89:107–110. doi: 10.1016/s0304-3959(00)00478-4. [DOI] [PubMed] [Google Scholar]
- 10.Burstein R, Cutrer FM, Yarnitsky D. The development of cutaneous allodynia during a migraine attack: clinical evidence for the sequential recruitment of spinal and supraspinal nociceptive neurons in migraine. Brain. 2000;123:1703–1709. doi: 10.1093/brain/123.8.1703. [DOI] [PubMed] [Google Scholar]
- 11.Burstein R, Jakubowski M. Analgesic triptan action in an animal model of intracranial pain: A race against the development of central sensitization. Ann Neurol. 2004;55:27–36. doi: 10.1002/ana.10785. [DOI] [PubMed] [Google Scholar]
- 12.Burstein R, Collins B, Jakubowski M. Defeating migraine pain with triptans: a race against the development of cutaneous allodynia. Ann Neurol. 2004;55:19–26. doi: 10.1002/ana.10786. [DOI] [PubMed] [Google Scholar]
- 13.Burstein R, Yamamura H, Malick A, Strassman AM. Chemical stimulation of the intracranial dura induces enhanced responses to facial stimulation in brain stem trigeminal neurons. Journal of Neurophysiology. 1998;79:964–982. doi: 10.1152/jn.1998.79.2.964. [DOI] [PubMed] [Google Scholar]
- 14.Burstein R, Yarnitsky D, Goor-Aryeh I, Ransil BJ, Bajwa ZH. An association between migraine and cutaneous allodynia. Annals Neurol. 2000;47:614–624. [PubMed] [Google Scholar]
- 15.Carmody J, Pawlak M, Messlinger K. Lack of a role for substance P in the control of dural arterial flow. Experimental Brain Research. 1996;111:424–428. doi: 10.1007/BF00228731. [DOI] [PubMed] [Google Scholar]
- 16.Cooper BY, Ahlquist M, Friedman RM, Labanc J. Properties of high-threshold mechanoreceptors in the goat oral mucosa. II. Dynamic and static reactivity in carrageenan-inflamed mucosa. Journal of Neurophysiology. 1991;66:1280–1290. doi: 10.1152/jn.1991.66.4.1280. [DOI] [PubMed] [Google Scholar]
- 17.Dai CF, Steyger PS, Wang ZM, Vass Z, Nuttall AL. Expression of Trk A receptors in the mammalian inner ear. Hear Res. 2004;187:1–11. doi: 10.1016/s0378-5955(03)00277-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dai Y, Moriyama T, Higashi T, Togashi K, Kobayashi K, Yamanaka H, Tominaga M, Noguchi K. Proteinase-activated receptor 2-mediated potentiation of transient receptor potential vanilloid subfamily 1 activity reveals a mechanism for proteinase-induced inflammatory pain. J Neurosci. 2004;24:4293–4299. doi: 10.1523/JNEUROSCI.0454-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daley ML, Pasupathy H, Griffith M, Robertson JT, Leffler CW. Detection of loss of cerebral vascular tone by correlation of arterial and intracranial pressure signals. IEEE Trans Biomed Eng. 1995;42:420–424. doi: 10.1109/10.376137. [DOI] [PubMed] [Google Scholar]
- 20.Davis KD, Meyer RA, Campbell JN. Chemosensitivity and sensitization of nociceptive afferents that innervate the hairy skin of monkey. Journal of Neurophysiology. 1993;69:1071–1081. doi: 10.1152/jn.1993.69.4.1071. [DOI] [PubMed] [Google Scholar]
- 21.DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- 22.Drummond PD. Scalp tenderness and sensitivity to pain in migraine and tension headache. Headache. 1987;27:45–50. doi: 10.1111/j.1526-4610.1987.hed2701045.x. [DOI] [PubMed] [Google Scholar]
- 23.Ebersberger A, Ringkamp M, Reeh PW, Handwerker HO. Recordings from brain stem neurons responding to chemical stimulation of the subarachnoid space. Journal of Neurophysiology. 1997;77:3122–3133. doi: 10.1152/jn.1997.77.6.3122. [DOI] [PubMed] [Google Scholar]
- 24.England S, Bevan S, Docherty RJ. PGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP-protein kinase A cascade. J Physiol (Lond) 1996;495(Pt 2):429–440. doi: 10.1113/jphysiol.1996.sp021604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Evans AR, Vasko MR, Nicol GD. The cAMP transduction cascade mediates the PGE2-induced inhibition of potassium currents in rat sensory neurones. J Physiol (Lond) 1999;516(Pt 1):163–178. doi: 10.1111/j.1469-7793.1999.163aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Foley KA, Cady R, Martin V, Adelman J, Diamond M, Bell CF, Dayno JM, Hu XH. Treating early versus treating mild: timing of migraine prescription medications among patients with diagnosed migraine. Headache. 2005;45:538–545. doi: 10.1111/j.1526-4610.2005.05107.x. [DOI] [PubMed] [Google Scholar]
- 27.Gobel H, Ernst M, Jeschke J, Keil R, Weigle L. Acetyl-salicylic acid activates antinociceptive brain-stem reflex activity in headache patients and in healthy subjects. Pain. 1992;48:187–195. doi: 10.1016/0304-3959(92)90058-J. [DOI] [PubMed] [Google Scholar]
- 28.Gold MS. Tetrodotoxin-resistant Na+ currents and inflammatory hyperalgesia. Proc Natl Acad Sci U S A. 1999;96:7645–7649. doi: 10.1073/pnas.96.14.7645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gold MS, Levine JD, Correa AM. Modulation of TTX-R INa by PKC and PKA and their role in PGE2-induced sensitization of rat sensory neurons in vitro. J Neuroscience. 1998;18:10345–10355. doi: 10.1523/JNEUROSCI.18-24-10345.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Halata Z, Cooper BY, Baumann KI, Schwegmann C, Friedman RM. Sensory nerve endings in the hard palate and papilla incisiva of the goat. Experimental Brain Research. 1999;129:218–228. doi: 10.1007/s002210050892. [DOI] [PubMed] [Google Scholar]
- 31.Hoogerwerf WA, Zou L, Shenoy M, Sun D, Micci MA, Lee-Hellmich H, Xiao SY, Winston JH, Pasricha PJ. The proteinase-activated receptor 2 is involved in nociception. J Neurosci. 2001;21:9036–9042. doi: 10.1523/JNEUROSCI.21-22-09036.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu HJ, Gereau RWt. ERK integrates PKA and PKC signaling in superficial dorsal horn neurons. II. Modulation of neuronal excitability. Journal of Neurophysiology. 2003;90:1680–1688. doi: 10.1152/jn.00341.2003. [DOI] [PubMed] [Google Scholar]
- 33.Hu JW, Sessle BJ, Raboisson P, Dallel R, Woda A. Stimulation of craniofacial muscle afferents induces prolonged facilitatory effects in trigeminal nociceptive brainstem neurones. Pain. 1992;48:53–60. doi: 10.1016/0304-3959(92)90131-T. [DOI] [PubMed] [Google Scholar]
- 34.Hunt SP, Pini A, Evan G. Induction of c-fos-like protein in spinal cord neurons following sensory stimulation. Nature. 1987;328:632–634. doi: 10.1038/328632a0. [DOI] [PubMed] [Google Scholar]
- 35.Iadarola MJ, Douglass J, Civelli O, Naranjo JR. Differential activation of spinal cord dynorphin and enkephalin neurons during hyperalgesia: evidence using cDNA hybridization. Brain Res Brain Res Rev. 1988;455:205–212. doi: 10.1016/0006-8993(88)90078-9. [DOI] [PubMed] [Google Scholar]
- 36.Ikeda H, Heinke B, Ruscheweyh R, Sandkuhler J. Synaptic plasticity in spinal lamina I projection neurons that mediate hyperalgesia. Science. 2003;299:1237–1240. doi: 10.1126/science.1080659. [DOI] [PubMed] [Google Scholar]
- 37.Ingram SL, Williams JT. Modulation of the hyper-polarization-activated current (Ih) by cyclic nucleotides in guinea-pig primary afferent neurons. J Physiol. 1996;492(Pt 1):97–106. doi: 10.1113/jphysiol.1996.sp021292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jakubowski M, Levy D, Goor-Aryeh I, Collins B, Bajwa Z, Burstein R. Terminating migraine with allodynia and ongoing central sensitization using parenteral administration of COX1/COX2 inhibitors. Headache. 2005;45:850–861. doi: 10.1111/j.1526-4610.2005.05153.x. [DOI] [PubMed] [Google Scholar]
- 39.Jakubowski M, Levy D, Kainz V, Zhang XC, Kosaras B, Burstein R. Sensitization of central trigeminovascular neurons: blockade by intravenous naproxen infusion. Neuroscience. 2007;148:573–583. doi: 10.1016/j.neuroscience.2007.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jakubowski M, Silberstein S, Ashkenazi A, Burstein R. Can allodynic migraine patients be identified interictally using a questionnaire? Neurology. 2005;65:1419–1422. doi: 10.1212/01.wnl.0000183358.53939.38. [DOI] [PubMed] [Google Scholar]
- 41.Jensen K. Extracranial blood flow, pain and tenderness in migraine. Clinical and experimental studies. Acta Neurol Scand Suppl. 1993;147:1–27. [PubMed] [Google Scholar]
- 42.Jensen K, Tuxen C, Olesen J. Pericranial muscle tenderness and pressure-pain threshold in the temporal region during common migraine. Pain. 1988;35:65–70. doi: 10.1016/0304-3959(88)90277-1. [DOI] [PubMed] [Google Scholar]
- 43.Jensen R, Rasmussen BK, Pedersen B, Lous I, Olesen J. Prevalence of oromandibular dysfunction in a general population. J Orofacial Pain. 1993;7:175–182. [PubMed] [Google Scholar]
- 44.Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci. 2002;22:478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 46.Ji RR, Rupp F. Phosphorylation of transcription factor CREB in rat spinal cord after formalin-induced hyperalgesia: relationship to c-fos induction. J Neurosci. 1997;17:1776–1785. doi: 10.1523/JNEUROSCI.17-05-01776.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ji RR, Woolf CJ. Neuronal plasticity and signal transduction in nociceptive neurons: implications for the initiation and maintenance of pathological pain. Neurobiol Dis. 2001;8:1–10. doi: 10.1006/nbdi.2000.0360. [DOI] [PubMed] [Google Scholar]
- 48.Klyachko VA, Ahern GP, Jackson MB. cGMP-mediated facilitation in nerve terminals by enhancement of the spike afterhyperpolarization. Neuron. 2001;31:1015–1025. doi: 10.1016/s0896-6273(01)00449-4. [DOI] [PubMed] [Google Scholar]
- 49.Koltzenburg M, Torebjork HE, Wahren LK. Nociceptor modulated central sensitization causes mechanical hyper-algesia in acute chemogenic and chronic neuropathic pain. Brain. 1994;117:579–591. doi: 10.1093/brain/117.3.579. [DOI] [PubMed] [Google Scholar]
- 50.Kurosawa M, Messlinger K, Pawlak M, Schmidt RF. Increase of meningeal blood flow after electrical stimulation of rat dura mater encephali: mediation by calcitonin gene-related peptide. British Journal of Pharmacology. 1995;114:1397–1402. doi: 10.1111/j.1476-5381.1995.tb13361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Levine JD, Reichling DB. Peripheral mechanisms of inflammatory pain. In: Wall PD, editor. Textbook of Pain. Churchill Livingstone; New-York: 1999. pp. 59–84. [Google Scholar]
- 52.Levy D, Strassman AM. Distinct sensitizing effects of the cAMP-PKA second messenger cascade on rat dural mechanonociceptors. J Physiol. 2002;538:483–493. doi: 10.1113/jphysiol.2001.013175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Levy D, Strassman AM. Modulation of Dural Nociceptor Mechanosensitivity by the Nitric Oxide – Cyclic GMP Signaling Cascade. Journal of Neurophysiology. 2004;92:766–772. doi: 10.1152/jn.00058.2004. [DOI] [PubMed] [Google Scholar]
- 54.Levy D, Jakubowski M, Burstein R. Disruption of communication between peripheral and central trigeminovascular neurons mediates the antimigraine action of 5HT 1B/1D receptor agonists. Proc Natl Acad Sci. 2004;101:4274–4279. doi: 10.1073/pnas.0306147101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liveing E. On megrim, sick headache. Arts and Boeve Publishers; Nijmegen: 1873. [Google Scholar]
- 56.Lous I, Olesen J. Evaluation of pericranial tenderness and oral function in patients with common migraine, muscle contraction headache and ‘combination headache’. Pain. 1982;12:385–393. doi: 10.1016/0304-3959(82)90183-X. [DOI] [PubMed] [Google Scholar]
- 57.Magerl W, Wilk SH, Treede RD. Secondary hyperalgesia and perceptual wind-up following intradermal injection of capsaicin in humans. Pain. 1998;74:257–268. doi: 10.1016/s0304-3959(97)00177-2. [DOI] [PubMed] [Google Scholar]
- 58.Malmberg AB, Chen C, Tonegawa S, Basbaum AI. Preserved acute pain and reduced neuropathic pain in mice lacking PKCgamma. Science. 1997;278:279–283. doi: 10.1126/science.278.5336.279. [DOI] [PubMed] [Google Scholar]
- 59.Mannion RJ, Costigan M, Decosterd I, Amaya F, Ma QP, Holstege JC, Ji RR, Acheson A, Lindsay RM, Wilkinson GA, Woolf CJ. Neurotrophins: peripherally and centrally acting modulators of tactile stimulus-induced inflammatory pain hypersensitivity. Proc Natl Acad Sci U S A. 1999;96:9385–9390. doi: 10.1073/pnas.96.16.9385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mantyh PW, Rogers SD, Honore P, Allen BJ, Ghilardi JR, Li J, Daughters RS, Lappi DA, Wiley RG, Simone DA. Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance P receptor. Science. 1997;278:275–279. doi: 10.1126/science.278.5336.275. [DOI] [PubMed] [Google Scholar]
- 61.Martin HA, Basbaum AI, Kwait GC, Goetzl EJ, Levine JD. Leukotriene and prostaglandin sensitization of cutaneous high threshold C- and A-delta mechanonociceptors in the hairy skin of rat hindlimb. Neuroscience. 1987;22:651–659. doi: 10.1016/0306-4522(87)90360-5. [DOI] [PubMed] [Google Scholar]
- 62.Mayer DJ, Mao J, Holt J, Price DD. Cellular mechanisms of neuropathic pain, morphine tolerance, and their interactions. Proc Natl Acad Sci U S A. 1999;96:7731–7736. doi: 10.1073/pnas.96.14.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McCarson KE, Krause JE. NK-1 and NK-3 type tachykinin receptor mRNA expression in the rat spinal cord dorsal horn is increased during adjuvant or formalin-induced nociception. J Neurosci. 1994;14:712–720. doi: 10.1523/JNEUROSCI.14-02-00712.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McMahon SB, Lewin GR, Wall PD. Central hyperexcitability triggered by noxious inputs. Curr Opin Neurobiol. 1993;3:602–610. doi: 10.1016/0959-4388(93)90062-4. [DOI] [PubMed] [Google Scholar]
- 65.Morisset V, Nagy F. Plateau potential-dependent windup of the response to primary afferent stimuli in rat dorsal horn neurons. Eur J Neurosci. 2000;12:3087–3095. doi: 10.1046/j.1460-9568.2000.00188.x. [DOI] [PubMed] [Google Scholar]
- 66.Murase K, Ryu PD, Randic M. Substance P augments a persistent slow inward calcium-sensitive current in voltage-clamped spinal dorsal horn neurons of the rat. Brain Res Brain Res Rev. 1986;365:369–376. doi: 10.1016/0006-8993(86)91652-5. [DOI] [PubMed] [Google Scholar]
- 67.Obreja O, Schmelz M, Poole S, Kress M. Interleukin-6 in combination with its soluble IL-6 receptor sensitises rat skin nociceptors to heat, in vivo. Pain. 2002;96:57–62. doi: 10.1016/s0304-3959(01)00420-1. [DOI] [PubMed] [Google Scholar]
- 68.Olesen J, Thomsen LL, Iversen H. Nitric oxide is a key molecule in migraine and other vascular headaches. Trends Pharmacol Sci. 1994;15:149–153. doi: 10.1016/0165-6147(94)90075-2. [DOI] [PubMed] [Google Scholar]
- 69.Ozawa T, Nakagawa T, Shige K, Minami M, Satoh M. Changes in the expression of glial glutamate transporters in the rat brain accompanied with morphine dependence and naloxone-precipitated withdrawal. Brain Res. 2001;905:254–258. doi: 10.1016/s0006-8993(01)02536-7. [DOI] [PubMed] [Google Scholar]
- 70.Potrebic S, Anh AH, Skinner K, Fields JL, Basbaum AI. Peptidergic nociceptors of both trigeminal and dorsal root ganglia express serotonin 1D receptors: implications for the selective antimigraine action of triptans. J Neurosci. 2003;23:10988–10997. doi: 10.1523/JNEUROSCI.23-34-10988.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pozo MA, Gallego R, Gallar J, Belmonte C. Blockade by calcium antagonists of chemical excitation and sensitization of polymodal nociceptors in the cat’s cornea. J Physiol (Lond) 1992;450:179–189. doi: 10.1113/jphysiol.1992.sp019122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Premkumar LS, Ahern GP. Induction of vanilloid receptor channel activity by protein kinase C. Nature. 2000;408:985–990. doi: 10.1038/35050121. [DOI] [PubMed] [Google Scholar]
- 73.Raith K, Hochhaus G. Drugs used in the treatment of opioid tolerance and physical dependence: a review. Int J Clin Pharmacol Ther. 2004;42:191–203. doi: 10.5414/cpp42191. [DOI] [PubMed] [Google Scholar]
- 74.Rasmussen BK, Jensen R, Olesen J. A population-based analysis of the diagnostic criteria of the International Headache Society. Cephalalgia. 1991;11:129–134. doi: 10.1046/j.1468-2982.1991.1103129.x. [DOI] [PubMed] [Google Scholar]
- 75.Ren K, Dubner R. NMDA receptor antagonists attenuate mechanical hyperalgesia in rats with unilateral inflammation of the hindpaw. Neurosci Lett. 1993;163:22–26. doi: 10.1016/0304-3940(93)90220-f. [DOI] [PubMed] [Google Scholar]
- 76.Reuter U, Bolay H, Jansen-Olesen I, Chiarugi A, Sanchez del Rio M, Letourneau R, Theoharides TC, Waeber C, Moskowitz MA. Delayed inflammation in rat meninges: implications for migraine pathophysiology. Brain. 2001;124:2490–2502. doi: 10.1093/brain/124.12.2490. [DOI] [PubMed] [Google Scholar]
- 77.Reynier-Rebuffel AM, Mathiau P, Callebert J, Dimitriadou V, Farjaudon N, Kacem K, Launay JM, Seylaz J, Abineau P. Substance P, calcitonin gene-related peptide, and capsaicin release serotonin from cerebrovascular mast cells. Am J Physiol. 1994;267:R1421–R1429. doi: 10.1152/ajpregu.1994.267.5.R1421. [DOI] [PubMed] [Google Scholar]
- 78.Ryu S, Liu B, Qin F. Low pH potentiates both capsaicin binding and channel gating of VR1 receptors. J Gen Physiol. 2003;122:45–61. doi: 10.1085/jgp.200308847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sachs D, Cunha FQ, Poole S, Ferreira SH. Tumour necrosis factor-alpha, interleukin-1beta and interleukin-8 induce persistent mechanical nociceptor hypersensitivity. Pain. 2002;96:89–97. doi: 10.1016/s0304-3959(01)00433-x. [DOI] [PubMed] [Google Scholar]
- 80.Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- 81.Schaible HG, Schmidt RF. Excitation and sensitization of fine articular afferents from cat’s knee joint by prostaglandin E2. J Physiol (Lond) 1988;403:91–104. doi: 10.1113/jphysiol.1988.sp017240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schepelmann K, Ebersberger A, Pawlak M, Oppmann M, Messlinger K. Response properties of trigeminal brain stem neurons with input from dura mater encephali in the rat. Neuroscience. 1999;90:543–554. doi: 10.1016/s0306-4522(98)00423-0. [DOI] [PubMed] [Google Scholar]
- 83.Selby G, Lance JW. Observations on 500 cases of migraine and allied vascular headache. J Neurol Neurosurg Psychiat. 1960:23–32. doi: 10.1136/jnnp.23.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Seybold VS, McCarson KE, Mermelstein PG, Groth RD, Abrahams LG. Calcitonin gene-related peptide regulates expression of neurokinin1 receptors by rat spinal neurons. J Neurosci. 2003;23:1816–1824. doi: 10.1523/JNEUROSCI.23-05-01816.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shepheard SL, Williamson DJ, Hill RG, Hargreaves RJ. The non-peptide neurokinin1 receptor antagonist, RP 67580, blocks neurogenic plasma extravasation in the dura mater of rats. British Journal of Pharmacology. 1993;108:11–12. doi: 10.1111/j.1476-5381.1993.tb13432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shore SE, Vass Z, Wys NL, Altschuler RA. Trigeminal ganglion innervates the auditory brainstem. J Comp Neurol. 2000;419:271–285. doi: 10.1002/(sici)1096-9861(20000410)419:3<271::aid-cne1>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 87.Simone DA, Sorkin LS, Oh U, Chung JM, Owens C, LaMotte RH, Willis WD. Neurogenic hyperalgesia: central neural correlates in responses of spinothalamic tract neurons. Journal of Neurophysiology. 1991;66:228–246. doi: 10.1152/jn.1991.66.1.228. [DOI] [PubMed] [Google Scholar]
- 88.Sivilotti LG, Thompson SW, Woolf CJ. Rate of rise of the cumulative depolarization evoked by repetitive stimulation of small-caliber afferents is a predictor of action potential windup in rat spinal neurons in vitro. Journal of Neurophysiology. 1993;69:1621–1631. doi: 10.1152/jn.1993.69.5.1621. [DOI] [PubMed] [Google Scholar]
- 89.Steen KH, Reeh PW, Anton F, Handwerker HO. Protons selectively induce lasting excitation and sensitization to mechanical stimulation of nociceptors in rat skin, in vitro. J Neurosci. 1992;12:86–95. doi: 10.1523/JNEUROSCI.12-01-00086.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Strassman AM, Raymond SA. Electrophysiological evidence for tetrodotoxin-resistant sodium channels in slowly conducting dural sensory fibers. Journal of Neurophysiology. 1999;81:413–424. doi: 10.1152/jn.1999.81.2.413. [DOI] [PubMed] [Google Scholar]
- 91.Strassman AM, Raymond SA, Burstein R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature. 1996;384:560–564. doi: 10.1038/384560a0. [DOI] [PubMed] [Google Scholar]
- 92.Su X, Gebhart GF. Mechanosensitive pelvic nerve afferent fibers innervating the colon of the rat are polymodal in character. Journal of Neurophysiology. 1998;80:2632–2644. doi: 10.1152/jn.1998.80.5.2632. [DOI] [PubMed] [Google Scholar]
- 93.Sugiura T, Tominaga M, Katsuya H, Mizumura K. Bradykinin lowers the threshold temperature for heat activation of vanilloid receptor 1. Journal of Neurophysiology. 2002;88:544–548. doi: 10.1152/jn.2002.88.1.544. [DOI] [PubMed] [Google Scholar]
- 94.Suzuki R, Furuno T, McKay DM, Wolvers D, Teshima R, Nakanishi M, Bienenstock J. Direct neurite-mast cell communication in vitro occurs via the neuropeptide substance P. J Immunol. 1999;163:2410–2415. [PubMed] [Google Scholar]
- 95.Tfelt-Hansen P, Lous I, Olesen J. Prevalence and significance of muscle tenderness during common migraine attacks. Headache. 1981;21:49–54. doi: 10.1111/j.1526-4610.1981.hed2102049.x. [DOI] [PubMed] [Google Scholar]
- 96.Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D. The cloned capsaicin receptor integrates multiple pain-producing stimuli [see comments] Neuron. 1998;21:531–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- 97.Torebjork HE, Lundberg LE, LaMotte RH. Central changes in processing of mechanoreceptive input in capsaicin-induced secondary hyperalgesia in humans. J Physiol (Lond) 1992;448:765–780. doi: 10.1113/jphysiol.1992.sp019069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vasko MR. Prostaglandin-induced neuropeptide release from spinal cord. Prog Brain Res. 1995;104:367–380. doi: 10.1016/s0079-6123(08)61801-4. [DOI] [PubMed] [Google Scholar]
- 99.Vass Z, Shore SE, Nuttall AL, Jancsó G, Brechtelsbauer PB, Miller JM. Trigeminal ganglion innervation of the cochlea – a retrograde transport study. Neurosci. 1997;79:605–615. doi: 10.1016/s0306-4522(96)00641-0. [DOI] [PubMed] [Google Scholar]
- 100.Vass Z, Shore SE, Nuttall AL, Miller JM. Endolym-phatic hydrops reduces retrograde labeling of trigemnial innervation to the cochlea. Exp Neurol. 1998;151:241–248. doi: 10.1006/exnr.1998.6813. [DOI] [PubMed] [Google Scholar]
- 101.Vass Z, Shore SE, Nuttall AL, Miller JM. Direct evidence of trigeminal innervation of the cochlear blood vessels. Neurosci. 1998;84:559–567. doi: 10.1016/s0306-4522(97)00503-4. [DOI] [PubMed] [Google Scholar]
- 102.Vass Z, Steyger PS, Hordichok AJ, Trune DR, Jancsó G, Nuttall AL. Capsaicin stimulation of the cochlea and electric stimulation of the trigeminal ganglion mediate vascular permeability in cochlear and vertebro-basilar arteries: a potential cause of inner ear dysfunction in headache. Neurosci. 2001;103:189–201. doi: 10.1016/s0306-4522(00)00521-2. [DOI] [PubMed] [Google Scholar]
- 103.Vass Z, Dai CF, Steyger PS, Jancsó G, Trune DR, Nuttall AL. Co-localization of the vanilloid capsaicin receptro and substance P in sensory nerve fibers innervating cochlear and vertebro-basilar arteries. Neurosci. 2004;124:919–927. doi: 10.1016/j.neuroscience.2003.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Waelkens J. Warning symptoms in migraine: characteristics and therapeutic implications. Cephalalgia. 1985;5:223–228. doi: 10.1046/j.1468-2982.1985.0504223.x. [DOI] [PubMed] [Google Scholar]
- 105.Wang JF, Khasar SG, Ahlgren SC, Levine JD. Sensitization of C-fibres by prostaglandin E2 in the rat is inhibited by guanosine 5′-O-(2-thiodiphosphate), 2′,5′-dideoxyadenosine and Walsh inhibitor peptide. Neuroscience. 1996;71:259–263. doi: 10.1016/0306-4522(95)00429-7. [DOI] [PubMed] [Google Scholar]
- 106.Watkins LR, Hutchinson MR, Johnston IN, Maier SF. Glia: novel counter-regulators of opioid analgesia. Trends Neurosci. 2005;28:661–669. doi: 10.1016/j.tins.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 107.Willis WD, Coggeshall RE. Sensory mechanisms of the spinal cord. Plenum Press; New York: 1991. pp. 132–148. [Google Scholar]
- 108.Wolff HG, Tunis MM, Goodell H. Studies on migraine. Arch Internal Medicine. 1953;92:478–484. doi: 10.1001/archinte.1953.00240220026006. [DOI] [PubMed] [Google Scholar]
- 109.Woolf CJ. Evidence for a central component of post-injury pain hypersensitivity. Nature. 1983;306:686–688. doi: 10.1038/306686a0. [DOI] [PubMed] [Google Scholar]
- 110.Woolf CJ. Somatic pain – pathogenesis and prevention. Br J Anaesth. 1995;75:169–176. doi: 10.1093/bja/75.2.169. [DOI] [PubMed] [Google Scholar]
- 111.Woolf CJ, Doubell TP. The pathophysiology of chronic pain–increased sensitivity to low threshold A beta-fibre inputs. Curr Opin Neurobiol. 1994;4:525–534. doi: 10.1016/0959-4388(94)90053-1. [DOI] [PubMed] [Google Scholar]
- 112.Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 113.Yano H, Wershil BK, Arizono N, Galli SJ. Substance P-induced augmentation of cutaneous vascular permeability and granulocyte infiltration in mice is mast cell dependent. J Clin Invest. 1989;84:1276–1286. doi: 10.1172/JCI114295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zheng J, Dai C, Steyger PS, Kim Y, Vass Z, Ren T, Nuttall AL. Vanilloid receptors in hearing: altered cochlear sensitivity by vanilloids and expression of TRPV1 in the organ of Corti. J Neurophysiol. 2003;90:444–455. doi: 10.1152/jn.00919.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]