

To the editor
Proteasome-associated auto-inflammatory syndrome (PRAAS) is caused by autosomal recessive mutations in the proteasome subunit β type 8 (PSMB8) gene. It includes Nakajo-Nishimura Syndrome (NNS),1 Japanese Autoinflammatory Syndrome with Lipodystrophy (JASL),2 Joint Contractures, Muscle Atrophy, Microcytic Anemia, and Panniculitis-Induced Lipodystrophy Syndrome (JMP)3 and Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated Temperature (CANDLE).4 Affected patients present with periodic fevers, rash, lipodystrophy, myositis, hyper-γ-globulinemia and autoantibodies at times.
We report a Hispanic male with a longstanding history of recurrent fevers and associated rash that has presented a diagnostic challenge over the past 19 years. During his infancy, extensive evaluations including multiple skin biopsies, a bone marrow biopsy and laboratory testing were inconclusive. The patient was lost to follow up since 2003. On physical examination in 2012, in addition to erythematous papules/nodules primarily on his upper and lower extremities, he was both physically and developmentally delayed and he had lipodystrophy on his face and upper body (Figure 1). Skin biopsies again revealed a dermal CD68 positive mononuclear infiltrate centering around unaltered adnexal structures. Subcutaneous lobules were atrophic with shrunken adipocytes. The epidermis was not involved and no vascular occlusion or leukocytoclasis was seen. Abnormal laboratory findings included elevated aldolase, CK, IL-6, IgG, IgM, IgE and Interferon-γ-inducible protein 10 (IP-10). Elevated ANA, ANCA, RF, CRP, TSH, ALT, and ALK were also noted. CT of the brain indicated basal ganglia calcification. An MRI of the upper thighs revealed patchy myositis. Based on the clinical presentations and laboratory findings, PRAAS was entertained. Direct DNA sequencing of PSMB8 was performed at the National Institutes of Health, and the patient was found to be heterozygous for the known founder mutation p.T75M and a novel missense mutation p.A92T. Since his mother only carries the p.T75M mutation and his father died several years ago, it is unclear if the p.A92T mutation was inherited from his father or if it occurred de novo. Therefore the diagnosis of PRAAS is further confirmed by genetic testing.
Figure 1.
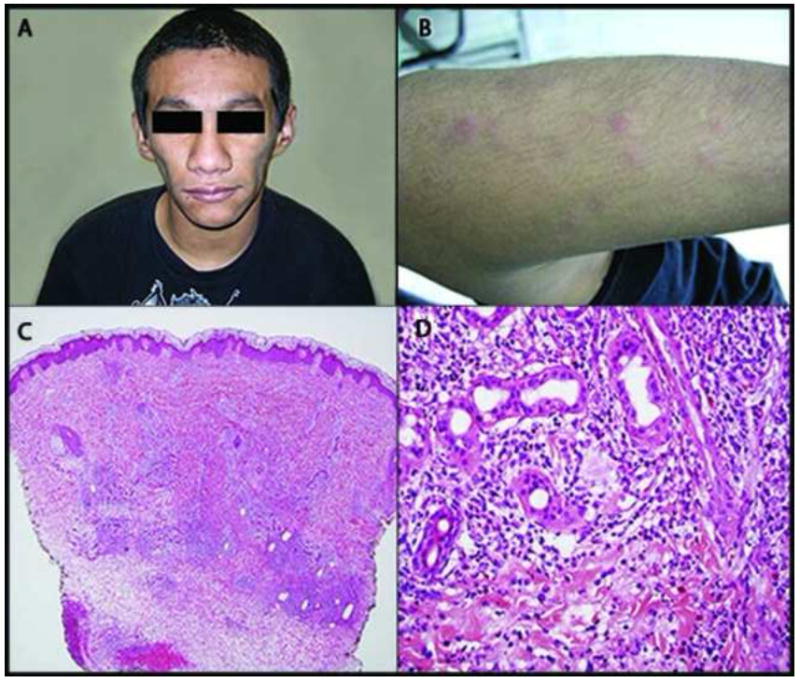
Proteasome-associated auto-inflammatory syndrome. Clinical photograph demonstrates facial lipoatrophy (A). The patient displayed periodic presence of erythematous papules on extensor surfaces (B). Hematoxylin and eosin stain. Lower magnification demonstrates a perivascular, peri-eccrine, and interstitial mixed infiltrate most prominent in the mid to deep reticular dermis (C). On higher magnification, the infiltrate can be seen to be composed predominately of histiocytes and neutrophils, with admixed lymphocytes and eosinophils (D).
Proteasomes are intracellular protease complexes that degrade polyubiquitinated proteins5 and they are involved in cell cycle regulation, gene repair, NF-κB and IFN pathway activation.4 Decreased proteasome activities caused by PSMB8 mutations evoke an inflammatory response that involves activation of mitogen-activated protein kinase, upregulation of the interferon pathway and subsequent elevation of IL-6,IP -10 and/or IFN.1 However, IL-1β and TNFα levels are normal. The table summarizes clinical symptoms, inflammatory disease manifestations and manifestations of organ damage that occurs with ongoing and longstanding inflammation.
Table.
Summary of Clinical Presentations and Laboratory Findings of PRAAS
| Our patient | CANDLE syndrome (n = 9) | NNS (n = 7) | JASL (n = 3) | JMP (n = 3) | |
|---|---|---|---|---|---|
| Clinical presentations | |||||
|
| |||||
| Age of onset | 3m | Early infancy | Infancy | 1M or 3 Y | N/A |
| Recurrent fever | Yes | Yes | Yes | Yes | No |
| Arthritis | Yes | Yes | Yes | Yes | Yes |
| Skin eruptions | Erythematous nodules | Annular plaques Violacious eyelid | Heliotrope-like periorbital rash in 4/7 patients Nodular erythema in all 7 patiens | Nodular erythema | Erythematous maculopapular and nodules |
| Low weight and height | Yes | 7/9 | 1/7 | ND | |
| Low IQ | Yes | ND | 1/7 | 1/3 | Yes |
| Seizures | No | ND | ND | ND | Yes |
| Macroglossia | No | ND | ND | Yes | Yes |
| Anemia | Yes | 8/9 | 6/7 | ND | ND |
| WBC | Low nl range | Mild leukocytosis at early age and leukopenia later | ND | ND | ND |
|
| |||||
| Inflammatory disease and autoimmunity | |||||
|
| |||||
| Autoantibodies | ↑↑ANA & ANCA | c-ANCA and ANA in 3/9 (two converted to normal) | ANCA, dsDNA and SS-A in 5/7 | Undetectable | nl |
| Elevated ESR/CRP | Yes | Yes | Yes | Yes | Yes |
| Elevated CPK | Yes | ND | 4/7 | Normal | ND |
| Cytokine levels | ↑↑ IL-6, nl TNFα, IL-1β, IP-10↑ | ↑ IL-6 and IP-10 in 3/9 | ↑↑ IL-6& IP-10, nl IFNγ. | ↑ IL-6 | ↑ IFNγ, ↑ IL-6, ↑ IL-8 |
| Hyper-γ-globulinemia | ↑↑ IgE & G; nl IgA &D | Normal | 7/7 | ↑↑ IgA & G 3/3 | ND |
|
| |||||
| Organs involved | |||||
|
| |||||
| Lipodystrophy | Yes | Yes | Yes | Yes | Yes |
| Hepatomegaly | Yes | 7/9 | 6/7 | Yes | Yes |
| Splenomegaly | Yes | 3/9 | |||
| Cardiac disease | No | ND | ND | 2/3 | ND |
| Hypertension | No | 2/9 | ND | No | ND |
| Myositis/muscle atrophy | Yes | Yes | Yes | Yes | Yes |
| Elevated LFTs | Yes | 8/9 | ND | ND | Yes |
| Dyslipidemia | No | 6/9 | 4/7 | No | Yes |
| Elevated TSH | Yes | 2/9 | ND | ND | ND |
| Basal ganglia calcification | Yes | 2/6 | 6/7 | 2/3 | 3/3 |
Nl, normal; ND, not described
Lack of effective treatment modalities contributes to a poor prognosis. Fever, skin lesions, and acute-phase reactant are responsive to oral glucocorticosteroids, but no therapy successfully halts the progression of the lipodystrophy and wasting. Inhibitors of IL-1, TNFα, and IL-6, show no or only temporary clinical improvement.4 The immunoproteasome inhibitor bortezomib induces histiocytoid Sweet syndrome.1 Since a JAK inhibitor tofactinib reduces upregulated STAT-1 phosphorylation in CANDLE patients, a compassionate study using the JAK 1/2 inhibitor baricitinib (Eli Lilly) is currently ongoing at the NIH (NCT01724580) and our patient is enrolled in the study.
Acknowledgments
Doctors Adriana Almeida de Jesus, Yin Liu, Peter Kim, Gina A Montealegre Sanchez, Yongqing Chen, and Raphaela Goldbach-Mansky are supported by the NIAMS intramural Research Program (IRP).
Acronyms and Abbreviations
- CANDLE
Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated Temperature
- IP-10
Interferon-γ- inducible protein 10
- IFN
Interferon
- JASL
Japanese autoinflammatory syndrome with lipodystrophy
- JMP
Joint Contractures, Muscle Atrophy, Microcytic Anemia, and Panniculitis-Induced Lipodystrophy Syndrome
- MAPK
mitogen-activated protein kinase
- NF-κB
nuclear factor-κB
- NNS
Nakajo-Nishimura Syndrome
- PSMB8
human proteasome subunit β type 8 gene
- PRAAS
proteasome-associated auto-inflammatory syndrome
Footnotes
The authors state that there are no conflicts of interest for this article.
The case discussed was previously presented at the Arkansas Dermatology State Meeting in April 2012, the American Society of Dermatopathology in October 2012, and the American College of Physicians Internal Medicine Poster Competition at UAMS in October 2012.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Arima K, Kinoshita A, Mishima H, Kanazawa N, Kaneko T, Mizushima T, Ichinose K, Nakamura H, Tsujino A, Kawakami A, Matsunaka M, Kasagi S, Kawano S, Kumagai S, Ohmura K, Mimori T, Hirano M, Ueno S, Tanaka K, Tanaka M, Toyoshima I, Sugino H, Yamakawa A, Tanaka K, Niikawa N, Furukawa F, Murata S, Eguchi K, Ida H, Yoshiura K. Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proc Natl Acad Sci U S A. 2011 Sep 6;108(36):14914–9. doi: 10.1073/pnas.1106015108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kitamura A, Maekawa Y, Uehara H, Izumi K, Kawachi I, Nishizawa M, Toyoshima Y, Takahashi H, Standley DM, Tanaka K, Hamazaki J, Murata S, Obara K, Toyoshima I, Yasutomo K. A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J Clin Invest. 2011 Oct;121(10):4150–60. doi: 10.1172/JCI58414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agarwal AK, Xing C, DeMartino GN, Mizrachi D, Hernandez MD, Sousa AB, Martínez de Villarreal L, dos Santos HG, Garg A. PSMB8 encoding the β5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am J Hum Genet. 2010 Dec 10;87(6):866–72. doi: 10.1016/j.ajhg.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Y, Ramot Y, Torrelo A, Paller AS, Si N, Babay S, Kim PW, Sheikh A, Lee CC, Chen Y, Vera A, Zhang X, Goldbach-Mansky R, Zlotogorski A. Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 2012 Mar;64(3):895–907. doi: 10.1002/art.33368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seifert U, Bialy LP, Ebstein F, Bech-Otschir D, Voigt A, Schröter F, Prozorovski T, Lange N, Steffen J, Rieger M, Kuckelkorn U, Aktas O, Kloetzel PM, Krüger E. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell. 2010 Aug 20;142(4):613–24. doi: 10.1016/j.cell.2010.07.036. [DOI] [PubMed] [Google Scholar]