

Abstract
Isosterism is commonly used in drug discovery and development to address stability, selectivity, toxicity, pharmacokinetics, and efficacy issues. A series of 14-O-substituted naltrexone derivatives were identified as potent mu opioid receptor (MOR) antagonists with improved selectivity over the kappa opioid receptor (KOR) and the delta opioid receptor (DOR), compared to naltrexone. Since esters are not metabolically very stable under typical physiological conditions, their corresponding amide analogs were thus synthesized and biologically evaluated. Unlike their isosteres, most of these novel ligands seem to be dually selective for the MOR and the KOR over the DOR. The restricted flexibility of the amide bond linkage might be responsible for their altered selectivity profile. However, the majority of the 14-N-substituted naltrexone derivatives produced marginal or no MOR stimulation in the 35S-GTP[γS] assay, which resembled their ester analogs. The current study thus indicated that the 14-substituted naltrexone isosteres are not bioisosteres since they have distinctive pharmacological profile with the regard to their opioid receptor binding affinity and selectivity.
Keywords: Naltrexone, Isosterism, Mu opioid receptor, Kappa opioid receptor, Antagonist
The antinociceptive actions and the addiction/abuse liability of most opiates are primarily mediated through the mu opioid receptor (MOR).1,3 Thus, blockade of the MOR represents a practical pharmacological intervention for opioid addiction treatment. However, the available non-peptidic, reversible MOR antagonists (Figure 1), such as naltrexone, failed the expectation,4 partially due to its lack of high MOR selectivity over both the kappa opioid receptor (KOR) and the delta opioid receptor (DOR).5 Some moderately potent ligands, e.g. cyprodime6, are in use. Compared with the high selectivity of GNTI for the KOR (Ki value ratios are mu/kappa≈120, delta/kappa≈250)7 and NTI for the DOR (Ki value ratios are mu/delta≈152, kappa/delta≈276)8, cyprodime has a moderate selectivity for the MOR over the DOR and KOR (Ki value ratios are kappa/mu≈45, delta/mu≈40)9. Another drawback of cyprodime is that it showed much lower affinity for the MOR than naloxone and naltrexone,6 which generally limits its application. Further structure-activity relationship studies of cyprodime derivatives did not generate any antagonists with improved selectivity for the MOR.10-15 -FNA16, clocinnamox17 and its derivatives18-24, have been reported as selective and irreversible antagonists for the MOR. However, the fact that they bear the capacity to bind covalently with the receptor largely limits their utility. In most cases, reversible antagonist would be preferred because they can “knock out” the receptors temporarily for pharmacological study and then can be washed out from the binding locus and “revive” the receptors.
Figure 1.

Representative non-peptidyl MOR antagonists.
A series of the 14-O-substituted naltrexone derivatives were originally designed as MOR antagonists based on the “message-address” concept and molecular modeling study. One of them (ONP, Figure 1) showed promising MOR selectivity without any apparent agonist activity on the receptor.25 Further pharmacological characterization (particularly some unrepeatable in vitro whole cell system assays and certain in vivo experimental observation) indicated that ONP was not metabolically stable. Therefore its ester bond at 14 position linkage was replaced with its isostere, the amide bond. We here report the chemical synthesis and biological evaluation of these novel ligands and compare their pharmacological profile with that of their ester analogs.
The synthesis of 14-N-substituted naltrexone derivatives 1 8 is shown in Scheme 1. Northebaine hydrochloride salt 11 was prepared from thebaine 9 by the method of Pohland and Sullivan.26 Reaction of 11 with cyclopropylcarbonyl chloride, followed by lithium aluminium hydride reduction afforded compound 13,27 which was then conjugated with the C-nirtrosoformate esters generated in situ from 1428 to give the Diels-Alder adduct 15.29 Compound 16 was obtained by catalytic hydrogenation of 15 in acetic acid/sodium acetate buffer using Pd/C as reported by Sebastian et al.30 Demethylation of 16 with BBr330 yielded 14-amino-17-cyclopropylmethyl-7,8-dihydronormorphinone (17), which was then coupled with either acyl chloride or acid to furnish the 14-N-substituted naltrexone derivatives 1 8 as described previously25, 31-33. All new ligands were obtained with reasonable yields (See Supplementary Information).
Scheme 1.

Synthetic route of 14-N-substituted naltrexone derivatives.
To determine the pharmacological properties of these novel ligands 1 8 as compared to their ester isosteres, the MOR, KOR and DOR competitive radioligand binding assay and the [35S]GTP S functional assay were performed using monocloned opioid receptor-expressing Chinese hamster ovary (CHO) cell membranes as reported previously31-34.
As seen in Table 1, all the amide isosteres displayed subnanomolar to low nanomolar binding affinity to the MOR, with pyridinyl series (compounds 1 3) showing slightly higher affinity than the quinolinyl series (compounds 5, 6). Similarly to their ester analogs, the presence of the nitrogen atom in the aromatic ring of these new ligands seemed to enhance MOR binding affinity, except for compound 6, compared to the corresponding control compounds 4 and 8. These findings are consistent with the original hypothesis that the nitrogen atom in the aromatic ring can act as a hydrogen bond acceptor in the alternative MOR address domain25. Meanwhile, compounds with the nitrogen atom located in the meta- or para- position bound slightly more potently to MOR than those with an ortho-nitrogen substitution (1 vs 2 or 3, 6 vs 7), whereas no such a trend was observed for the ester counterparts. Furthermore, the relatively low MOR binding affinity of the phenyl-, 3-isoquinolinyl-, and 2-naphthalenyl substitution in the ester analogs was significantly improved in compounds 4, 5, and 8, respectively, indicating a positive contribution of the amide bond for the ligand-receptor interactions.
Table 1.
The binding affinity, selectivity, and MOR [35S]GTP S functional assay results of 14-N-substituted naltrexone derivativesa
| Compd | R |
Ki (nM) |
Selectivity |
MOR 35S-GTP[γS] Binding |
||||
|---|---|---|---|---|---|---|---|---|
| [3H]NLX (μ) | [3H]DPN (κ) | [3H]NTI (δ) | κ/μ | δ/μ | EC50 (nM) | % Emax of DAMGO | ||
| NTXb | N/A | 0.34 ± 0.03 | 0.90 ± 0.11 | 95.46 ± 6.09 | 2.6 | 281 | 0.38 ± 0.10 | 7.18 ± 0.57 |
| 1 |
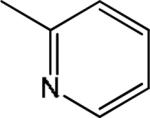
|
1.51 ± 0.34 | 0.36 ± 0.01 | 94.54 ± 6.48 | 0.24 | 63 | N.D.b | 0.90 ± 0.42c |
| 2 |
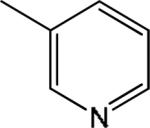
|
0.75 ± 0.28 | 0.16 ± 0.01 | 39.88 ± 0.50 | 0.21 | 53 | N.D.b | 5.09 ± 0.57c |
| 3 |
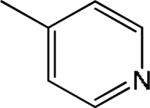
|
0.82 ± 0.33 | 0.33 ± 0.01 | 10.86 ± 1.31 | 0.40 | 13 | 1.67 ± 0.99 | 7.74 ± 0.69 |
| 4 |
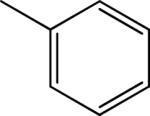
|
4.34 ± 0.70 | 0.12 ± 0.001 | 57.32 ± 4.33 | 0.03 | 13 | 7.20 ± 1.74 | 5.79 ± 1.35 |
| 5 |
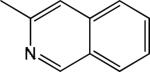
|
3.50 ± 1.87 | 0.27 ± 0.02 | 25.07 ± 1.84 | 0.07 | 7.2 | N.D.b | 2.84 ± 1.62c |
| 6 |
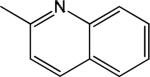
|
9.09 ± 4.94 | 0.26 ± 0.004 | 15.13 ± 0.63 | 0.03 | 1.7 | 38.85 ± 17.98 | 34.40 ± 5.43 |
| 7 |
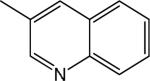
|
1.13 ± 0.25 | 0.13 ± 0.02 | 1.48 ± 0.05 | 0.12 | 1.3 | 2.81 ± 0.29 | 15.76 ± 5.61 |
| 8 |
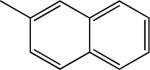
|
6.22 ± 4.01 | 0.33 ± 0.02 | 10.54 ± 1.35 | 0.05 | 1.7 | 11.13 ± 5.11 | 16.52 ± 1.97 |
The values are the means ± S.E.M. of three independent experiments. The percentage Emax of DAMGO is the Emax of the compound compared to of the stimulation produced by 3 μM DAMGO (normalized to 100%). Naltrexone (NTX) was tested as a control compound under same assay conditions. NLX, naloxone; DPN, diprenorphine; NTI, naltrindole; N/A, not applicable.
N.D. = not determined because dose dependent stimulation was not produced and the data could not be fit by non-linear regression.
Percentage stimulation relative to DAMGO that was produced at the maximum concentration of 10 μM of test compound.
In addition, the replacement of the ester bond with the amide bond considerably increased the KOR binding affinity of all the 14-N-substituted isosteres, with the Ki values in the subnanomolar range as compared to the double or triple digit nanomolar Ki values for the ester analogs.25 Not only did the pyridinyl and quinolinyl series bind with equal affinity to the KOR, the presence/absence of the nitrogen atom in the aromatic ring also did not significantly affect the KOR binding, which supported the original hypothesis that an alternative MOR “address” domain composed of hydrogen bonding interaction is absent in the KOR binding pocket.6 It thus appeared that the introduction of the amide bond linkage could be the major cause of the enhanced KOR binding of 14-N-substituted isosteres. As a matter of fact, compounds 4 and 6 displayed at least 30-fold KOR selectivity over the MOR, whereas their ester counterparts are more MOR selective.25
Although the presence of the amide bond also improved the DOR binding affinity of most of the 14-N-substituted isosteres compared to their ester analogs, all of these new ligands bound to the DOR with at least modestly lower affinity than to both the MOR and KOR. Compounds with one aromatic ring had lower DOR binding affinity than the corresponding analogs with two aromatic rings, when taking the substitution effect of the aromatic nitrogen atom into account (1 vs 5/6, 2 vs 7). The position of the nitrogen atom also seemed to affect DOR binding affinity of the 14-N-substituted isosteres, with ortho-substitution exhibiting the lowest affinity for both the pyridinyl and quinolinyl series.
Collectively, it appeared that isosterism had a substantial effect on opioid receptor binding affinity and selectivity for the 14-O-substituted and 14-N-substituted naltrexone derivatives. The replacement of the ester bond with the amide bond facilitated binding affinity to all three opioid receptors, with a general rank order of KOR > DOR > MOR. Figure 2 illustrates the possible cause of the different opioid receptor selectivity profiles for the 14-substituted naltrexone isosteres. The anticipated lower flexibility of the aryl group due to the presence of the amide bond, is postulated as the reason for the difference in activity between amide and ester analogs.
Figure 2.

Schematic demonstration of the potential cause of the different opioid receptor selectivity profiles of the 14-substituted naltrexone isosteres. Replacement of the ester linkage with the amide linkage may decrease the flexibility of the side chain for new ligands 1 8 due to the resonance effect of the amide bond.
The [35S]GTP S functional assay (Table 1) revealed that compounds 1, 2, and 5 acted as neutral MOR antagonists, which resembled their corresponding ester isosteres. Compounds 3, 4, 7, and 8 were MOR partial agonists with low efficacy (percentage Emax of DAMGO < 20%) and moderate to high potency, whereas compound 6 behaved as a MOR partial agonist with moderate efficacy and potency. In contrast, all the ester counterparts acted as MOR neutral antagonists except for the 3-quinolinyl substituted one (isostere of compound 7).25 It thus seemed that the presence of the amide group also played some role in MOR activation by the 14-N-substituted naltrexone derivatives.
In conclusion, a series of 14-N-substituted naltrexone derivatives were synthesized as metabolically stable isosteres of the corresponding 14-O-substituted analogs. The isosterism employed here significantly altered the opioid receptor selectivity profile while producing a less profound impact on their functional activity. Among the newly synthesized 14-N-substituted naltrexone derivatives, compounds 1 and 2 showed the highest KOR/MOR selectivity over the DOR and neutral MOR antagonism, and were thus identified as new leads for future optimization. The current study indicates that the 14-substituted naltrexone isosteres seem not to act as bioisosteres because they have distinctive pharmacological profiles with regard to their opioid receptor binding affinity and selectivity.
Supplementary Material
Acknowledgement
We are grateful for Drs. Lee-Yuan Liu-Chen (Temple University) and Ping-Yee Law (University of Minnesota) for the generous gift of opioid receptor expressing CHO cell lines. O.E. thanks Joanna C. Jacob and Jordan O. Cox for their technical guidance on the biological assays. The work was funded by PHS grants from NIH/NIDA, DA024022 (YZ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Supplementary data (chemical synthesis and compounds characterization) associated with this article can be found, in the online version, at.
References and notes
- 1.Fiellin DA, Kleber H, Trumble-Hejduk JG, McLellan AT, Kosten TR. J. Subst. Abuse Treat. 2004;27:153. doi: 10.1016/j.jsat.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 2.Schmidhammer H. Prog. Med. Chem. 1998;35:83. [PubMed] [Google Scholar]
- 3.Zimmerman DM, Leander JD. J. Med. Chem. 1990;33:895. doi: 10.1021/jm00165a002. [DOI] [PubMed] [Google Scholar]
- 4.a Minozzi S, Amato L, Vecchi S, Davoli M, Kirchmayer U, Verster A. Cochrane Database Syst. Rev. 2006:CD001333. doi: 10.1002/14651858.CD001333.pub2. [DOI] [PubMed] [Google Scholar]; b Lobmaier P, Kornor H, Kunoe N, Bjørndal A. Cochrane Database Syst. Rev. 2008:CD006140. doi: 10.1002/14651858.CD006140.pub2. [DOI] [PubMed] [Google Scholar]
- 5.a Miotto K, McCann M, Basch J, Rawson R, Ling W. Am J Addict. 2002;11:151. doi: 10.1080/10550490290087929. [DOI] [PubMed] [Google Scholar]; b Ritter AJ. Aust. N. Z. J. Psychiatry. 2002;36:224. doi: 10.1046/j.1440-1614.2002.01012.x. [DOI] [PubMed] [Google Scholar]
- 6.Schmidhammer H, Burkard WP, Eggstin-Aeppli L, Smith CFC. J. Med. Chem. 1989;32:418. doi: 10.1021/jm00122a021. [DOI] [PubMed] [Google Scholar]
- 7.Jones RM, Hjorth SA, Schwartz TW, Portoghese PS. J. Med. Chem. 1998;41:4911. doi: 10.1021/jm9805182. [DOI] [PubMed] [Google Scholar]
- 8.Portoghese PS, Sultana M, Nagase H, Takemori AE. J. Med. Chem. 1988;31:281. doi: 10.1021/jm00397a001. [DOI] [PubMed] [Google Scholar]
- 9.Marki A, Monory K, Otvos F, Toth G, Krassnig R, Schmidhammer H, Traynor JR, Roques BP, Maldonado R, Borsodi A. Eur J Pharmacol. 1999;383:209. doi: 10.1016/s0014-2999(99)00610-x. [DOI] [PubMed] [Google Scholar]
- 10.Schmidhammer H, Jennewein HK, Krassnig R, Traynor JR, Patel D, Bell K, Froschauer G, Mattersberger K, Jachs-Ewinger C, Jura P, Fraser GL, Klinini VN. J Med Chem. 1995;38:3071. doi: 10.1021/jm00016a010. [DOI] [PubMed] [Google Scholar]
- 11.Schmidhammer H, Jennewein HK, Smith CF. Arch Pharm (Weinheim) 1991;324:209. doi: 10.1002/ardp.19913240404. [DOI] [PubMed] [Google Scholar]
- 12.Schmidhammer H, Smith CF, Erlach D, Koch M, Krassnig R, Schwetz W, Wechner C. J Med Chem. 1990;33:1200. doi: 10.1021/jm00166a018. [DOI] [PubMed] [Google Scholar]
- 13.Schmidhammer H, Smith CF, Erlach D, Koch M, Krassnig R, Schwetz W, Wechner C. Prog. Clin. Biol. Res. 1990;328:37. [PubMed] [Google Scholar]
- 14.Spetea M, Schullner F, Moisa RC, Berzetei-Gurske IP, Schraml B, Dorfler C, Aceto MD, Harris LS, Coop A, Schmidhammer H. J Med Chem. 2004;47:3242. doi: 10.1021/jm031126k. [DOI] [PubMed] [Google Scholar]
- 15.Lewis JW, Smith CFC, McCarthy PS, Kobylecki RJ, Myers M, Haynes AS, Lewis CJ, Waltham K. NIDA Res. Monogr. 1988;90:136. [PubMed] [Google Scholar]
- 16.Portoghese PS, Takemori AE. NIDA Research Monograph. 1986;69:157. [PubMed] [Google Scholar]
- 17.Burke TF, Woods JH, Lewis JW, Medzihradsky F. J. Pharmacol. Exp. Ther. 1994;271:715. [PubMed] [Google Scholar]
- 18.Husbands SM, Sadd J, Broadbear JH, Woods JH, Martin J, Traynor JR, Aceto MD, Bowman ER, Harris LS, Lewis JW. J Med Chem. 1998;41:3493. doi: 10.1021/jm9810248. [DOI] [PubMed] [Google Scholar]
- 19.Broadbear JH, Sumpter TL, Burke TF, Husbands SM, Lewis JW, Woods JH, Traynor JR. J. Pharmacol. Exp. Ther. 2000;294:933. [PubMed] [Google Scholar]
- 20.Nieland NP, Moynihan HA, Carrington S, Broadbear J, Woods JH, Traynor JR, Husbands SM, Lewis JW. J Med Chem. 2006;49:5333. doi: 10.1021/jm0604777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rennison D, Moynihan H, Traynor JR, Lewis JW, Husbands SM. J Med Chem. 2006;49:6104. doi: 10.1021/jm060595u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rennison D, Neal AP, Cami-Kobeci G, Aceto MD, Martinez-Bermejo F, Lewis JW, Husbands SM. J Med Chem. 2007;50:5176. doi: 10.1021/jm070255o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nieland NP, Rennison D, Broadbear JH, Purington L, Woods JH, Traynor JR, Lewis JW, Husbands SM. J Med Chem. 2009;52:6926. doi: 10.1021/jm901074a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moynihan HA, Derrick I, Broadbear JH, Greedy BM, Aceto MD, Harris LS, Purington LC, Thomas MP, Woods JH, Traynor JR, Husbands SM, Lewis JW. J Med Chem. 2012;55:9868. doi: 10.1021/jm301096s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li G, Aschenbach LCK, He H, Selley DE, Zhang Y. Bioorg. Med. Chem. Lett. 2009;19:1825. doi: 10.1016/j.bmcl.2008.12.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pohland A, Sullivan HR. 1964 U.S. Patent 3,342,824.
- 27.Bentley KW, Bower JD, Lewis JW. J. Chem. Soc. C. 1969;19:2569. doi: 10.1039/j39690002569. [DOI] [PubMed] [Google Scholar]
- 28.Kirby GW, McGuigan H, Mackinnon JWM, McLean D, Sharma RP. J. Chem. Soc. Perkin Trans. 1. 1985:1437. [Google Scholar]
- 29.Kirby GW, Mclean D. J. Chem. Soc. Perkin Trans. 1. 1985:1443. [Google Scholar]
- 30.Sebastian A, Bidlack JM, Jiang Q, Deecher D, Teitler M, Glick SD, Archer S. J. Med. Chem. 1993;36:3154. doi: 10.1021/jm00073a015. [DOI] [PubMed] [Google Scholar]
- 31.Li G, Aschenbach LCK, Chen J, Cassidy MP, Stevens DL, Gabra BH, Selley DE, Dewey WL, Westkaemper RB, Zhang Y. J. Med. Chem. 2009;52:1416. doi: 10.1021/jm801272c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan Y, Elbegdorj O, Chen J, Akubathini SK, Beletskaya IO, Selley DE, Zhang Y. Bioorg. Med. Chem. Lett. 2011;21:5625. doi: 10.1016/j.bmcl.2011.06.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan Y, Elbegdorj O, Chen J, Akubathini SK, Zhang F, Stevens DL, Beletskaya IO, Scoggins KL, Zhang Z, Gerk PM, Selley DE, Akbarali HI, Dewey WL, Zhang Y. J. Med. Chem. 2012;55:10118. doi: 10.1021/jm301247n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuan Y, Li G, He H, Stevens DL, Kozak P, Scoggins KL, Mitra P, Gerk PM, Selley DE, Dewey WL, Zhang Y. ACS Chem. Neurosci. 2011;2:346. doi: 10.1021/cn2000348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.