

Summary
Age-related loss of muscle mass and force (sarcopenia) contributes to disability and increased mortality. Ryanodine receptor 1 (RyR1) is the skeletal muscle sarcoplasmic reticulum calcium release channel required for muscle contraction. RyR1 from aged (24 months) rodents were oxidized, cysteine-nitrosylated, and depleted of the channel stabilizing subunit calstabin1, compared to RyR1 from younger (3–6 months) adults. This RyR1 channel complex remodeling resulted in “leaky” channels with increased open probability leading to intracellular calcium leak in skeletal muscle. Similarly, six-month old mice harboring leaky RyR1-S2844D mutant channels exhibited skeletal muscle defects comparable to 24-month old wild type mice. Treating aged mice with S107, stabilized binding of calstabin1 to RyR1, reduced intracellular calcium leak, decreased reactive oxygen species (ROS), and enhanced tetanic Ca2+ release, muscle specific force and exercise capacity. Taken together these data indicate that leaky RyR1 contribute to age-related loss of muscle function.
Introduction
A hallmark of aging is the progressive decline in skeletal muscle function, characterized by reduced force generating capacity and loss of muscle mass (Rolland et al., 2008; Vijg and Campisi, 2008). Moreover, age-dependent deterioration of muscle function is not restricted to mammals as it is also observed in many animals including the nematode Caenorhabditis elegans (C. elegans) (Herndon et al., 2002). Indeed, loss of muscular strength is highly predictive of frailty, disability and mortality with increased age (Marzetti and Leeuwenburgh, 2006; Metter et al., 2002; Rantanen et al., 1999). Progressive development of muscle dysfunction is common in aged humans and in animal models of aging (Marzetti and Leeuwenburgh, 2006), and affects as many as 30–50% of 80 year olds leading to profound loss of function in the elderly (Rolland et al., 2008).
Much attention has been focused on understanding how to reverse age-related muscle wasting, but there are no established treatments for sarcopenia at this time (Marzetti and Leeuwenburgh, 2006; Saini et al., 2009; Thomas, 2007). In contrast, improving specific force production, which is also significantly reduced in aged muscle (Brooks and Faulkner, 1988; Gonzalez et al., 2000), has received less attention.
The observed loss of specific force in aged muscle suggests that the Ca2+ dependent process known as excitation-contraction (EC) coupling may be impaired in aging. During muscle contraction, membrane depolarization activates voltage-sensing Ca2+ channels in the transverse tubules (Cav1.1) that in turn activate the sarcoplasmic reticulum (SR) Ca2+ release channel, ryanodine receptor 1 (RyR1). The subsequent rise in cytoplasmic [Ca2+]cyt is critical for activation of actin-myosin cross-bridging, shortening of the sarcomere and muscle contraction (Allen et al., 2008; Andersson and Marks, 2010). The RyR1 is a homotetrameric macromolecular protein complex that includes four RyR1 monomers (~565,000 Daltons each), the RyR1 channel stabilizing subunit calstabin1 (FK506 binding protein 12, FKBP12), kinases, a phosphatase (PP1), phosphodiesterase (PDE4D3), and calmodulin (Zalk et al., 2007). Moreover, maladaptive cAMP-dependent protein kinase A (PKA)-mediated phosphorylation and redox-dependent modifications (cysteine-nitrosylation and oxidation) of the RyR1 have been linked to impaired Ca2+ handling and contractile dysfunction in chronic muscle fatigue, heart failure and muscular dystrophy (Allen et al., 2008; Bellinger et al., 2009; Bellinger et al., 2008; Zalk et al., 2007). Defective SR Ca2+ release has been reported in age-dependent muscle weakness (Gonzalez et al., 2003; Jimenez-Moreno et al., 2008) and maladaptive modifications of the RyR1 macromolecular complex have been implicated in this condition (Russ et al., 2010). However, the mechanism underlying impaired SR Ca2+ release in aging muscle remains to be elucidated.
Oxidative stress occurs in aged muscle (Jang et al. 2010; Moylan and Reid, 2007) and stress-induced protein oxidation increases with age (Jackson, 2009; Muller et al., 2007). Two important redox-dependent cellular mediators are cysteine-nitrosylation (SNO) and carbonyl modifications of proteins. Both nitrosylation and oxidation have been shown to affect skeletal muscle RyR1 function and Ca2+ signaling (Aracena-Parks et al., 2006; Barreiro and Hussain; Hidalgo, 2005). Jang et al showed that ablation of the antioxidant enzyme superoxide dismutase 1 (SOD1) increased superoxide levels in murine skeletal muscle resulting in reduced specific force and accelerated age-dependent muscle pathology (Jang et al. 2010). Increased levels of reactive oxygen species (ROS) in the aged muscle are associated with altered cellular Ca2+ handling. Mice with a malignant hyperthermia mutation (Y522S) in RyR1 exhibit SR Ca2+ leak, which promotes mitochondrial dysfunction and oxidative stress-mediated changes of the RyR1 (Durham et al., 2008). Moreover, oxidation-dependent modifications of RyR1 can increase SR Ca2+ leak and a vicious cycle between RyR1-mediated SR Ca2+ leak and mitochondrial ROS production has been proposed (Durham et al., 2008). Furthermore, SNO-modification of the RyR1 has been shown to disrupt the interaction between RyR1 and calstabin1 resulting in channels that can leak SR Ca2+ leading to reduced SR Ca2+ release and muscle function (Bellinger et al., 2009; Zalk et al., 2007).
In the present study, we explored the role of RyR1 dysfunction as an underlying mechanism for defective Ca2+ handling in age-dependent and stress-induced muscle weakness. Fast twitch muscles from aged mice (~2 years old) exhibited reduced specific force and cytoplasmic Ca2+ transients. Moreover, RyR1 from aged rodents were oxidized, cysteine-nitrosylated and depleted of calstabin1 resulting in “leaky” RyR1 manifested as increased single channel open probability and Ca2+ spark frequency. Six month old mice genetically engineered to express constitutively “leaky” RyR1 channels (RyR1-S2844D) exhibited defective muscle function comparable that observed in 24-month old WT mice. Mice with a muscle specific deletion of calstabin1 had reduced muscle specific force and impaired exercise capacity. Furthermore, wild-type (WT) flexor digitorum brevis (FDB) muscle fibers that were acutely treated with rapamycin to dissociate calstabin1 from RyR1 (Ahern et al., 1997; Brillantes et al., 1994) exhibited increased resting mitochondrial [Ca2+], loss of mitochondrial membrane potential, increased ROS and reactive nitrogen species (RNS) production. Muscle from aged mice with mitochondrially targeted catalase that exhibit reduced mitochondrial ROS (Lee et al., 2010; Schriner et al., 2005) showed markedly reduced RyR1 oxidation. Taken together these data suggest that RyR1 mediated SR Ca2+ leak causes mitochondrial Ca2+ overload and increased ROS production, which in turn further exacerbates the RyR1 SR Ca2+ leak by oxidizing the channel. Thus, age-related loss of muscle function and muscular dystrophy, in which RyR1 are also “leaky” (Bellinger et al., 2009), likely share a common mechanism that contributes to impaired exercise capacity. Treatment with the RyR-stabilizing compound, S107, preserved the RyR1-calstabin1 interaction, and restored tetanic Ca2+ release, muscle specific force and exercise capacity in aged mice but not in calstabin1 deficient mice, suggesting that S107 inhibits intracellular Ca2+ leak by restoring the binding of calstabin1 to RyR1.
Results
Oxidation-dependent remodeling of RyR1 and defective SR Ca2+ release in aged muscle
We examined EDL muscles from aged (24 month old) C57/bl6 mice to explore the potential role of RyR1 dysfunction in aging skeletal muscle. EDL from aged mice exhibited reduced specific force compared to EDL from young (3–6 month old) controls (mean specific force at 70 Hz tetanic contraction ± SEM: aged, 251 ± 21 kNm−2, vs. young, 388 ± 14 kNm−2; n = 5 (aged), 7 (young), P < 0.001; Figure 1A-1C). Isolated fast twitch muscle FDB fibers from aged mice also exhibited significantly reduced tetanic Ca2+ transients compared to FDB fibers from young mice (mean ΔF/F0 at 70 Hz tetanic contraction ± SEM: aged, 4.4 ± 0.6, vs. young, 6.8 ± 0.9; n = 8 aged, 10 young, P < 0.05; Figure 1D-1F).
Figure 1. Impaired force production and reduced SR Ca2+ release in EDL muscle from aged mice.

(A and B) Tetanic contractions of EDL muscle from young (A) and aged (B) mice (force normalized to cross-sectional area). (C) Average force at the indicated stimulation frequencies in EDL muscles from young and aged mice (mean, ± SEM, n = 5 (young), 7 (aged), P < 0.05 among groups at all stimulation frequencies). (D and E) Normalized fluo-4 fluorescence in FDB muscle fibers during a 70 Hz tetanic stimulation. (F) Peak Ca2+ responses in FDB fibers stimulated at 70 Hz (fibers taken from the same animals as in A and B; mean, ± SEM, n = 8 (young), 10 (aged), * P < 0.05). (G) Immunoblots of immunoprecipitated RyR1 from young and aged mice. (H) Bar graphs show quantification of the immunoblots in G (mean, ± SEM, n = 2, ** P < 0.01, ***P < 0.001). DNP: 2,4- dinitrophenylhydrazone. P*RyR1: Phosphorylated RyR1 (at serine 2844). See also Figure S1 and 2A.
ROS were measured in FDB fibers using a cell permeant form of the fluorescent indicator 2′ ,7′ 7orm of the fluorescent ind(Aydin et al., 2009; Durham et al., 2008). Muscle fibers from aged mice exhibited oxidative stress with significantly increased levels of DCF fluorescence compared to fibers from young adult mice (Figures S1A and S1B). To determine whether the observed reductions in muscle specific force and tetanic Ca2+ release were associated with oxidation-dependent remodeling of the RyR1 macromolecular complexes, RyR1 from aged and young adult murine muscles were immunoprecipitated and immunoblotted for components of the RyR1 complex (Bellinger et al., 2009). Skeletal muscle RyR1 complexes from aged mice exhibited significantly increased nitrosylation and oxidation, and depletion of calstabin1, compared to channels from young adult mice (Figure 1G and 1H). Similar remodeling of the RyR1 complex was observed in skeletal muscles from 24-month old rats (Figure S2A and S2B). Aging is known to be associated with oxidative stress and mitochondrial abnormalities (Haigis and Yankner, 2010). EDL muscle from 24-month old WT mice examined by electron microscopy exhibited a significant increase in mitochondria with disorganized or absent cristae compared to EDL muscle from young adult mice (Figure S3A-B,E).
RyR1 Ca2+ leak causes mitochondrial dysfunction in skeletal muscle
Transient increases of mitochondrial [Ca2+] enhance ATP production (Brookes et al., 2004; Duchen, 2000), whereas prolonged and excessively elevated mitochondrial [Ca2+] impair mitochondrial function due to dissipation of the mitochondrial membrane potential (Δψm) and increased ROS production (Brookes et al., 2004; Duchen, 2000). To test whether RyR1 mediated SR Ca2+ leak can lead to mitochondrial dysfunction we measured changes in mitochondrial/cytosol [Ca2+] and mitochondrial membrane potential in FDB muscle fibers in the presence of rapamycin (15 µM), which causes SR Ca2+ leak by disrupting RyR1-calstabin1 interactions (Ahern et al., 1997; Brillantes et al., 1994). FDB fibers loaded with Rhod-2 were used to measure mitochondrial Ca2+ (Aydin et al., 2009; Bruton et al., 2003). Fibers were co-stained with Mitotracker Green to confirm that Rhod-2 was loaded into mitochondria (Figure S4A-C). Rapamycin treatment caused a time-dependent increase in the Rhod-2 signal, indicating Ca2+ accumulation in the mitochondria (Figure 2A). We next measured mitochondrial membrane potential in FDB fibers using the fluorescent indicator tetra-metyl rhodamine ethyl ester (TMRE) (Aydin et al., 2009; Duchen, 2004; Nagy et al., 2011). Rapamycin induced a progressive reduction in the TMRE signal (Figure 2B). At the end of each experiment, an uncoupler of the mitochondrial membrane potential (carbonyl cyanide p-trifluoromethoxyphenylhydrazone; FCCP) was added to the muscle fiber resulting in a further reduction in TMRE fluorescence (Figure 2B). Rapamycin has other activities in addition to disrupting RyR1-calstabin1 interactions including inhibition of mTOR signaling. FK506 similarly depletes calstabin1 from RyR1 channels, but does not inhibit mTOR (Brillantes et al., 1994) and caused a reduction in the TMRE signal similar to rapamycin (Figure S4D). Moreover, rapamycin had no effect on mitochondrial membrane potential in FDB muscle from mice with a skeletal muscle targeted deficiency in FKBP12 (calstabin1) (Tang et al., 2004) (Figure S4E). Together, these results indicate that inhibiting RyR1-calstabin1 binding leads to Ca2+ leak and mitochondrial dysfunction. Mitochondrial dysfunction is typically associated with increased ROS production. We used the fluorescent indicator MitoSOX Red to measure mitochondrial superoxide (O2*−) production (Aydin et al., 2009). Rapamycin caused an increase in the MitoSOX Red signal (control: 114±6%, N = 5, rapamycin: 171±8%, N = 10, P < 0.01; Figure 2C). At the end of each experiment the electron transport chain inhibitor Antimycin A (10 µM) was applied as a positive control (Mukhopadhyay et al., 2007). Ca2+ leak and ROS production in muscle are associated with increased reactive nitrogen species (RNS) (Durham et al., 2008) and increased nitrosylation of the RyR1 was observed in skeletal muscle from aged mice (Fig. 1G). We loaded FDB fibers with the fluorescent RNS indicator DAF-FM (DAF) to examine the effects of leaky RyR1 on RNS production. In the presence of rapamycin, DAF fluorescence increased significantly compared to untreated controls (rapamycin: 121±3%, baseline: 102±5%, P < 0.001, N=6; Figure 2D), consistent with increased RNS production. Taken together, our data indicate that acute induction of RyR1-mediated SR Ca2+ leak leads to defective mitochondrial function associated with elevated ROS and RNS production.
Figure 2. Effects of SR Ca2+ leak on mitochondrial membrane potential, ROS and RNS production in skeletal muscle fibers.

(A) Rapamycin-induced increase in mitochondrial Ca2+ measured with the fluorescent indicator Rhod-2. The Rhod-2 signal was measured from three mitochondria rich regions in each cell (mitochondria rich regions were confirmed using mitotracker green, see Fig. S4) and normalized to baseline; * P < 0.05 indicates significant difference of the control rapamycin (N = 7) compared to S107 (N = 6) and control no rapamycin (N = 4) groups (ANOVA). In the S107 rapamycin group FDB fibers were incubated with S107 (5 µM) for 2–4 hrs before starting the experiment. (B) Changes in mitochondrial membrane potential (measured with TMRE fluorescence) with respect to different interventions. Arrow indicates onset of rapamycin (for groups control rapamycin and S107 rapamycin) or repetitive twitch stimulation without rapamycin (twitching). The dashed line indicates application of FCCP (300 nM). * P < 0.05 indicates significant difference (ANOVA) among the groups control rapamycin (n = 5) and control no rapamycin (n = 3) or group twitching (n = 6) or S107 rapamycin (n = 5). (C) Mitochondrial superoxide production in FDB fibers measured with MitoSOX Red. Arrow indicates when rapamycin was applied. The dashed line indicates application of Antimycin A (10 µM) as a positive control for superoxide production. Control rapamycin (n = 8), Control no rapamycin (n = 5), S107 rapamycin (n = 5). * P < 0.05 indicates significant difference between the control rapamycin group and no rapamycin or S107 groups (ANOVA). (D) The effect of rapamycin-induced Ca2+ leak on RNS production in FDB fibers measured with the RNS indicator DAF. Arrow indicates application of rapamycin. The NO donor S-nitroso-N-acetylpenicillamine (SNAP; 100 nM) was applied as a positive control at the end of each experiment (indicated by dashed line). Control rapamycin (n = 6), control no rapamycin (n = 6), S107 rapamycin (n = 5). * P < 0.05 indicates significant difference (ANOVA) for the control rapamycin group compared to the S107 and no rapamycin groups. All data are shown as mean ± SEM. See also Figure S4.
The small molecule S107 inhibits SR Ca2+ leak by reducing the stress-induced depletion of calstabin from the RyR channel complex (Andersson and Marks, 2010; Bellinger et al., 2009; Lehnart et al., 2008). Therefore, we sought to determine whether the rapamycin-induced Ca2+ leak and the consequent detrimental effects on mitochondrial membrane potential could be prevented by treatment with S107. FDB fibers from young WT mice were incubated with S107 (5 µM) for 2–3 hours before the start of the experiment (Shan et al., 2010). The rapamycin-induced increase in Rhod-2 fluorescence as well as the loss of mitochondrial membrane potential and increase in MitoSOX Red and DAF signals were prevented by S107 (Figure 2A-D) while intermittent twitch stimulation of the FDB fibers (which causes large but transient increases in cytoplasmic [Ca2+]) did not alter the mitochondrial membrane potential (Figure 2B). These data indicate that pathologic SR Ca2+ leak, but not action potential-mediated SR Ca2+ release, have detrimental effects on mitochondrial function and that the effects of pathologic Ca2+-leak on mitochondrial membrane potential could be prevented by S107 treatment.
Inhibiting RyR1 Ca2+ leak improves muscle force and exercise capacity
Aged mice were housed individually in cages equipped with running wheels, and voluntary running time and distance were continuously recorded. Half of the aged mice (n = 13) were supplied with S107 (~50 mg kg−1 d−1) in their drinking water for a 4-week period, while the other half (n = 14) served as the control group. Water consumption was not different between the S107 and vehicle groups (average daily consumption in ml, aged + S107, 8.4 ± 0.63; aged, 7.9 ± 0.4; ± SEM, P = NS). The S107 treated mice exhibited significantly increased running distance (aged + S107, 94 ± 14 km; aged, 57 ± 7 km; mean total distance in 4 weeks, ±SEM, P < 0.05; Figure 3A) and average speed (S107, 155 ± 20 m/h; vehicle, 88 ± 11 m/h; mean speed over the entire treatment period, ± SEM, P < 0.01). Moreover, the S107 treated mice exhibited more episodes at higher running speeds (Figure 3B). Thus, S107-treated aged mice exhibited increased exercise capacity and exercised at higher intensity levels compared to controls.
Figure 3. Improved exercise capacity, muscle specific force, and increased calstabin1 in the RyR1 complex following S107 treatment of aged mice.

(A) Daily voluntary running distance in aged mice ± S107 treatment (mean, ± SEM, n = 13 aged + S107, n = 14 aged, * P < 0.05, ANOVA). The arrow indicates start of the S107 treatment. (B) Histogram showing the distribution of the number of 5-minute episodes in which the mice ran at a given speed. The insert shows the tail of the histogram at expanded time scale. Note the increased number of high-speed episodes in S107 treated mice compared to control. (C and D) 70 Hz tetanic contractions in isolated EDL muscles from aged and S107 treated aged mice. (E) Average specific force in EDL muscles from the same mice as in A (mean ±SEM, n = 6 young, 7 aged, * P < 0.05, *** P < 0.001). (F) Immunoblot of immunoprecipitated RyR1 from aged murine skeletal muscle (aged EDL muscles taken from the mice in A and E). (G) Quantification of the immunoblot in F. S107 reduced depletion of calstabin1 from the RyR1 complex in skeletal muscle from aged mice (mean, ±SEM, n=3, ** P < 0.01, compared to young). See also Figure S1 and S2.
To determine the mechanism underlying the improved exercise capacity in S107 treated aged mice we measured muscle force production. There was a significant increase in specific force at all stimulation frequencies in the EDL muscles from the S107 treated mice (mean force at 70 Hz tetanic contraction ± SEM: aged, 201 ± 21 kNm−2, aged + S107, 320 ± 19 kNm−2; P < 0.001; Figure 3C-E). The average tetanic forces for EDL muscles from untreated aged mice at 70 Hz in the experiments shown in Figs. 1 and 3 were not significantly different (aged mice Fig. 1: 250 ± 21 kN/m2, N = 5; aged mice Fig. 3: 196 ± 19 kN/m2, N = 6; P = NS; T-test). The cross-sectional area of EDL muscle fibers from aged mice were slightly smaller compared to young mice (Figure S5). There was no difference in the fiber size between S107 treated versus untreated aged mice (Figure S5). Furthermore, EDL muscle mass was not different between the S107 and vehicle treated aged mice (aged + S107, 12.7 ± 0.6 mg; aged, 13.8 ± 0.6 mg; P = 0.22).
EDL muscles used in the force measurements were also analyzed for post-translational modifications of RyR1. There were no differences in the levels of nitrosylation, oxidation or PKA phosphorylation of RyR1 from muscles from mice treated with S107 vs. control. This suggests that the oxidative protein modifications are irreversible, as has been reported (Palmese et al., 2011), and protein nitrosylation may have a half-life longer than the 4 weeks of S107 treatment, which has also been reported (Hess et al., 2005). Muscles from the S107 treated mice did, however, show significantly more calstabin1 in the RyR1 complexes compared to skeletal muscle from control mice (Figures 3F and 3G).
To determine whether the improvement in exercise and muscle specific force were associated with improved SR Ca2+ release, Ca2+ responses to tetanic stimulation were recorded in FDB myocytes from S107 treated and control mice. Tetanic Ca2+ transients were significantly increased in FDB myocytes from S107 treated mice compared to those from the control group (mean ΔF/F0 at 70 Hz tetanic contraction ± SEM: aged, 3.7 ± 0.3; S107, 5.9 ± 0.5; n = 13, P < 0.01; Figure 4A-C). Thus, the improved tetanic SR Ca2+ release in muscles from S107 treated aged mice accounts for the observed increase in muscle force production (Figure 3C-E). In order to assess the effects of S107 treatment on the single-channel properties of isolated skeletal muscle RyR1 channels, SR membranes were prepared from EDL muscles and fused to planar lipid membrane bilayers and Ca2+ fluxes through RyR1 channels were recorded as previously described (Brillantes et al., 1994) using conditions that simulate resting muscle (150 nM Ca2+ on the cis, “cytosolic” side of the channel). The open probability (Po) of skeletal muscle RyR1 channels from young mice was low, as expected for normal skeletal muscle RyR1 channels (Figure 4D-E). In contrast, skeletal muscle RyR1 channels from the aged mice exhibited a significantly increased Po, whereas channels from S107 treated aged mice displayed normal, low Po (Figure 4D-E). Taken together, these data suggest that S107 treatment improves exercise capacity in aged mice by reducing the loss of calstabin1 from the channel complexes, and restoring normal (non-leaky) channel function which in turn results in improved tetanic Ca2+ and muscle specific force production.
Figure 4. S107 reduces SR Ca2+ leak resulting in enhanced tetanic SR Ca2+ release in skeletal muscle from aged mice.

(A and B) Ca2+ transients Fluo-4 fluorescence in FDB muscle fibers during a 70 Hz tetanic stimulation in control mice (A) and mice treated with S107 (B). (C) Peak tetanic Ca2+ amplitudes in the two treatment groups (muscle fibers were taken from the same animals as in Figure 3A-B; mean, ±SEM, n = 10–13, ** P < 0.01). (D) Single channel current traces of skeletal RyR1 channels isolated from young, aged, and aged + S107 treated mice. Single channel currents were measured at 150 nM cytosolic [Ca2+] using Ca2+ as a charge carrier at 0 mV. Channel openings are shown as upward deflections; the closed (c-) state of the channel is indicated by horizontal bars in the beginning of each trace. Tracings from over three minutes of recording for each condition showing channel activity at two time scales (5 s in upper trace and 500 ms in lower trace) as indicated by dimension bars, and the respective Po, To (average open time) and Tc (average closed time) are shown above each 5 s trace. The activity of the channel indicated by the thick black bar is shown on the expanded time scale (the 500 ms trace below). (E) Bar graph summarizing Po at 150 nM cytosolic [Ca2+] in young n = 4, aged n = 5, and aged + S107 treated n = 5 channels (mean, ± SEM, * P < 0.05 (ANOVA)).
To further test the hypothesis that the RyR1 channels in aged muscle are leaky we recorded spontaneous releases of SR Ca2+, i.e. Ca2+ sparks (Bellinger et al., 2009; Shirokova and Niggli, 2008; Ward et al., 2003), in permeabilized EDL muscle fiber bundles from S107 treated and muscles from untreated adult and aged mice. Ca2+ spark frequency was significantly increased in muscle fibers from aged compared to young mice examined under identical conditions by blinded observers (Figure 5A-B). Furthermore, S107 treatment in vivo resulted in significantly reduced Ca2+ spark frequencies in EDL muscle from aged mice (Figure 5A-B). Thus, both the increased Ca2+ spark frequency and RyR1 Po in muscle from the aged mice support a model in which leaky RyR1 channels are associated with defective SR Ca2+ release and reduced muscle force production. These defects can be reversed using S107 which inhibits the RyR1-mediated SR Ca2+ leak by preventing depletion of the calstabin1 subunit from the channel complex, resulting in stabilization of the closed state of the channel in resting muscle (Brillantes et al., 1994).
Figure 5. Elevated Ca2+ spark frequency is reversed by S107 in EDL muscle from aged WT mice and RyR1-S2844D mice but not in calstabin1 KO mice.

(A) Line scans of Fluo-4 fluorescence from permeablized EDL muscle fibers (young: upper panel; aged: middle panel; aged +S107: lower panel) showing Ca2+ spark activity. The heat diagram indicates the normalized change in fluorescence intensity (ΔF/F0). (B) Bar graph showing average Ca2+ spark frequency (the number of sparks examined were: 1219 in the young mice, n = 530 line scans from 32 fibers and 6 animals; 7389 in the vehicle-treated aged mice, n = 505 line scans from 30 fibers and 6 animals; 3713 in aged mice treated with S107, n = 414 line scans from 25 fibers and 5 animals; 673 in the untreated RyR1-S2844D mice, n = 240 line scans from 15 fibers and 3 animals; 2405 in S107 treated RyR1-S2844D mice, n = 210 line scans from 14 cells and 3 animals; mean, ± SEM, *** P < 0.001 (ANOVA).
S107 requires calstabin1 to reduce Ca2+ leak and improve muscle function
(Shan et al., 2010)Extreme exercise and heart failure are both associated with “leaky” RyR1 due to phosphorylation of RyR1 at serine 2844 resulting in skeletal muscle weakness (Bellinger et al., 2008; Reiken et al., 2003). To further test whether “leaky” RyR1 can cause muscle weakness we developed a knock-in mouse model with a “leaky” RyR1 due to substitution of aspartic acid for serine 2844 (RyR1-S2844D mouse). Compared to WT muscles, EDL muscles from 6-month-old RyR1-S2844D mice displayed increased Ca2+ spark frequency (Figure 5B) and increased single RyR1 channel open probability under resting conditions (Figure S6A-C). These abnormalities observed in muscle from 6-month-old RyR1-S2844D mice were comparable to those observed in 24-month old WT muscle, consistent with “leaky” RyR1. RyR1 from RyR1-S2844D mice were progressively oxidized, nitrosylated and depleted of calstabin1 by 6 months of age (Figure 6A-B), again comparable to changes observed in RyR1 complexes from 24 month old WT mice. EDL muscles from the RyR1-S2844D mouse exhibited a significant increase in mitochondria with abnormal morphology (Figure S3C,E). Muscle specific force and action potential-triggered Ca2+ transient amplitudes were reduced in the 6-month-old RyR1-S2844D mice compared to WT mice (Figure 6C and 6D). Four weeks of S107 treatment in vivo reduced the elevated Ca2+ spark frequency (Figure 5B) and improved muscle force in 6-month-old RyR1-S2844D mice, both to levels comparable to those observed in muscle from young WT mice (Figure 6C). In summary, the SR Ca2+ leak and impaired muscle force production observed in skeletal muscle from 6-month old RyR1-S2844D mice were comparable to those found in 24-month old WT muscle.
Figure 6. Improved muscle function and exercise capacity in S107 treated mice requires calstabin1.

(A) Immunoblot of immunoprecipitated RyR1 from WT, 1 month old (1 m), 6 month old (6 m) RyR1-S2844D mice and 6 month old (6 m) RyR1-S2844D mice that was treated with S107 (from the same animals as in (C)). (B) Quantification of band intensities in A (mean ± SEM, n = 3, ** P < 0.01 compared to WT, ## P < 0.01 compared to S2844D 1 m, ANOVA). RyR1 from RyR1-S2844D mice are progressively oxidized (DNP) and depleted of calstabin1 with age. (C) EDL muscle force-frequency curves in 6 month old RyR1-S2844D mice and young WT mice. S107 treatment (4 weeks) significantly increased muscle force in the RyR1-S2844D mice (mean ± SEM). (D) Peak Ca2+ transient amplitudes at 70 Hz tetanic stimulation [peak Fluo-4 fluorescence (F) was normalized to resting fluorescence (F0), ΔF/F0]. (E) EDL muscle from muscle-specific calstabin1 KO mice produce significantly less force compared to young WT. S107 treatment (4 weeks) did not restore EDL muscle force in muscle-specific calstabin1 KO mice. (F) Daily voluntary running distance in young WT mice ± S107 treatment and in muscle-specific calstabin1 KO mice ± S107 treatment (mean, ± SEM; * P < 0.05 (ANOVA). The arrow indicates start of the S107 treatment. (G) Immunoblot of immunoprecipitated muscle RyR1 from young WT, aged (18 month) WT, young transgenic mice with mitochondrial targeted overexpression of catalase (MCAT) and aged (18 month) MCAT mice. (H) Quantification of band intensities in G (mean ± SEM, n = 4 all groups; *** P < 0.001, ## P < 0.01 compared to aged WT (ANOVA). The pooled data in the figure are mean ± SEM; * P < 0.05, *** P < 0.001 (ANOVA); the number of samples are indicated in parentheses in the figure legend. See also Figure S2.
To further test whether the beneficial effects of S107 in aged mice can be attributed to the restored RyR1-calstabin1 binding with the consequent reduction in SR Ca2+ leak, we conducted a series of experiments in which we treated muscle specific calstabin1 deficient (calstabin1 KO) mice with S107. In agreement with the results from Tang et al (Tang et al., 2004), we found that muscle specific calstabin1 KO mice exhibited reduced EDL muscle specific force (Figure 6E) compared to WT mice. Calstabin1 KO muscles had a “leaky RyR1” phenotype as indicated by enhanced frequency of Ca2+ sparks compared to young WT muscle (Figure 5B). Electron microscopy of EDL muscle from the muscle specific calstabin1 KO mouse revealed a significant increase in mitochondria with abnormal morphology (Figure S3D,E). Muscle specific calstabin1 KO FDB muscle fibers loaded with the ROS indicator DCF had increased fluorescence compared to the young WT, consistent with oxidative stress (Figure S1). However, S107 treatment, which reduced DCF fluorescence in aged WT mice, did not alter the DCF signal in muscle specific calstabin1 KO muscle, suggesting that the ability of S107 to reduce SR Ca2+ leak and oxidative stress requires calstabin1 (Figure S1). Consistent with a reduction in muscle specific force, action potential triggered Ca2+ release measured in Fluo-4 loaded FDB fibers from muscle specific calstabin1 KO mice exhibited reduced Ca2+ transient amplitudes compared to young WT (Figure 6D). Moreover, exercise capacity was reduced in the calstabin1 KO mice compared to WT (Fig 6E). Treatment with S107 in the drinking water (~50 mg kg−1 d−1) for 4 weeks did not restore specific force, Ca2+ transient amplitudes, Ca2+ spark frequency or the DCF signal in calstabin1 KO muscle (Figures 5B, 6D, 6F, S1A and S1B). Furthermore, exercise capacity in young WT or calstabin1 KO mice was not improved by S107 treatment (Figure 6F). Taken together, these data indicate that the beneficial effects of S107 on Ca2+ handling and muscle function observed in both aged mice and in RyR1-S2844D mice require calstabin1 in skeletal muscle. Moreover, RyR1 from calstabin1 KO mice were oxidized and nitrosylated (Figure S2C,D). This suggests that ROS, produced downstream of the SR Ca2+ leak, feeds back to the RyR1 where it causes oxidative modifications.
To directly test the hypothesis that mitochondrial-derived ROS causes age-dependent RyR1 oxidation we examined RyR1 oxidation in muscles from young (3 month) and aged (18 month) transgenic mice with mitochondrial targeted overexpression of catalase (MCAT) (Lee et al., 2010; Schriner et al., 2005). RyR1 oxidation was substantially reduced, nitrosylation slightly reduced and calstabin1 binding was preserved in samples from aged MCAT mice compared to age-matched WT controls (Fig. 6G,H). We treated SR microsomes from WT muscle with oxidizing (H2O2) and nitrosylating (Noc-12) compounds alone and in combination to examine the effects of these modifications in more detail. Either nitrosylation or oxidation of the RyR1 led to partial depletion of calstabin1, whereas a combination of these two post-translational modifications led to more extensive depletion (Figure S2E-F). Thus, oxidation and nitrosylation of RyR1 appear to work additively with respect to calstabin1 depletion.
Discussion
In the present study we show that oxidized RyR1 in muscle from aged mice are depleted of calstabin1 resulting in leaky channels, reduced tetanic Ca2+, decreased muscle specific force and impaired exercise capacity. To confirm that “leaky” RyR1 can cause the defects in function observed in aged muscle we generated a “leaky RyR1” model (RyR1-S2844D mice) and used muscle specific calstabin1 deficient mice. Both strains prematurely develop a skeletal muscle phenotype similar to that observed in 24 month old WT mice. Moreover, the small molecule rycal drug S107, that preserves RyR1-calstabin 1 binding and stabilizes RyR1 channels, reduced Ca2+ spark frequency, improved tetanic Ca2+ release, restored muscle specific force and improved exercise capacity in aged WT mice. S107 had no beneficial effects in muscle specific calstabin1 KO mice indicating that the mechanism of action of the drug involves calstabin1.
Our present study is consistent with previous reports showing impaired Ca2+ release in aged muscle (Jimenez-Moreno et al., 2008), reduced SR Ca2+ release in SR vesicles (Russ et al., 2010) and reduced caffeine-induced release of the SR Ca2+ store (Romero-Suarez et al., 2010). In addition to the pathologic SR Ca2+ leak via remodeled RyR1 that we report in the present study, other defects such as uncoupling between the voltage sensor and RyR1 (Jimenez-Moreno et al., 2008) may also contribute to muscle weakness in aging.
Mitochondria are the primary source of superoxide (O2*−) and, in the presence of NO, O2*− facilitates the production of reactive nitrogen species (RNS) and protein nitrosylation (Szabo et al., 2007). Skeletal muscle RyR1 is sensitive to redox changes (Xia et al., 2000) and we have previously shown that leaky RyR1 in muscular dystrophy and in muscle fatigue after extreme exercise are cysteine-nitrosylated and depleted of calstabin1. In this study we show that the skeletal muscle RyR1 from aged mice are oxidized and nitrosylated. Moreover, the leaky RyR1 from RyR1-S2844D mice become progressively oxidized with age. Furthermore, acute induction of SR Ca2+ leak by with rapamycin or FK506 or using muscle specific calstabin1 deficient mice, increased ROS and RNS production. Taken together these findings suggest that SR Ca2+ leak may exacerbate mitochondrial dysfunction by causing mitochondrial Ca2+ overload which in turn leads to increased RNS and ROS production. The increase in oxidative stress would further promote RyR1 leak by further oxidizing the channel and depleting it of the stabilizing subunit calstabin1.
The 4-week S107 treatment partially reduced oxidative stress in the aged muscle (Figure S1) but did not reduce oxidation or cysteine-nitrosylation of the RyR1. Oxidation-dependent carbonyl modifications of proteins have been reported to be irreversible (Palmese et al., 2011). Thus, inhibiting SR Ca2+ leak would decrease additional oxidation of the RyR1 but would not necessarily reverse the oxidation of RyR1. SNO protein modification is critically dependent on local production of NO, e.g. by NO synthases (NOS). The stability of SNO protein modifications are highly variable and can be influenced by multiple factors including the protein topology, redox state and pH in the vicinity of the affected protein (Hess et al., 2005). Moreover, while the effects of Ca2+ overload on mitochondrial dysfunction and increased ROS production are multi-factorial, some of them are believed to be irreversible (Feissner et al., 2009; Jekabsone et al., 2003). The half-life of the mitochondria is 2–4 weeks (Kowald, 2001; Menzies and Gold, 1971) and the half-life of the RyR1 protein is ~10 days in muscle from aged rats (Ferrington et al., 1998). Thus, the continued ROS production from mitochondria and the slow turnover of the RyR1 protein contribute to the observation that RyR1 oxidation is not reduced during a four week course of S107 treatment, despite inhibition of the intracellular Ca2+ leak. Moreover, ROS generation from non-mitochondrial sources, e.g. non-phagocytic NAD(P)H oxidase (NOX), could contribute as they have been reported to activate RyR channels(Hidalgo et al., 2006; Xia et al., 2003). Our results, however, are consistent with a “vicious cycle” whereby SR Ca2+ leak and mitochondrial ROS are locally amplified and lead to progressive age-dependent muscle dysfunction (Figure 7). Moreover, our data show that despite persistent RyR1 oxidation and nitrosylation, S107 treatment inhibits the loss of calstabin1 from RyR1 resulting in stabilization of the channel closed state. Thus, S107 inhibits the pathologic SR Ca2+ leak, as long as calstabin1 is present and ameliorates age-dependent loss of muscle function, despite persistent oxidation and nitrosylation of RyR1.
Figure 7. Model of RyR1-mediated SR Ca2+ leak and mitochondrial dysfunction in aging skeletal muscle.
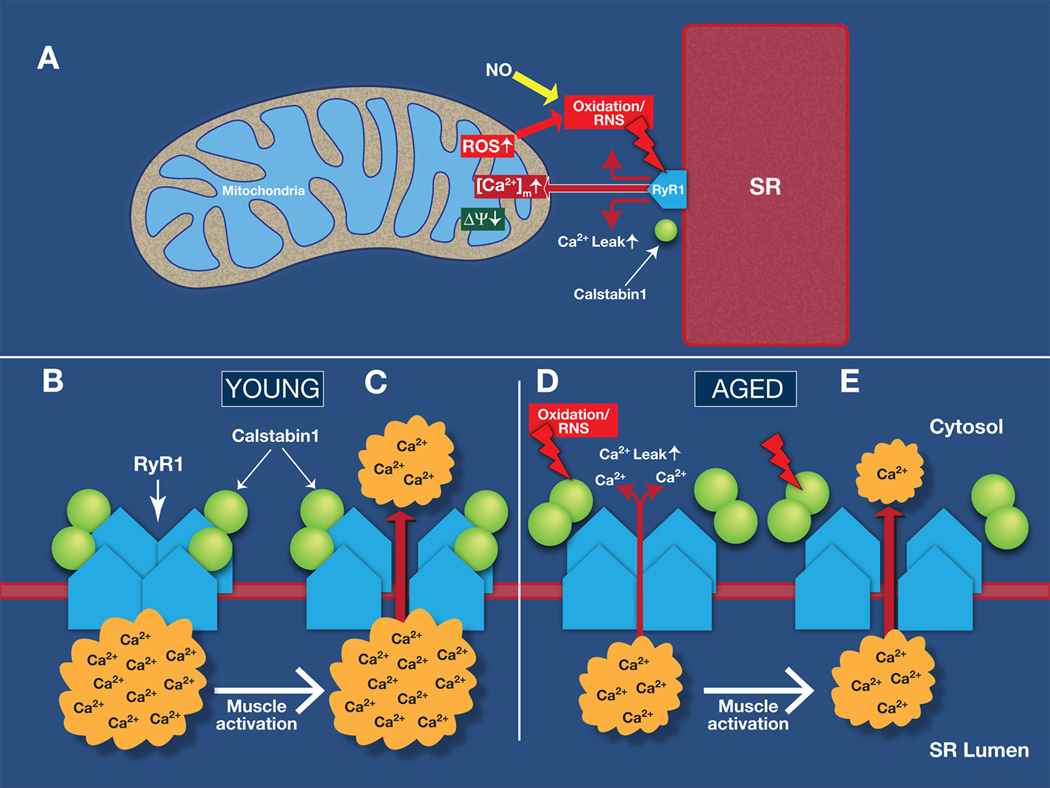
(A) Sarcoplasmic reticulum (SR) Ca2+ leak due to oxidation-dependent modifications of RyR1 exacerbates mitochondrial dysfunction and production of reactive oxygen species (ROS). This causes remodeling of RyR1 resulting in SR Ca2+ leak, which impairs muscle force production. (B-C) The RyR1 from young mice are not “leaky”, the SR Ca2+ stores are filled and activation of the myocyte leads to SR Ca2+ release which triggers muscle contraction. (D-E) In aging, ROS and reactive nitrogen species (RNS)–mediated remodeling of RyR1 results in dossociation of the RyR1 stabilizing subunit calstabin1 and SR Ca2+ leak. Under these conditions, muscle activation will lead to reduced SR Ca2+ release and impaired muscle force. Ryanodine receptor type 1: RyR1 Reactive oxygen species: ROS. Sarcoplasmic reticulum: SR. Mitochondrial [Ca2+]: [Ca2+]m. Mitochondrial membrane potential: Δψm.
Our data showing increased Ca2+ spark frequency in skeletal muscle from aged WT, RyR1-2844D and muscle-specific calstabin1 deficient mice are at odds with a previous study that reported reduced Ca2+ spark activity in aged skeletal muscle (Weisleder et al., 2006). The discrepancy between our results and those of Weisleder et al could be due to differences in methodology since Weisleder et al used intact muscle fibers treated with a hypotonic solution that causes the muscle cells to swell resulting in Ca2+ sparks (Weisleder et al., 2006). In contrast, we used saponin permeabilized muscle fibers in order to control the intracellular conditions (e.g. pH, [Ca2+], [Mg2+] and the Ca2+ dye concentration) which is not possible using intact muscle fibers (Isaeva et al., 2005; Rios et al., 1999; Shirokova and Niggli, 2008). We have previously used this method to study pathological conditions where RyR1 exhibited a leaky phenotype (Bellinger et al., 2009; Reiken et al., 2003; Ward et al., 2003). In addition, the increased Ca2+ spark frequency is consistent with the increased RyR1 Po observed in the aged muscle.
Much of the focus in the field of aging is on therapeutics that target anabolic pathways with hormones including testosterone, growth hormone, and insulin-like growth factor-1 (Lynch, 2008; Rolland et al., 2008) to improve muscle mass. Some studies have demonstrated increased muscle mass without increased muscle strength or power (Lynch, 2008; Rolland et al., 2008). Inhibition of the endogenous negative regulator of myogenesis, myostatin (growth differentiation factor 8), leads to a dramatic increase of muscle mass in mice and cattle (Lynch, 2008). However, muscular dystrophy patients that were treated with an anti-myostatin recombinant human antibody failed to improve muscle power (Wagner et al., 2008). Thus, increasing skeletal muscle mass is not necessarily accompanied by improved muscle function. Indeed, treating aged mice with S107 enhances muscle strength without increasing the size of the muscle, at least during the 4 week period of treatment examined in the present study.
Despite the important role of oxidative stress in aging-dependent pathologies, the use of dietary antioxidants as an experimental treatment for sarcopenia has not demonstrated improvement in muscle function (Kim et al.). A complicating factor is that systemic antioxidants could impair beneficial effects of ROS (Jackson, 2009; Vijg and Campisi, 2008). By inhibiting the SR Ca2+ leak via RyR1 which is due to oxidation, S107 appears to be able to improve muscle function without blocking systemic ROS-dependent signaling and may represent a promising therapeutic option for reducing age-dependent loss of muscle function.
The regulatory mechanisms of aging are likely multifactorial and several signaling pathways seem to contribute, e.g. changes in insulin/insulin like growth factor (IGF), sirtuin, AMP kinase and inflammatory signaling (Kenyon, 2010; Marzetti and Leeuwenburgh, 2006). Moreover, mitochondrial dysfunction is strongly implicated in the aging mechanism (Balaban et al., 2005; Larsson, 2010). In the present study, we show that “leaky” RyR1, mitochondrial dysfunction and oxidative stress conspire to produce age-dependent muscle weakness and reduced exercise capacity.
Experimental procedures
A detailed description is found in the supporting materials.
Animal models
Aged mice (23–26 months; C57BL/6) came from the National Institute of Aging and young mice (3–6 month, C57BL/6) came from our own breeding. Generation of RyR1-S2844D mice (Figure S7) is described in the supplement. Muscle-specific cal1 −/− (calstabin1) mice were bred in the lab after a breeding pair was generously provided to us by Dr Susan Hamilton (Baylor College of Medicine, Houston) (Tang et al., 2004). Muscle lysates from young and aged mice with mitochondrial targeted overexpression of catalase (MCAT) were generously provided to us by Dr Gerald Shulman (Yale University) who used mice provided by Dr Peter S Rabinovitch (University of Washington, Seattle). The mice in the drug treatment experiments were randomized into two groups. The first group received S107 (50 mg/kg/day) in the drinking water while the second group received water only. Voluntary exercise (running wheel) intensity was recorded continuously. All experiments were performed by blinded observers and were conducted in accordance to Columbia University IACUC regulations.
Force and Ca2+ measurements
Extensor digitorum longus (EDL) and flexor digitorum longus (FDB) muscles were dissected. Force measurement was done on whole EDL muscle using a system from Aurora Scientific (Ontario, Canada). For Ca2+ spark measurements, saponin permeabilized fiber bundles from EDL were loaded with the fluorescent Ca2+ indicator Fluo-3 and the frequency of spontaneous SR Ca2+ release was imaged using a confocal microscope (Zeiss, LSM 5 live) in line scan mode. Intact muscle fibers from enzymatically dissociated FDB muscles (Aydin et al., 2009) were loaded with the Ca2+ indicator Fluo-4 AM. The FDB fibers were electrically stimulated to tetanic contraction and the Fluo-4 signal was imaged using a confocal microscope (Zeiss, LSM 5 live).
Measurements of mitochondrial Ca2+, mitochondrial membrane potential, ROS and RNS
Enzymatically isolated FDB muscle fibers were loaded with the fluorescent indicators Rhod-2 and the mitochondrial marker Mitotracker green, TMRE, MitoSOX Red and DAF-FM to measure mitochondrial Ca2+, mitochondrial membrane potential, ROS and RNS respectively.
RyR1 Immunoblotting
Muscle samples were prepared as described (Bellinger et al., 2009). Immunoblots were developed with anti-RyR (Affinity Bioreagents, Bolder, CO 1:2,000), anti-phospho-RyR1-pSer2844 (1:5000), an anti-Cys-NO antibody (Sigma, St. Louis, MO, 1:2,000), or an anti-calstabin antibody (1:2,500). To determine channel oxidation, the carbonyl groups in the protein side chains within the immunoprecipitate are derivatized to 2,4-dinitrophenylhydrazone (DNP-hydrazone) by reaction with 2,4- dinitrophenylhydrazine (DNPH). The DNP signal associated with RyR is determined using an anti-DNP antibody.
Single RyR1 channel
Crude SR vesicles containing RyR1 were fused to planar lipid bilayers. Conductance through the RyR1 was measured using Ca2+ as the charge carrier. RyR1 openings were quantified and open probability (Po) was calculated.
Histology and electron microscopy
Preparation of tissue samples and microscopy was performed according to established methods as described in the supporting experimental procedures.
Supplementary Material
Highlights.
RyR1 in skeletal muscle is oxidized, nitrosylated and depleted of calstabin1 with age
Maladaptation of the RyR1 channel leads to SR Ca2+ leak and muscle weakness
Vicious cycle: Ca2+ leak raises mitochondrial ROS, which oxidizes RyR1 enhancing leak
The RyR1 stabilizing drug S107 fixes Ca2+ leak and improves exercise capacity in aging
Acknowledgements
This project was supported by grants from the NHLBI to A.R.M., and the Swedish Research Council to D.A. The authors thank Dr Susan Hamilton (Baylor College of Medicine, Houston, TX, USA) for providing the muscle-specific FKBP12 (calstabin1)-deficient mice. We thank Dr Peter S Rabinovitch (University of Washington, Seattle) and Dr Gerald Shulman (Yale University) for generously providing us with muscles from MCAT mice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: A.R. Marks is a consultant for a start-up company, ARMGO Pharma Inc., that is targeting RyR1 to improve exercise capacity in muscle diseases
References
- Ahern GP, Junankar PR, Dulhunty AF. Subconductance states in single-channel activity of skeletal muscle ryanodine receptors after removal of FKBP12. Biophys J. 1997;72:146–162. doi: 10.1016/S0006-3495(97)78654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen DG, Lamb GD, Westerblad H. Skeletal muscle fatigue: cellular mechanisms. Physiological Reviews. 2008;88:287–332. doi: 10.1152/physrev.00015.2007. [DOI] [PubMed] [Google Scholar]
- Andersson DC, Marks AR. Fixing ryanodine receptor Ca leak - a novel therapeutic strategy for contractile failure in heart and skeletal muscle. Drug Discov Today Dis Mech. 2010;7:e151–e157. doi: 10.1016/j.ddmec.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aracena-Parks P, Goonasekera SA, Gilman CP, Dirksen RT, Hidalgo C, Hamilton SL. Identification of cysteines involved in S-nitrosylation, S-glutathionylation, and oxidation to disulfides in ryanodine receptor type 1. J Biol Chem. 2006;281:40354–40368. doi: 10.1074/jbc.M600876200. [DOI] [PubMed] [Google Scholar]
- Aydin J, Andersson DC, Hanninen SL, Wredenberg A, Tavi P, Park CB, Larsson NG, Bruton JD, Westerblad H. Increased mitochondrial Ca2+ and decreased sarcoplasmic reticulum Ca2+ in mitochondrial myopathy. Human Molecular Genetics. 2009;18:278–288. doi: 10.1093/hmg/ddn355. [DOI] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Barreiro E, Hussain SN. Protein carbonylation in skeletal muscles: impact on function. Antioxid Redox Signal. 12:417–429. doi: 10.1089/ars.2009.2808. [DOI] [PubMed] [Google Scholar]
- Bellinger AM, Reiken S, Carlson C, Mongillo M, Liu X, Rothman L, Matecki S, Lacampagne A, Marks AR. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med. 2009;15:325–330. doi: 10.1038/nm.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinger AM, Reiken S, Dura M, Murphy PW, Deng SX, Landry DW, Nieman D, Lehnart SE, Samaru M, Lacampagne A, et al. Remodeling of ryanodine receptor complex causes "leaky" channels: a molecular mechanism for decreased exercise capacity. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:2198–2202. doi: 10.1073/pnas.0711074105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasova E, Moschella MC, Jayaraman T, Landers M, Ehrlich BE, Marks AR. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. 1994;77:513–523. doi: 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. American Journal of Physiology Cell Physiology. 2004;287:C817–C833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- Brooks SV, Faulkner JA. Contractile properties of skeletal muscles from young, adult and aged mice. J Physiol. 1988;404:71–82. doi: 10.1113/jphysiol.1988.sp017279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruton JD, Dahlstedt AJ, Abbate F, Westerblad H. Mitochondrial function in intact skeletal muscle fibres of creatine kinase deficient mice. Journal of Physiology. 2003;552:393–402. doi: 10.1113/jphysiol.2003.050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria and Ca2+ in cell physiology and pathophysiology. Cell Calcium. 2000;28:339–348. doi: 10.1054/ceca.2000.0170. [DOI] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria in health and disease: perspectives on a new mitochondrial biology. Mol Aspects Med. 2004;25:365–451. doi: 10.1016/j.mam.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Durham WJ, Aracena-Parks P, Long C, Rossi AE, Goonasekera SA, Boncompagni S, Galvan DL, Gilman CP, Baker MR, Shirokova N, et al. RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell. 2008;133:53–65. doi: 10.1016/j.cell.2008.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feissner RF, Skalska J, Gaum WE, Sheu SS. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front Biosci. 2009;14:1197–1218. doi: 10.2741/3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrington DA, Krainev AG, Bigelow DJ. Altered turnover of calcium regulatory proteins of the sarcoplasmic reticulum in aged skeletal muscle. J Biol Chem. 1998;273:5885–5891. doi: 10.1074/jbc.273.10.5885. [DOI] [PubMed] [Google Scholar]
- Gonzalez E, Messi ML, Delbono O. The specific force of single intact extensor digitorum longus and soleus mouse muscle fibers declines with aging. J Membr Biol. 2000;178:175–183. doi: 10.1007/s002320010025. [DOI] [PubMed] [Google Scholar]
- Gonzalez E, Messi ML, Zheng Z, Delbono O. Insulin-like growth factor-1 prevents age-related decrease in specific force and intracellular Ca2+ in single intact muscle fibres from transgenic mice. J Physiol. 2003;552:833–844. doi: 10.1113/jphysiol.2003.048165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis MC, Yankner BA. The aging stress response. Mol Cell. 2010;40:333–344. doi: 10.1016/j.molcel.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herndon LA, Schmeissner PJ, Dudaronek JM, Brown PA, Listner KM, Sakano Y, Paupard MC, Hall DH, Driscoll M. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature. 2002;419:808–814. doi: 10.1038/nature01135. [DOI] [PubMed] [Google Scholar]
- Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- Hidalgo C. Cross talk between Ca2+ and redox signalling cascades in muscle and neurons through the combined activation of ryanodine receptors/Ca2+ release channels. Philos Trans R Soc Lond B Biol Sci. 2005;360:2237–2246. doi: 10.1098/rstb.2005.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo C, Sanchez G, Barrientos G, Aracena-Parks P. A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S -glutathionylation. Journal of Biological Chemistry. 2006;281:26473–26482. doi: 10.1074/jbc.M600451200. [DOI] [PubMed] [Google Scholar]
- Isaeva EV, Shkryl VM, Shirokova N. Mitochondrial redox state and Ca2+ sparks in permeabilized mammalian skeletal muscle. J Physiol. 2005;565:855–872. doi: 10.1113/jphysiol.2005.086280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MJ. Strategies for reducing oxidative damage in ageing skeletal muscle. Adv Drug Deliv Rev. 2009;61:1363–1368. doi: 10.1016/j.addr.2009.07.018. [DOI] [PubMed] [Google Scholar]
- Jang YC, Lustgarten MS, Liu Y, Muller FL, Bhattacharya A, Liang H, Salmon AB, Brooks SV, Larkin L, Hayworth CR, et al. Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. Faseb J. 2010;24:1376–1390. doi: 10.1096/fj.09-146308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jekabsone A, Ivanoviene L, Brown GC, Borutaite V. Nitric oxide and calcium together inactivate mitochondrial complex I and induce cytochrome c release. J Mol Cell Cardiol. 2003;35:803–809. doi: 10.1016/s0022-2828(03)00137-8. [DOI] [PubMed] [Google Scholar]
- Jimenez-Moreno R, Wang ZM, Gerring RC, Delbono O. Sarcoplasmic reticulum Ca2+ release declines in muscle fibers from aging mice. Biophys J. 2008;94:3178–3188. doi: 10.1529/biophysj.107.118786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- Kim JS, Wilson JM, Lee SR. Dietary implications on mechanisms of sarcopenia: roles of protein, amino acids and antioxidants. J Nutr Biochem. 21:1–13. doi: 10.1016/j.jnutbio.2009.06.014. [DOI] [PubMed] [Google Scholar]
- Kowald A. The mitochondrial theory of aging. Biol Signals Recept. 2001;10:162–175. doi: 10.1159/000046885. [DOI] [PubMed] [Google Scholar]
- Larsson NG. Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem. 2010;79:683–706. doi: 10.1146/annurev-biochem-060408-093701. [DOI] [PubMed] [Google Scholar]
- Lee HY, Choi CS, Birkenfeld AL, Alves TC, Jornayvaz FR, Jurczak MJ, Zhang D, Woo DK, Shadel GS, Ladiges W, et al. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab. 2010;12:668–674. doi: 10.1016/j.cmet.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnart SE, Mongillo M, Bellinger A, Lindegger N, Chen BX, Hsueh W, Reiken S, Wronska A, Drew LJ, Ward CW, et al. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest. 2008;118:2230–2245. doi: 10.1172/JCI35346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch GS. Update on emerging drugs for sarcopenia - age-related muscle wasting. Expert Opin Emerg Drugs. 2008;13:655–673. doi: 10.1517/14728210802544476. [DOI] [PubMed] [Google Scholar]
- Marzetti E, Leeuwenburgh C. Skeletal muscle apoptosis, sarcopenia and frailty at old age. ExpGerontol. 2006;41:1234–1238. doi: 10.1016/j.exger.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Menzies RA, Gold PH. The turnover of mitochondria in a variety of tissues of young adult and aged rats. J Biol Chem. 1971;246:2425–2429. [PubMed] [Google Scholar]
- Metter EJ, Talbot LA, Schrager M, Conwit R. Skeletal muscle strength as a predictor of all-cause mortality in healthy men. J Gerontol A Biol Sci Med Sci. 2002;57:B359–B365. doi: 10.1093/gerona/57.10.b359. [DOI] [PubMed] [Google Scholar]
- Moylan JS, Reid MB. Oxidative stress, chronic disease, and muscle wasting. Muscle & Nerve. 2007;35:411–429. doi: 10.1002/mus.20743. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay P, Rajesh M, Yoshihiro K, Hasko G, Pacher P. Simple quantitative detection of mitochondrial superoxide production in live cells. Biochemical & Biophysical Research Communications. 2007;358:203–208. doi: 10.1016/j.bbrc.2007.04.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H. Trends in oxidative aging theories. Free Radic Biol Med. 2007;43:477–503. doi: 10.1016/j.freeradbiomed.2007.03.034. [DOI] [PubMed] [Google Scholar]
- Nagy E, Andersson DC, Caidahl K, Eriksson MJ, Eriksson P, Franco-Cereceda A, Hansson GK, Back M. Upregulation of the 5-lipoxygenase pathway in human aortic valves correlates with severity of stenosis and leads to leukotriene-induced effects on valvular myofibroblasts. Circulation. 2011;123:1316–1325. doi: 10.1161/CIRCULATIONAHA.110.966846. [DOI] [PubMed] [Google Scholar]
- Palmese A, De Rosa C, Marino G, Amoresano A. Dansyl labeling and bidimensional mass spectrometry to investigate protein carbonylation. Rapid Commun Mass Spectrom. 2011;25:223–231. doi: 10.1002/rcm.4863. [DOI] [PubMed] [Google Scholar]
- Rantanen T, Guralnik JM, Foley D, Masaki K, Leveille S, Curb JD, White L. Midlife hand grip strength as a predictor of old age disability. Jama. 1999;281:558–560. doi: 10.1001/jama.281.6.558. [DOI] [PubMed] [Google Scholar]
- Reiken S, Lacampagne A, Zhou H, Kherani A, Lehnart SE, Ward C, Huang F, Gaburjakova M, Gaburjakova J, Rosemblit N, et al. PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: defective regulation in heart failure. Journal of Cell Biology. 2003;160:919–928. doi: 10.1083/jcb.200211012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios E, Stern MD, Gonzalez A, Pizarro G, Shirokova N. Calcium release flux underlying Ca2+ sparks of frog skeletal muscle. J Gen Physiol. 1999;114:31–48. doi: 10.1085/jgp.114.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland Y, Czerwinski S, Abellan Van Kan G, Morley JE, Cesari M, Onder G, Woo J, Baumgartner R, Pillard F, Boirie Y, et al. Sarcopenia: its assessment, etiology, pathogenesis, consequences and future perspectives. J Nutr Health Aging. 2008;12:433–450. doi: 10.1007/BF02982704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Suarez S, Shen J, Brotto L, Hall T, Mo C, Valdivia HH, Andresen J, Wacker M, Nosek TM, Qu CK, et al. Muscle-specific inositide phosphatase (MIP/MTMR14) is reduced with age and its loss accelerates skeletal muscle aging process by altering calcium homeostasis. Aging (Albany NY) 2010;2:504–513. doi: 10.18632/aging.100190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ DW, Grandy JS, Toma K, Ward CW. Ageing, but not yet senescent, rats exhibit reduced muscle quality and sarcoplasmic reticulum function. Acta Physiol (Oxf) 2010 doi: 10.1111/j.1748-1716.2010.02191.x. [DOI] [PubMed] [Google Scholar]
- Saini A, Faulkner S, Al-Shanti N, Stewart C. Powerful signals for weak muscles. Ageing Res Rev. 2009;8:251–267. doi: 10.1016/j.arr.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- Shan J, Betzenhauser MJ, Kushnir A, Reiken S, Meli AC, Wronska A, Dura M, Chen BX, Marks AR. Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J Clin Invest. 2010 doi: 10.1172/JCI37649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirokova N, Niggli E. Studies of RyR function in situ. Methods. 2008;46:183–193. doi: 10.1016/j.ymeth.2008.09.017. [DOI] [PubMed] [Google Scholar]
- Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- Tang W, Ingalls CP, Durham WJ, Snider J, Reid MB, Wu G, Matzuk MM, Hamilton SL. Altered excitation-contraction coupling with skeletal muscle specific FKBP12 deficiency. FASEB J. 2004;18:1597–1599. doi: 10.1096/fj.04-1587fje. [DOI] [PubMed] [Google Scholar]
- Thomas DR. Loss of skeletal muscle mass in aging: examining the relationship of starvation, sarcopenia and cachexia. Clin Nutr. 2007;26:389–399. doi: 10.1016/j.clnu.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Vijg J, Campisi J. Puzzles, promises and a cure for ageing. Nature. 2008;454:1065–1071. doi: 10.1038/nature07216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KR, Fleckenstein JL, Amato AA, Barohn RJ, Bushby K, Escolar DM, Flanigan KM, Pestronk A, Tawil R, Wolfe GI, et al. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol. 2008;63:561–571. doi: 10.1002/ana.21338. [DOI] [PubMed] [Google Scholar]
- Ward CW, Reiken S, Marks AR, Marty I, Vassort G, Lacampagne A. Defects in ryanodine receptor calcium release in skeletal muscle from post-myocardial infarct rats. FASEB Journal. 2003;17:1517–1519. doi: 10.1096/fj.02-1083fje. [DOI] [PubMed] [Google Scholar]
- Weisleder N, Brotto M, Komazaki S, Pan Z, Zhao X, Nosek T, Parness J, Takeshima H, Ma J. Muscle aging is associated with compromised Ca2+ spark signaling and segregated intracellular Ca2+ release. J Cell Biol. 2006;174:639–645. doi: 10.1083/jcb.200604166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia R, Stangler T, Abramson JJ. Skeletal muscle ryanodine receptor is a redox sensor with a well defined redox potential that is sensitive to channel modulators. J Biol Chem. 2000;275:36556–36561. doi: 10.1074/jbc.M007613200. [DOI] [PubMed] [Google Scholar]
- Xia R, Webb JA, Gnall LL, Cutler K, Abramson JJ. Skeletal muscle sarcoplasmic reticulum contains a NADH-dependent oxidase that generates superoxide. Am J Physiol Cell Physiol. 2003;285:C215–C221. doi: 10.1152/ajpcell.00034.2002. [DOI] [PubMed] [Google Scholar]
- Zalk R, Lehnart SE, Marks AR. Modulation of the ryanodine receptor and intracellular calcium. AnnuRevBiochem. 2007;76:367–385. doi: 10.1146/annurev.biochem.76.053105.094237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.