

Abstract
Ozone (O3), a commonly encountered environmental pollutant, has been shown to induce pulmonary fibrosis in different animal models; the underlying mechanism, however, remains elusive. To investigate the molecular mechanism underlying O3-induced pulmonary fibrosis, 6- to 8-week-old C57BL/6 male mice were exposed to a cyclic O3 exposure protocol consisting of 2 days of filtered air and 5 days of O3 exposure (0.5 ppm, 8 h/day) for 5 and 10 cycles with or without intraperitoneal injection of IN-1233, a specific inhibitor of the type 1 receptor of transforming growth factor beta (TGF-β), the most potent profibrogenic cytokine. The results showed that O3 exposure for 5 or 10 cycles increased the TGF-β protein level in the epithelial lining fluid (ELF), associated with an increase in the expression of plasminogen activator inhibitor 1 (PAI-1), a TGF-β-responsive gene that plays a critical role in the development of fibrosis under various pathological conditions. Cyclic O3 exposure also increased the deposition of collagens and alpha smooth muscle actin (α-SMA) in airway walls. However, these fibrotic changes were not overt until after 10 cycles of O3 exposure. Importantly, blockage of the TGF-β signaling pathway with IN-1233 suppressed O3-induced Smad2/3 phosphorylation, PAI-1 expression, as well as collagens and α-SMA deposition in the lung. Our data demonstrate for the first time that O3 exposure increases TGF-β expression and activates TGF-β signaling pathways, which mediates O3-induced lung fibrotic responses in vivo.
Keywords: Ozone, TGF-β, PAI-1, airway fibrosis
Introduction
Ozone (O3) is an extremely reactive gas formed in the troposphere when nitrogen oxides and volatile organic compounds, released mainly from fossil fuel combustion, react in the presence of sunlight. O3 is one of the most abundant ambient urban pollutants with over half of the population in the United States (56%) living in areas with unhealthful levels of ozone (American Lung Association State of the Air 2010). In addition, intentionally generated O3 is used widely in many industrial settings such as purification of drinking water, waste treatment, deodorization of air and sewage gases, bleaching of waxes, oils, wet paper, and textile. Therefore, many workers in different industries including pulp mill workers, outdoor construction workers, copy machine operators, and steel mill welders are exposed to relatively high concentrations of O3 through their work environment.
Although O3 exposure causes a myriad of health effects involving different organ systems, the lung is considered to be the major target. Both clinical and epidemiological studies have shown that O3 induces airway obstruction, noninfectious rhinitis, adult-onset asthma, and decreased lung function. Animal studies have also shown that O3 exposure increases the expression of various inflammatory cytokines and chemokines, causes hyperplasia and hypertrophy in airway and alveolar epithelial cells, and disrupts normal lung development. Collagens are the major components of extracellular matrix (ECM) and increased collagen production/ deposition is a pathological hallmark of fibrosis. Several studies have shown that O3 increases collagen synthesis and deposition in the lung, indicating that O3 induces lung fibrosis (Last et al., 1994; Parks and Roby, 1994; Chang et al., 1995; Stockstill et al., 1995). Importantly, it has been reported that O3-induced lung fibrotic changes persist several months after exposure ceased (Last et al., 1981; Pickrell et al., 1987; Reiser et al., 1987; Sun et al., 1988), suggesting a long-term health impact of O3 exposure. Nonetheless, although it has been well-documented that O3 induces lung fibrosis, the underlying mechanism is unclear. It should also be pointed out that in most of these studies, animals (rats in most cases) were exposed to ozone continuously for many months (Last et al., 1984; Shima and Adachi, 1991; Dormans et al., 1999; van Bree et al., 2001) or 8 h a day for almost their entire life (20–24 months) (Last et al., 1984, 1994; Chang et al., 1995; Stockstill et al., 1995). Such exposure regimens do not resemble environmental or occupational exposure scenarios. Whether exposure to ozone under conditions that mimic environmental/occupational exposure situations can also induce lung fibrosis and the molecular mechanism underlying O3-induced lung fibrosis, however, remain to be determined.
Transforming growth factor beta (TGF-β) is the most potent and ubiquitous profibrogenic cytokine. TGF-β mRNA and protein expression are increased in various lung fibrotic diseases (Bergeron et al., 2003; Fahy et al., 2003; Tzortzaki et al., 2007) and in lung fibrosis models induced by various agents (Williams et al., 1993; Coker et al., 1997; Rube et al., 2000; Churg et al., 2009). Overexpression of TGF-β in transgenic mice or administration of TGF-β to the lung by adenovirus-mediated gene transfer techniques lead to lung fibrosis (Sime et al., 1997; Kolb et al., 2002; Lee et al., 2004; Vicencio et al., 2004). On the other hand, treatment of animals with TGF-β-binding proteins or anti-TGF-β antibodies ameliorates lung fibrosis induced by different stimuli (Yamaguchi et al., 1990; Bonniaud et al., 2005; Higashiyama et al., 2007). All these lines of evidence indicate that TGF-β plays a vital role in the development of lung fibrosis. Nonetheless, although studies have shown that O3 exposure induces the expression of TGF-β in human blood cells and in fibroblasts co-cultured with bronchoepithelial cells (Bocci et al., 1994; Lang et al., 1998; Valacchi and Bocci, 1999), whether O3 increases TGF-β production in the lung in vivo and whether TGF-β mediates O3-induced lung fibrosis, however, are unknown.
In this study, we showed that cyclic O3 exposure increased the expression of TGF-β1, which was followed by deposition of collagens and alpha smooth muscle actin (α-SMA) in airway walls and in the lung. Furthermore, we showed that inhibition of TGF-β signaling with a specific inhibitor to TGF-β type 1 receptor suppressed O3-induced Smad phosphorylation, PAI-1 expression, and airway fibrosis. The results demonstrate for the first time that O3 induces TGF-β expression in vivo and that increased TGF-β-expression mediates O3- induced airway fibrosis.
Materials and methods
Animals and O3 exposure regimen
Six- to eight-week-old C57BL/6 male mice (Charles River, Wilmington, MA) were exposed to a series of O3 exposure cycles consisting of 2 days of filtered air (FA) and 5 days of O3 exposure (0.5 ppm, 8 h/day) for 5 or 10 cycles at the UAB Inhalation Exposure Facility. Animals were exposed to O3 or FA in 0.8-m3 stainless steel chambers that employed 30 vol changes/h turnover rates. O3 was generated from medical grade O2 using a silent arc electrode and bled into the chamber inflow (~22°C; 50% relative humidity) using mass flow controllers. Chamber supply air (O3 and FA chambers) was conditioned via passage through sequential course filter, activated charcoal, and HEPA filter units. O3 concentrations in the Chamber were continuously monitored using a ThermoEnvironmental model 49 Photometric O3 Analyzer. FA (unexposed) controls were treated similarly in all aspects except for O3 in the chambers, and done in parallel. Animals were allowed free access to water, whereas food was withheld during exposure to prevent ingestion of constituents oxidized by O3, which could introduce confounders. The animals were sacrificed after cessation of O3 exposure at the end of the fifth and 10th cycles. All procedures involving animals were approved by the Institutional Animal Care and Use Committees at the University of Alabama at Birmingham.
To determine whether TGF-β mediates O3-induced lung fibrosis, 6–8-week-old C57BL/6 male mice were divided into four groups: group 1 mice were exposed to FA and intraperitoneally (i.p.) injected with 100 μL of sterile saline; group 2 mice were exposed to O3 and i.p. injected with 100 μL of sterile saline; group 3 mice were exposed to FA and i.p. injected with IN-1233, a selective inhibitor of TGF-β type I receptor (Kim et al., 2010), at a dose of 20 mg/kg (dissolved in 100 μL of sterile saline); group 4 mice were exposed to ozone and i.p. injected with the same amount of IN-1233. Exposures were conducted as described above for 10 cycles of air or O3. IN-1233 administration (i.p. injection) was conducted every morning before O3 exposure initiated, started on the first day of O3 exposure and continued to the end of the 10th cycle (discontinued during 2 days of FA exposure period in each cycle).
Bronchoalveolar lavage
Mice were anesthetized and tracheas cannulated with a 22G 1 ½-in bead-tipped needle. Bronchoalveolar lavage (BAL) was performed with 0.8 mL saline and the resulting BAL fluid (BALF) was centrifuged at 500 g for 10 min. The supernatants were collected for ELISA analyses of TGF-β1 and PAI-1.
Lung tissue collection and collagen staining
After lavage, the pulmonary artery was cannulated and the vascular bed was perfused using 310 mOsm PBS. The left main stem bronchus was cannulated; the lung was resected and inflated under constant pressure of 25 cm H2O with 4% paraformaldehyde. Sections were cut 4–6 μm thickness and used for collagen and immunohistochemical staining. Masson’s trichrome staining technique was employed to reveal the collagen deposition. In brief, the sections were deparaffinized and hydrated through gradient alcohol, subsequently fixed in Bouin’s mordant fluid, and the sections sequentially stained using Weigert’s hematoxylin, Biebrich Scarlet-Acid Fuchsin, and Aniline Blue solutions. Quantification of collagen in the airway was done using computer-assisted stereology technique as described before (Fanucchi et al., 1997). The collagen mass (V/S) as measured by volume (μm3) of bronchiolar epithelial cells per unit area (μm2) of basement membrane was estimated from point and intercept counts with the cycloid grid by the equation = (Po × l/p)/(2 × Ibl), where Po represents the points counted for collagen, Ibl represents the number of intercepts of the basal lamina, and l/p = (No. of curves on grid × length of curve)/points on grid.
Measurement of urea concentration in the BALF and plasma
The concentrations of urea in the plasma and BALF samples were measured using a commercially available kit from Teko Diagnostics (cat # B550-400) according to manufactures’ protocol. In brief, 1 mL of the assay reagent was warmed to 37°C for 10 min. After 10 min, 10 μL of BALF or plasma sample was added to it, and absorbance was read at 340 nm for 5 min with 30 s interval. The dilution factor of epithelial lining fluid (ELF) from each mouse was calculated by dividing plasma urea concentration with BALF urea concentration from the same mouse.
ELISA analyses of TGF-β and PAI-1 proteins in the ELF
The amount of TGF-β protein in the BALF was measured using an ELISA kit from e-Biosciences (San Diego, CA) according to the protocol provided by the manufacturer. Total TGF-β was measured after activation of latent TGF-β with HCl. PAI-1 antigen in the BALF was measured using a commercially available ELISA kit from Molecular Innovations (Southfield, MI), which detects active and inactive PAI-1 as well as PAI-1–PA complexes, as we have described previously (Vayalil et al., 2005). The amounts of TGF-β and PAI-1 proteins in ELF were calculated based on standard curves generated with each batch of samples and normalized with dilution factor of ELF as discussed above.
Western analyses of proteins
Lung tissues were homogenized in 500 μL of 0.25 M sucrose solution containing 1 mM EDTA, 20 mM Tris–HCl (pH 7.4), and both protease and phosphatase inhibitors. The homogenates were centrifuged at 3000 g for 10 min at 4°C and the resulting supernatants centrifuged again at 13,000 g for 20 min at 4°C. Protein concentrations were measured using the commercially available BCA protein assay kit (Pierce–Thermo Scientific, Rockford, IL). Forty micrograms of protein were resolved on 10% SDS-PAGE gels. The separated proteins were electrophoretically transferred onto polyvinylidene difluoride (PVDF) membranes and blocked with 5% nonfat dry milk. The membranes were probed at 4°C overnight with the following antibodies: α-SMA Biocare Medical LLC (Concord, CA) at 1:1000 dilution, PAI-1 Molecular Innovations (Novi, MI) at 1:2000 dilution, pSmad2/3 Santa Cruz (Paso Robels, CA) at 1:200 dilution, Smad2/3 Cell Signaling (Danvers, MA) at 1:200 dilution, and β-actin Sigma (Saint Louise, Missouri) at 1: 5000 dilution, and then probed with corresponding HRP-tagged secondary antibodies. Protein bands were detected using ECL solution Pierce (Atlanta, GA). Densitometric analyses of the protein bands were performed using a Bio-Rad Chemi-Doc-XRS Imaging system and semiquantified with Quantity One software.
Immunohistochemical staining of α-SMA
Sections were deparaffinized and hydrated through graded alcohol series. Endogenous peroxidase activity was quenched with 3% hydrogen peroxide solution in methanol. After rinsing in PBS, sections were blocked with 10% normal horse serum and probed with primary antibody (α-SMA 1:200 dilution) at 4°C overnight. Sections were subsequently washed and incubated with biotin-conjugated secondary antibody (Vector, Burlingame, CA) for 1 h at room temperature. After rinsing, the sections were incubated in avidin–biotin enzyme reagent (Vector) for 1 h at room temperature, washed, and incubated with DAB chromophoric solution (Scytek Labs, Logan, UT) for 5 min at room temperature, then counterstained with Harris’s hematoxylin (Sigma). α-SMA immunostaining was semiquantified using Axiovision and AV4 Mod Automeasure software Carl Zeiss MicroImaging, Inc (Thornwood, NY) and the results were expressed as percentage of area.
Statistical analysis
Data are presented as mean ± SE and evaluated by Student’s t-test or two-way ANOVA followed by Fisher LSD test. P < 0.05 was considered significant.
Results
Ozone exposure increased TGF-β expression in the lung, which was followed by an increase in collagen deposition in airway walls.
To determine whether TGF-β was involved in O3- induced lung fibrosis, we first assessed the amount of TGF-β protein in the ELF harvested from air and O3- exposed mice by ELISA. The results showed that mice exposed to O3 for five cycles had significantly higher levels of TGF-β1 protein in the ELF than mice exposed to air (Figure 1A) and that such an increase persisted even after 10 cycles of O3 exposure. Interestingly, trichrome staining showed that although O3 exposure for 10 cycles increased collagen deposition in the airways (Figure 1B) no significant change in collagen accumulation in the lung was observed in mice exposed to O3 for five cycles (data not shown). These results showed for the first time that O3 exposure induced a sustained elevation of TGF-β expression in the lung in vivo. The results also suggest that increased TGF-β expression may mediate O3-induced lung fibrosis.
Figure 1.

Ozone exposure increased transforming growth factor beta (TGF-β) protein levels in the epithelial lining fluid (ELF) and collagen deposition in the airway walls in mice. (A) Bronchoalveolar lavage fluid (BALF) was harvested after 5 and 10 cycles of air or O3 exposure and the amounts of total TGF-β protein determined by ELISA. Results were normalized with a dilution factor to determine ELF concentrations as described in the “Materials and methods” section and expressed as the mean ± SEM. *Significantly different from filtered air exposure controls (P < 0.05, n = 10). (B) Collagen deposition (blue) in the lung after 10 cycles of O3 or filtered air exposure was assessed by trichrome staining and the amounts of collagen in the airway walls were quantified using computer-assisted stereology technique as described in the “Materials and methods” section. *Significantly different from the filtered air-exposed control (P < 0.05, n = 6).
Effect of ozone exposure on lung PAI-1 protein levels
Plasminogen activator inhibitor 1 (PAI-1), a physiological inhibitor of urokinase type and tissue type plasminogen activator (uPA and tPA), plays an important role in the regulation (suppression) of ECM turnover and is induced by TGF-β (Liu, 2008). Therefore, we examined the impact of cyclic O3 exposure on lung PAI-1 protein levels by western analysis. The results showed that PAI-1 protein level was significantly increased in mouse lung after five cycles of O3 exposure and remained elevated after 10 cycles of O3 exposure (Figure 2). The results further suggest that O3 exposure activates TGF-β signaling pathways and that increased PAI-1 expression may contribute to O3-induced lung fibrosis.
Figure 2.

Ozone exposure increased PAI-1 protein levels in mouse lung. PAI-1 protein levels in the lung after 5 and 10 cycles of filtered air or O3 exposure were determined by western blot techniques. Upper panel: representative western blots; Lower panel: densitometry analysis of the PAI-1 bands using Bio-Rad Chemi-Doc-XRS Imaging system with Quantity One software. The results were normalized to β-actin. *Significantly different from the filtered air-exposed controls (P < 0.05, n = 6).
Effect of ozone exposure on α-SMA deposition in the lung
One important mechanism whereby TGF-β promotes ECM production is by activation of fibroblasts, forming myofibroblasts, the major producers of ECM. To reveal fibroblast activation, expression of α-SMA, a marker of myofibroblasts, was assessed by immunohistochemical staining and western analysis. Both immunohistochemical staining and western data showed that α-SMA deposition in the lung was significantly increased in mice exposed to O3 for 10 cycles (Figure 3A and 3B). However, no significant change in α-SMA expression was detected after five cycles of O3 exposure (Fig 3B). The data further suggest that the increase in TGF-β expression mediates O3-induced lung fibrosis at least in part by activating fibroblasts.
Figure 3.

Ozone exposure induced alpha smooth muscle actin (α-SMA) expression in the lung. (A) Immunohistochemistry staining of α-SMA after 10 cycles of filtered air or O3 exposure. The results were semiquantified using Axiovision and AV4 Mod Automeasure software and expressed as percentage of area. (B) Western analysis of α-SMA protein. Intensities of the bands were semiquantified using Bio-Rad Chemi-Doc-XRS Imaging system with Quantity One software. The data were normalized to β-actin. *Significantly different from the filtered air-exposed controls (P < 0.05, n = 6).
Effect of TGF-β type I receptor inhibitor on O3-induced lung fibrosis
TGF-β signaling occurs through a heteromeric complex of types I and II receptors (TβRI and TβRII, respectively). IN-1233 is a specific inhibitor of TβRI/ALK5, which has been reported to block TGF-β signaling in different animal models (Long et al., 2009; Kim et al., 2010). To further investigate whether TGF-β mediates O3-induced lung fibrosis, 6–8-week-old mice were exposed to O3 for 10 cycles as described above and, at the same time, treated with IN-1233 through intraperitoneal injection. The results showed that O3 exposure increased Smad2/3 phosphorylation in the lung (Figure 4). Administration of IN-1233, on the other hand, blocked O3-induced Smad2/3 phosphorylation although IN-1233 alone had no significant effect on the baseline levels of Smad2/3 phosphorylation. Most importantly, the results showed that IN-1233 administration blocked O3-induced collagen accumulation in the airways (Figure 5), PAI-1 expression in ELF (Figure 6), and α-SMA accumulation in the lung (Figure 7). IN-1233 alone had no significant effect on collagen accumulation, PAI-1, or α-SMA expression (Figures 5, 6, and 7), nor did it block O3-induced TGF-β expression (data not shown). However, IN-1233 inhibited the expression of collagen, PA-1, and α-SMA in O3-exposed group more than in FA-exposed mice. The mechanism underlying such enhancement of O3 on IN-1233 effect is unclear at moment. Nonetheless, the results further demonstrate that induction of TGF-β expression and activation of TGF-β signaling pathways mediate O3-induced lung fibrotic responses.
Figure 4.

Inhibitor of transforming growth factor beta (TGF-β) type I receptor blocked O3-induced Smad2/3 phosphorylation. The levels of phosphorylated Smad2/3 (pSmad2/3) and non-phosphorylated Smad2/3 were examined by western blotting. Top panel: representative western blots; bottom panel, semiquantified data of pSmad2/3 and Smad2/3 bands. (a) Significantly different from the filtered air alone exposed control and (b) significantly different from filtered air plus IN-1233 treated group (P < 0.05, n = 3).
Figure 5.

Inhibitor of transforming growth factor beta (TGF-β) type I receptor suppressed O3-induced collagen accumulation in the airway walls. Collagen deposition in the lung was assessed by trichrome staining and the amounts of collagens in the airway walls were quantified using computer-assisted stereology technique as described in the “Materials and methods” section. Top panel, representative trichrome staining pictures; bottom panel, semiquantitative trichrome staining data. (a) Significantly different from the filtered air alone exposed control and (b) significantly different from filtered air plus IN-1233-treated group (P < 0.05, n = 7).
Figure 6.

Inhibitor of transforming growth factor beta (TGF-β) type I receptor suppressed O3-induced PAI-1 expression. PAI-1 protein levels in epithelial lining fluid (ELF) were determined by ELISA and normalized with a dilution factor to determine ELF concentrations as described in the “Materials and methods” section. Results were expressed as the mean ± SEM. *Significantly different from the filtered air exposure controls (P < 0.05, n = 6).
Figure 7.
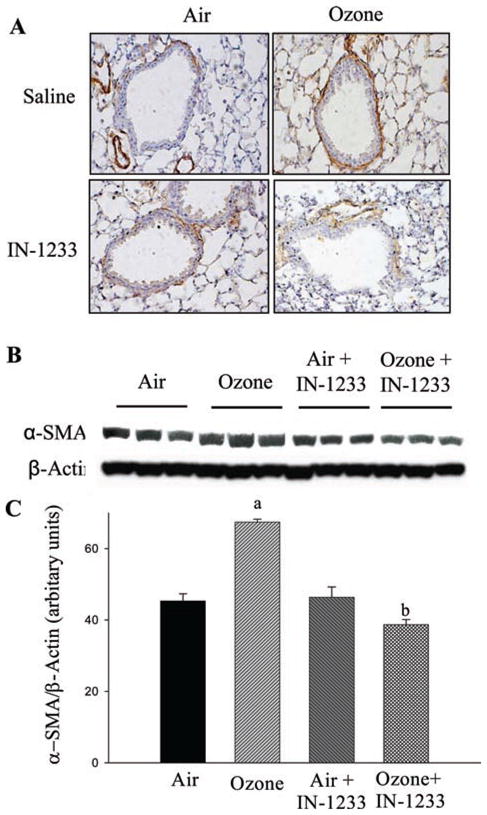
Inhibitor of transforming growth factor beta (TGF-β) type I receptor suppressed O3-induced alpha smooth muscle actin (α-SMA) expression. (A) Immunostaining of α-SMA in the lung. (B) Western analysis of α-SMA in the lung. (C) Summary of semiquantified α-SMA western data using Bio-Rad Chemi-Doc- XRS Imaging system with Quantity One software, normalized by β-actin band. (a) Significantly different from the filtered air control group and (b) significantly different from filtered air plus IN-1233-treated group (P < 0.05, n = 3).
Discussion
It has been reported that continuous or cyclic (8 h/day, 5 days/week) exposure to O3, an extremely reactive gas and one of the most abundant ambient urban pollutants, for an extended period of time (up to 24 months) induces lung fibrosis in different animal models. Whether exposure to O3 under the conditions that mimic environmental/ occupational settings also induces lung fibrosis and what is the molecular mechanism whereby O3 induces lung fibrosis, however, are unclear. The results presented in this study showed for the first time that a cyclic O3 exposure protocol that resembles certain environmental/ occupational exposure situations induced the expression of TGF-β1, the most potent profibrogenic cytokine, which was followed by lung (airway) fibrosis. Most importantly, we showed that blocking the TGF-β signaling pathway with a specific inhibitor to TGF-β receptor 1 attenuated O3-induced fibrotic responses and lung fibrosis in mice. Together, these data suggest that TGF-β1 plays a critical role in O3-induced lung fibrosis. This study is of significance as it not only reveals the potential adverse health effects of O3 exposure under physiologically relevant conditions but also sheds new light on the mechanism whereby O3 induces lung fibrosis.
TGF-β is a potent and ubiquitous profibrogenic cytokine and plays a critical role in the development of fibrosis under various pathological conditions. Although in vitro studies using cultured cells have shown that O3 increases TGF-β production (Bocci et al., 1994; Lang et al., 1998; Valacchi and Bocci, 1999), whether this is the case in vivo is unknown. In the present study, we showed for the first time that TGF-β protein levels were significantly increased in the ELF of mice exposed to O3 and that such induction persisted for a long time (even after 10 cycles of O3 exposure, Figure 1). The molecular mechanism underlying the induction of TGF-β1 by O3 is unknown at the moment. It has been reported that reactive oxygen species (ROS) stimulate the expression and secretion of TGF-β in different types of cells (Leonarduzzi et al., 1997; Bellocq et al., 1999; Saito et al., 2005; Shvedova et al., 2008; Sullivan et al., 2008; Zhao et al., 2008). Leonarduzzi et al. (1997) reported that treatment of human and murine macrophages with 4-hydroxy-2,3-nonenal (HNE), a major aldehyde end product of membrane lipid oxidation, induced both mRNA and protein expression of TGF-β1. It has also been reported that in human alveolar epithelial cells, ROS generated by xanthine/xanthine oxidase system induced TGF-β expression through a transcription mechanism, whereas reactive nitrogen species (RNS) increased TGF-β expression by a posttranscription mechanism (Bellocq et al., 1999). Whether O3 increased TGF-β1 protein level in mouse lung as shown in Figure 1 through a transcription or posttranscription mechanism is unknown. Lang et al. (1998) reported that O3 increased the expression of TGF-β1 mRNA in human fibroblasts that were co-cultured with human airway epithelial cells, suggesting that O3 increases TGF-β protein level at least in part by inducing its mRNA expression.
The steady-state levels of ECM represent a net balance between its production/deposition and degradation. ECM degradation is mediated mainly by the matrix metalloproteinases (MMPs) and the PAs/plasmin system (Laurent, 1987; Davidson, 1990; Arthur, 1994; Liu, 2008). Plasmin is converted from the zymogen plasminogen by tissue type and urokinase type plasminogen activators (tPA and uPA), whereas the activities of tPA and uPA, under physiological conditions, are controlled by PAI-1. Therefore, PAI-1 plays an important role in controlling ECM degradation. PAI-1 is of particular interest because its expression is elevated in variety of fibrotic disorders and in experimental fibrosis models (Idell et al., 1989; Bertozzi et al., 1990; Günther et al., 2000a, b). It has also been reported that knockout of the PAI-1 gene reduces whereas overexpression of PAI-1 enhances the sensitivity of animals to lung fibrosis induced by various stimuli (Eitzman et al., 1996; Hattori et al., 2000; Chuang-Tsai et al., 2003), further suggesting a pivotal role of PAI-1 in the development of lung fibrosis. In previous studies, we have shown that TGF-β1 induced PAI-1 expression in fibroblast, which contributed importantly to TGF-β1- mediated collagen accumulation by inhibiting collagen degradation (Vayalil et al., 2005, 2007; Liu et al., 2010). In this study, we showed for the first time that PAI-1 expression was increased in the lung after five cycles of O3 exposure, which was followed by increases in collagen accumulation in airway walls after 10 cycles of O3 exposure, suggesting that increased PAI-1 expression may contribute to O3-induced lung fibrosis. The molecular mechanism underlying the induction of PAI-1 in O3- exposed mice is unclear. As TGF-β is a potent inducer of PAI-1, it is hypothesized that O3 exposure increases PAI-1 expression through induction of TGF-β. This hypothesis is supported by the results from the TGF-β inhibitor study, in which we showed that inhibition of TGF-β signaling with TGF-β type I receptor inhibitor suppressed O3- induced PAI-1 expression. Together, these data suggest that induction of PAI-1, probably due to increased TGF-β expression/activity, may contribute to the development of lung fibrosis in O3-exposed animals through inhibition of ECM degradation as we have shown previously in fibroblasts.
The Smad pathway mediates the induction of many TGF-β-responsive genes including PAI-1. TGF-β signaling through the Smad pathway has been well-described in the past. Following the binding of TGF-β to the type II receptor (TβR-II), the type I receptor (TβR-I, also called ALK5) is phosphorylated, which further phosphorylates Smad2 or Smad3. Phosphorylated Smad2 and Smad3 form a heteromeric complex with Smad4, which translocates to the nucleus and binds to the promoters of TGF-β-targeted genes. In this study, we showed that ozone increases TGF-β protein level, which was followed by increases in Smad2/3 phosphorylation and PAI-1 expression. IN-1233, on the other hand, blocked ozone-induced Smad2/3 phosphorylation and PAI-1 expression. These data further suggest that IN-1233 inhibited ozone-induced lung fibrosis by blocking TGF-β signaling pathways.
In summary, this study demonstrated for the first time that exposure to a cyclic O3 exposure protocol that resembles certain environmental/occupational exposure situations increased the expression of TGF-β and PAI-1 in mouse lung and that increased TGF-β1 expression mediated O3-induced airway wall fibrosis. These results shed new light on the molecular mechanism whereby O3 induces lung fibrosis.
Acknowledgments
The authors would like to thank the following people for their technical assistance with ozone exposure and sample collection/processing: Jo Anne Balanay, Wentan Huang, and Kishor Gangani.
Footnotes
Declaration of interest
The work was supported by grants from National Institute of Environmental Health Sciences R01 ES011831 (RML) and P01 ES011617 (EMP, MF, and CB), National Heart, Lung, and Blood Institute R01 HL088141 (RML) and R01 HL54696 (EMP), and the American Lung Association (RML).
References
- Arthur MJ. Degradation of matrix proteins in liver fibrosis. Pathol Res Pract. 1994;190:825–833. doi: 10.1016/S0344-0338(11)80985-4. [DOI] [PubMed] [Google Scholar]
- Bellocq A, Azoulay E, Marullo S, Flahault A, Fouqueray B, Philippe C, Cadranel J, Baud L. Reactive oxygen and nitrogen intermediates increase transforming growth factor-beta1 release from human epithelial alveolar cells through two different mechanisms. Am J Respir Cell Mol Biol. 1999;21:128–136. doi: 10.1165/ajrcmb.21.1.3379. [DOI] [PubMed] [Google Scholar]
- Bergeron A, Soler P, Kambouchne M, Loiseau P, Milleron B, Valeyre D, Hance AJ, Tazi A. Cytokine profiles in idiopathic pulmonary fibrosis suggest an important role for TGF-beta and IL-10. Eur Respir J. 2003;22:69–76. doi: 10.1183/09031936.03.00014703. [DOI] [PubMed] [Google Scholar]
- Bertozzi P, Astedt B, Zenzius L, Lynch K, LeMaire F, Zapol W, Chapman HA., Jr Depressed bronchoalveolar urokinase activity in patients with adult respiratory distress syndrome. N Engl J Med. 1990;322:890–897. doi: 10.1056/NEJM199003293221304. [DOI] [PubMed] [Google Scholar]
- Bocci V, Luzzi E, Corradeschi F, Silvestri S. Studies on the biological effects of ozone: 6. Production of transforming growth factor 1 by human blood after ozone treatment. J Biol Regul Homeost Agents. 1994;8:108–112. [PubMed] [Google Scholar]
- Bonniaud P, Margetts PJ, Kolb M, Schroeder JA, Kapoun AM, Damm D, Murphy A, Chakravarty S, Dugar S, Higgins L, Protter AA, Gauldie J. Progressive Transforming Growth Factor {beta}1-induced Lung Fibrosis is Blocked by an orally Active ALK5 Kinase Inhibitor. Am J Respir Crit Care Med. 2005;171:889–898. doi: 10.1164/rccm.200405-612OC. [DOI] [PubMed] [Google Scholar]
- Chang LY, Stockstill BL, Menache MG, Mercer RR, Crapo JD. Consequences of prolonged inhalation of ozone on F344/N rats: collaborative studies. Part VIII: Morphometric analysis of structural alterations in alveolar regions. Res Rep Health Eff Inst. 1995:3–39. discussion 99. [PubMed] [Google Scholar]
- Chuang-Tsai S, Sisson TH, Hattori N, Tsai CG, Subbotina NM, Hanson KE, Simon RH. Reduction in fibrotic tissue formation in mice genetically deficient in plasminogen activator inhibitor-1. Am J Pathol. 2003;163:445–452. doi: 10.1016/S0002-9440(10)63674-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churg A, Zhou S, Preobrazhenska O, Tai H, Wang R, Wright JL. Expression of Profibrotic Mediators in Small Airways versus Parenchyma after Cigarette Smoke Exposure. Am J Respir Cell Mol Biol. 2009;40:268–276. doi: 10.1165/rcmb.2007-0367OC. [DOI] [PubMed] [Google Scholar]
- Coker RK, Laurent GJ, Shahzeidi S, Lympany PA, du Bois RM, Jeffery PK, McAnulty RJ. Transforming growth factors-beta 1, -beta 2, and -beta 3 stimulate fibroblast procollagen production in vitro but are differentially expressed during bleomycin-induced lung fibrosis. Am J Pathol. 1997;150:981–991. [PMC free article] [PubMed] [Google Scholar]
- Davidson JM. Biochemistry and turnover of lung interstitium. Eur Respir J. 1990;3:1048–1063. [PubMed] [Google Scholar]
- Dormans JA, van Bree L, Boere AJ, Marra M, Rombout PJ. Interspecies differences in time course of pulmonary toxicity following repeated exposure to ozone. Inhal Toxicol. 1999;11:309–329. doi: 10.1080/089583799197113. [DOI] [PubMed] [Google Scholar]
- Eitzman DT, McCoy RD, Zheng X, Fay WP, Shen T, Ginsburg D, Simon RH. Bleomycin-induced pulmonary fibrosis in transgenic mice that either lack or overexpress the murine plasminogen activator inhibitor-1 gene. J Clin Invest. 1996;97:232–237. doi: 10.1172/JCI118396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahy RJ, Lichtenberger F, McKeegan CB, Nuovo GJ, Marsh CB, Wewers MD. The acute respiratory distress syndrome: a role for transforming growth factor-beta 1. Am J Respir Cell Mol Biol. 2003;28:499–503. doi: 10.1165/rcmb.2002-0092OC. [DOI] [PubMed] [Google Scholar]
- Fanucchi MV, Buckpitt AR, Murphy ME, Plopper CG. Naphthalene cytotoxicity of differentiating Clara cells in neonatal mice. Toxicol Appl Pharmacol. 1997;144:96–104. doi: 10.1006/taap.1997.8119. [DOI] [PubMed] [Google Scholar]
- Gunther A, Mosavi P, Heinemann S, Ruppert C, Muth H, Markart P, Grimminger F, Walmrath D, Temmesfeld-Wollbruck B, Seeger W. Alveolar fibrin formation caused by enhanced procoagulant and depressed fibrinolytic capacities in severe pneumonia. Comparison with the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2000a;161:454–462. doi: 10.1164/ajrccm.161.2.9712038. [DOI] [PubMed] [Google Scholar]
- Gunther A, Mosavi P, Ruppert C, Heinemann S, Temmesfeld B, Velcovsky HG, Morr H, Grimminger F, Walmrath D, Seeger W. Enhanced tissue factor pathway activity and fibrin turnover in the alveolar compartment of patients with interstitial lung disease. Thromb Haemost. 2000b;83:853–860. [PubMed] [Google Scholar]
- Hattori N, Degen JL, Sisson TH, Liu H, Moore BB, Pandrangi RG, Simon RH, Drew AF. Bleomycin-induced pulmonary fibrosis in fibrinogen-null mice. J Clin Invest. 2000;106:1341–1350. doi: 10.1172/JCI10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashiyama H, Yoshimoto D, Kaise T, Matsubara S, Fujiwara M, Kikkawa H, Asano S, Kinoshita M. Inhibition of activin receptor-like kinase 5 attenuates bleomycin-induced pulmonary fibrosis. Exp Mol Pathol. 2007;83:39–46. doi: 10.1016/j.yexmp.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Idell S, James KK, Levin EG, Schwartz BS, Manchanda N, Maunder RJ, Martin TR, McLarty J, Fair DS. Local abnormalities in coagulation and fibrinolytic pathways predispose to alveolar fibrin deposition in the adult respiratory distress syndrome. J Clin Invest. 1989;84:695–705. doi: 10.1172/JCI114217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Song HY, Park JH, Yoon HJ, Park HG, Kim DK. IN-1233, an ALK-5 inhibitor: prevention of granulation tissue formation after bare metallic stent placement in a rat urethral model. Radiology. 2010;255:75–82. doi: 10.1148/radiol.09090670. [DOI] [PubMed] [Google Scholar]
- Kolb M, Bonniaud P, Galt T, Sime PJ, Kelly MM, Margetts PJ, Gauldie J. Differences in the fibrogenic response after transfer of active transforming growth factor-beta1 gene to lungs of “fibrosis-prone” and “fibrosis-resistant” mouse strains. Am J Respir Cell Mol Biol. 2002;27:141–150. doi: 10.1165/ajrcmb.27.2.4674. [DOI] [PubMed] [Google Scholar]
- Lang DS, Jorres RA, Mucke M, Siegfried W, Magnussen H. Interactions between human bronchoepithelial cells and lung fibroblasts after ozone exposure in vitro. Toxicol Lett. 1998:96–97. 13–24. doi: 10.1016/s0378-4274(98)00045-9. [DOI] [PubMed] [Google Scholar]
- Last JA, Gelzleichter TR, Harkema J, Hawk S. Consequences of prolonged inhalation of ozone on Fischer-344/N rats: collaborative studies. Part I: Content and cross-linking of lung collagen. Res Rep Health Eff Inst. 1994:1–29. discussion 31–40. [PubMed] [Google Scholar]
- Last JA, Hesterberg TW, Reiser KM, Cross CE, Amis TC, Gunn C, Steffey EP, Grandy J, Henrickson R. Ozone-induced alterations in collagen metabolism of monkey lungs: use of biopsy-obtained lung tissue. Toxicol Appl Pharmacol. 1981;60:579–585. doi: 10.1016/0041-008x(81)90345-8. [DOI] [PubMed] [Google Scholar]
- Last JA, Reiser KM, Tyler WS, Rucker RB. Long-term consequences of exposure to ozone. I. Lung collagen content. Toxicol Appl Pharmacol. 1984;72:111–118. doi: 10.1016/0041-008x(84)90254-0. [DOI] [PubMed] [Google Scholar]
- Laurent GJ. Dynamic state of collagen: pathways of collagen degradation in vivo and their possible role in regulation of collagen mass. Am J Physiol. 1987;252:C1–9. doi: 10.1152/ajpcell.1987.252.1.C1. [DOI] [PubMed] [Google Scholar]
- Lee CG, Cho SJ, Kang MJ, Chapoval SP, Lee PJ, Noble PW, Yehualaeshet T, Lu B, Flavell RA, Milbrandt J, Homer RJ, Elias JA. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor beta1-induced pulmonary fibrosis. J Exp Med. 2004;200:377–389. doi: 10.1084/jem.20040104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonarduzzi G, Scavazza A, Biasi F, Chiarpotto E, Camandola S, Vogel S, Dargel R, Poli G. The lipid peroxidation end product 4-hydroxy-2,3-nonenal up-regulates transforming growth factor beta1 expression in the macrophage lineage: a link between oxidative injury and fibrosclerosis. Faseb J. 1997;11:851–857. doi: 10.1096/fasebj.11.11.9285483. [DOI] [PubMed] [Google Scholar]
- Liu RM. Oxidative stress, plasminogen activator inhibitor 1, and lung fibrosis. Antioxid Redox Signal. 2008;10:303–319. doi: 10.1089/ars.2007.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RM, Choi J, Wu JH, Gaston Pravia KA, Lewis KM, Brand JD, Mochel NS, Krzywanski DM, Lambeth JD, Hagood JS, Forman HJ, Thannickal VJ, Postlethwait EM. Oxidative modification of nuclear mitogen-activated protein kinase phosphatase 1 is involved in transforming growth factor beta1-induced expression of plasminogen activator inhibitor 1 in fibroblasts. J Biol Chem. 2010;285:16239–16247. doi: 10.1074/jbc.M110.111732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long L, Crosby A, Yang X, Southwood M, Upton PD, Kim D-K, Morrell NW. Altered Bone Morphogenetic Protein and Transforming Growth Factor-{beta} Signaling in Rat Models of Pulmonary Hypertension: Potential for Activin Receptor-Like Kinase-5 Inhibition in Prevention and Progression of Disease. Circulation. 2009;119:566–576. doi: 10.1161/CIRCULATIONAHA.108.821504. [DOI] [PubMed] [Google Scholar]
- Parks WC, Roby JD. Consequences of prolonged inhalation of ozone on F344/N rats: collaborative studies. Part IV: Effects on expression of extracellular matrix genes. Res Rep Health Eff Inst. 1994:3–20. discussion 21–29. [PubMed] [Google Scholar]
- Pickrell JA, Hahn FF, Rebar AH, Horoda RA, Henderson RF. Changes in collagen metabolism and proteinolysis after repeated inhalation exposure to ozone. Exp Mol Pathol. 1987;46:159–167. doi: 10.1016/0014-4800(87)90062-1. [DOI] [PubMed] [Google Scholar]
- Reiser KM, Tyler WS, Hennessy SM, Dominguez JJ, Last JA. Long-term consequences of exposure to ozone. II. Structural alterations in lung collagen of monkeys. Toxicol Appl Pharmacol. 1987;89:314–322. doi: 10.1016/0041-008x(87)90151-7. [DOI] [PubMed] [Google Scholar]
- Rube CE, Uthe D, Schmid KW, Richter KD, Wessel J, Schuck A, Willich N, Rube C. Dose-dependent induction of transforming growth factor beta (TGF-beta) in the lung tissue of fibrosis-prone mice after thoracic irradiation. Int J Radiat Oncol Biol Phys. 2000;47:1033–1042. doi: 10.1016/s0360-3016(00)00482-x. [DOI] [PubMed] [Google Scholar]
- Saito K, Ishizaka N, Aizawa T, Sata M, Iso-o N, Noiri E, Mori I, Ohno M, Nagai R. Iron chelation and a free radical scavenger suppress angiotensin II-induced upregulation of TGF-beta1 in the heart. Am J Physiol Heart Circ Physiol. 2005;288:H1836–1843. doi: 10.1152/ajpheart.00679.2004. [DOI] [PubMed] [Google Scholar]
- Shima M, Adachi M. Changes of procoagulant and fibrinolytic activities in the alveoli of rats exposed to ozone. Nippon Eiseigaku Zasshi. 1991;46:724–733. doi: 10.1265/jjh.46.724. [DOI] [PubMed] [Google Scholar]
- Shvedova AA, Kisin ER, Murray AR, Kommineni C, Castranova V, Fadeel B, Kagan VE. Increased accumulation of neutrophils and decreased fibrosis in the lung of NADPH oxidase-deficient C57BL/6 mice exposed to carbon nanotubes. Toxicol Appl Pharmacol. 2008;231:235–240. doi: 10.1016/j.taap.2008.04.018. [DOI] [PubMed] [Google Scholar]
- Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest. 1997;100:768–776. doi: 10.1172/JCI119590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockstill BL, Chang LY, Menache MG, Mellick PW, Mercer RR, Crapo JD. Bronchiolarized metaplasia and interstitial fibrosis in rat lungs chronically exposed to high ambient levels of ozone. Toxicol Appl Pharmacol. 1995;134:251–263. doi: 10.1006/taap.1995.1191. [DOI] [PubMed] [Google Scholar]
- Sullivan DE, Ferris M, Pociask D, Brody AR. The latent form of TGFbeta(1) is induced by TNFalpha through an ERK specific pathway and is activated by asbestos-derived reactive oxygen species in vitro and in vivo. J Immunotoxicol. 2008;5:145–149. doi: 10.1080/15476910802085822. [DOI] [PubMed] [Google Scholar]
- Sun JD, Pickrell JA, Harkema JR, McLaughlin SI, Hahn FF, Henderson RF. Effects of buthionine sulfoximine on the development of ozone-induced pulmonary fibrosis. Exp Mol Pathol. 1988;49:254–266. doi: 10.1016/0014-4800(88)90038-x. [DOI] [PubMed] [Google Scholar]
- Tzortzaki EG, Antoniou KM, Zervo MI, Lambiri I, Koutsopoulos A, Tzanakis N, Plataki M, Maltezakis G, Bouros D, Siafakas NM. Effects of antifibrotic agents on TGF-[beta]1, CTGF and IFN-[gamma] expression in patients with idiopathic pulmonary fibrosis. Respiratory Medicine. 2007;101:1821–1829. doi: 10.1016/j.rmed.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Valacchi G, Bocci V. Studies on the biological effects of ozone: 10. Release of factors from ozonated human platelets. Mediators Inflamm. 1999;8:205–209. doi: 10.1080/09629359990360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bree L, Dormans JA, Boere AJ, Rombout PJ. Time study on development and repair of lung injury following ozone exposure in rats. Inhal Toxicol. 2001;13:703–718. doi: 10.1080/08958370126868. [DOI] [PubMed] [Google Scholar]
- Vayalil PK, Iles KE, Choi J, Yi A-K, Postlethwait EM, Liu R-M. Glutathione suppresses TGF-beta-induced PAI-1 expression by inhibiting p38 and JNK MAPK and the binding of AP-1, SP-1, and Smad to the PAI-1 promoter. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1281–1292. doi: 10.1152/ajplung.00128.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vayalil PK, Olman M, Murphy-Ullrich JE, Postlethwait EM, Liu RM. Glutathione restores collagen degradation in TGF-beta-treated fibroblasts by blocking plasminogen activator inhibitor-1 expression and activating plasminogen. Am J Physiol Lung Cell Mol Physiol. 2005;289:L937–945. doi: 10.1152/ajplung.00150.2005. [DOI] [PubMed] [Google Scholar]
- Vicencio AG, Lee CG, Cho SJ, Eickelberg O, Chuu Y, Haddad GG, Elias JA. Conditional overexpression of bioactive transforming growth factor-beta1 in neonatal mouse lung: a new model for bronchopulmonary dysplasia? Am J Respir Cell Mol Biol. 2004;31:650–656. doi: 10.1165/rcmb.2004-0092OC. [DOI] [PubMed] [Google Scholar]
- Williams AO, Flanders KC, Saffiotti U. Immunohistochemical localization of transforming growth factor-beta 1 in rats with experimental silicosis, alveolar type II hyperplasia, and lung cancer. Am J Pathol. 1993;142:1831–1840. [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Mann DM, Ruoslahti E. Negative regulation of transforming growth factor-beta by the proteoglycan decorin. Nature. 1990;346:281–284. doi: 10.1038/346281a0. [DOI] [PubMed] [Google Scholar]
- Zhao W, Zhao T, Chen Y, Ahokas RA, Sun Y. Oxidative stress mediates cardiac fibrosis by enhancing transforming growth factor-beta1 in hypertensive rats. Mol Cell Biochem. 2008;317:43–50. doi: 10.1007/s11010-008-9803-8. Inhalation Toxicology Downloaded from informahealthcare.com by University Of Alabama Birmingham on 06/21/11 For personal use only. [DOI] [PubMed] [Google Scholar]