

Abstract
Superoxide dismutases rely on protein structural elements to adjust the redox potential of the metallocenter to an optimum value near 300 mV (vs. NHE), to provide a source of protons for catalysis, and to control the access of anions to the active site. These aspects of the catalytic mechanism are examined herein for recombinant preparations of the nickel-dependent SOD (NiSOD) from Streptomyces coelicolor, and for a series of mutants that affect a key tyrosine residue, Tyr9 (Y9F-, Y62F-, Y9FY62F- and D3A-NiSOD). Structural aspects of the nickel sites are examined by a combination of EPR and x-ray absorption spectroscopies, and by single crystal x-ray diffraction at ~ 1.9 Å resolution in the case of Y9F- and D3A-NiSODs. The functional effects of the mutations are examined by kinetic studies employing pulse radiolytic generation of O2− and by redox titrations. These studies reveal that although the structure of the nickel center in NiSOD is unique, the ligand environment is designed to optimize the redox potential at 290 mV and results in the oxidation of 50% of the nickel centers in the oxidized hexamer. Kinetic investigations show that all of the mutant proteins have considerable activity. In the case of Y9F-NiSOD, the enzyme shows saturation behavior that is not observed in WT-NiSOD and suggests that release of peroxide is inhibited. The crystal structure of Y9F-NiSOD reveals an anion binding site that is occupied by either Cl− or Br− and is located close to, but not within bonding distance of the nickel center. The structure of D3A-NiSOD reveals that in addition to affecting the interaction between subunits, this mutation repositions Y9 and leads to altered chemistry with peroxide. Comparisons with Mn(SOD) and Fe(SOD) reveal that although different strategies are employed to adjust the redox potential and supply of protons, NiSOD has evolved a similar strategy to control the access of anions to the active site.
Superoxide dismutases (SODs) are enzymes that catalyze the conversion of superoxide to molecular oxygen and hydrogen peroxide (Equation 1–3), and are thus part of cellular defenses against damage due to reactive oxygen species (ROS)(1–4).
| (1) |
| (2) |
| (3) |
SODs utilize one-electron redox-active metal centers to carry out catalysis. There are four known SODs categorized by the metal present: (1) Cu/Zn, (2) Mn, (3) Fe and (4) Ni. These four SODs fall into three protein classes based on amino acid sequence homology, with Fe and Mn in the same class. In addition to having no sequence homology with other SODs(5), NiSOD has other notable differences: Nickel is the only metal found in a SOD where the aquated ion does not react with O2−(1), presumably because the redox potential of Ni(OH2)62+/3+ (> 2 V)(6) lies outside of the potentials for oxidation and reduction of O2−. In SODs, the protein component serves at least three important functions with regard to catalysis: to adjust the redox potential of the metallocenter(7–10), to provide a source of protons(11, 12), and to control the access of anions to the active site(13–16). In NiSOD, the mechanism by which the protein tunes the redox potential of nickel is also distinct from other SODs in that it is the only metal center that employs cysteine thiolate ligands(17–21), which at first glance would seem to be incompatible with a catalytic center that produces oxygen and peroxide(22–24).
Crystal structures of NiSOD reveal a five-coordinate pyramidal site that is associated with an oxidized Ni(III) center (Figure 1), and a four-coordinate planar site associated with a reduced Ni(II) center(17, 21). The nickel ligands derive from three amino acid residues: the N-terminal amino group of His1, an amidate N-donor from Cys2 and sidechain thiolates from Cys2 and Cys6. The fifth ligand in the Ni(III) site is provided by the imidazole sidechain of His1 (His-on), which is unbound in the Ni(II) (His-off) site(17, 21).
Figure 1.

Active site from one monomer of the homohexameric NiSOD from Streptomyces coelicolor, showing the first nine residues and Tyr62(17). The nickel is shown in the five-coordinate pyramidal oxidized His-on form. PDB code 1T6U. Image was generated in PyMOL (75).
There are four tyrosine residues in the amino acid sequence of Streptomyces coelicolor NiSOD, two of which (Tyr9 and Tyr62) are conserved in nearly all NiSODs (see supporting information). Tyr9 is positioned near the vacant coordination site opposite the His1 imidazole ligand in the His-on structure, (O-Ni = 5.47 Å) and is involved in H-bonding with two ordered water molecules in the active site, neither of which is a nickel ligand (O-Ni = 2.57, 3.15 Å). These water molecules are also H-bonded to amide protons from Cys6 and Asp3. The other conserved tyrosine, Tyr62, is near Tyr9 and ~13 Å away from the nickel center.
The SOD proteins supply protons for the production of H2O2 and control anion access to the active site(7–10). NiSOD lacks an aqua ligand that has been associated with redox tuning and proton supply(8) in MnSOD and FeSOD, but the thiolate ligands may serve an analogous role(25, 26). Anion access is controlled in SODs using a combination of size constraint(27), electrostatic steering(28–30) and, in the cases of MnSOD(12, 31, 32), and FeSOD(15, 33) a “gatekeeper” tyrosine and neighboring phenylalanine(27). Herein, we probe the role of the conserved tyrosine residues and the aspartate (Asp3) in the Ni hook domain, for insights into the NiSOD mechanism using a combination of mutagenesis to systematically perturb the tyrosine residues, crystallographic and spectroscopic studies to assess the structural ramifications of the mutations, and kinetics using pulse-radiolytic generation of superoxide to examine function. The mutation of Tyr9 led to the serendipitous isolation and the first crystallographic characterization of an anion complex of NiSOD (Cl− and Br−). In addition to intersubunit interactions, Asp3 also affects the position of Tyr9 in a way that changes the function of the active site. The results point to a role for Tyr9 in the mechanism for regulating anion access that has many features in common with other SODs, particularly MnSOD. Thus, NiSOD represents a fascinating case of molecular convergent evolution.
Experimental
Molecular Biological Methods
The sodN gene from H15 (the native gene has a 14 amino acid leader sequence that is cleaved in vivo leaving H15 as the first amino acid after post translation modification) forward was cloned out of the pET3a-SODN vector using PCR amplification with the primers 5′-GGTATTGAGGGTCGCCACTGCGACCTGCCCTG –3′ (5′SODN15) and 5′-AGAGGAGAGTTAGAGCCCTTTGTTAGCAGCCGG-3′ (3′SODNU). The PCR product was purified from a 1% agarose gel. Treatment of the PCR product with T4 DNA polymerase created overhangs complementary to the Ligation Independent Cloning (LIC) vector pET30 Xa/LIC (Novagen™). The sodN gene was annealed into the vector directly after the factor Xa recognition site (IEGR) so that upon cleavage with factor Xa, H1 would be the N-terminal residue. This 47 amino acid N-terminal extension also contains a His tag used during purification to isolate the fusion peptide (vide infra). The insert was annealed to the vector and NovaBlue (Novagen™) competent cells were transformed with the pET30 Xa/LIC plasmid containing the sodN gene. The NovaBlue cells were plated on LB media containing 34 μg/mL kanamycin (kan) and grown overnight at 37 °C. Plasmid mini preps (Qiagen™) were performed after single colonies were grown in 3 mL of LB kan media overnight. BL21 (DE3) pLysS (Novagen™) competent cells were transformed with the plasmid pET-30Xa/LIC-SODNH15 (pSODNH15) and were plated and grown overnight at 37 °C on LB media containing kan and 30 μg/mL chloramphenicol (cam). Single colonies were grown overnight in 3 mL cultures (LB kan/cam) and diluted 1:100 in 100 mL of fresh media. Cells were grown at 37 °C to an OD at 600 nm of 0.6 and induced with isopropyl-β-D-thiogalactoside (IPTG), final concentration 0.8 mM. After 3 hours the cells were harvested via centrifugation, and SDS-PAGE was run on cell extract to determine expression of the target fusion protein. Plasmids producing proteins of the appropriate molecular weight (18 kDa) were submitted to the University of Massachusetts DNA sequencing facility to confirm the presence of the His-tagged WT gene.
Single colonies of cells containing the pSODNH15 plasmid were grown in 10 mL culture (LB cam/kan) overnight at 37 °C with shaking and then added to 1 liter of pre-warmed fresh media. Cultures were grown to an OD of 0.6 at 600 nm and induced (0.8 mM IPTG) for 3 hours. Cells were harvested by centrifugation, resuspended in 50 mL of Ni-NTA binding buffer (10 mM imidazole, 50 mM sodium phosphate, 300 mM sodium chloride pH 8.0) with 1 mM phenylmethylsulfonyl fluoride (PMSF), and frozen at −80 °C. Cells were thawed, 100 μl of DNAse I solution (10 mg/mL DNAse I, 10 mM magnesium chloride, 20 mM Tris pH 7.5, 40 % glycerol) was added and incubated at 37 °C until the viscosity of the solution was significantly reduced. All chromatographic purifications employed an AKTA-FPLC system (Amersham Biosciences). The cell lysate was then loaded onto a column (Pharmacia HR10) containing Ni-NTA His•Bind Superflow™ resin (Novagen™). Non-specifically or weakly bound proteins were removed by using a step gradient of 0–30% elute buffer (250 mM imidazole, 50 mM sodium phosphate, 300 mM sodium chloride, pH 8.0). Following a wash step, the fusion protein was eluted from the column with 100% elute buffer. The eluted protein was then dialyzed into Factor Xa buffer (5 mM calcium chloride, 50 mM Tris, 100 mM sodium chloride, pH 8.0) that contained 0.25 mM PMSF to prepare the sample for N-terminal processing.
N-terminal processing (required to remove leader sequence, which contains the His·Tag for purification) was accomplished by cleaving the fusion protein at the IEGR site using Factor Xa. The cleavage reaction was performed at 4 °C until the reaction was complete, as monitored by SDS-PAGE. Subsequently, the reaction was dialyzed against 20 mM Tris·HCl pH 8.0 to remove excess salt. Anion exchange chromatography (Pharmacia FPLC-Mono Q) using a 350 mM sodium chloride gradient over 10 column volumes at a slow flow rate of 0.5 mL/min gave pure apo-NiSOD in the first fractions off the column, as confirmed by the molecular weights of the products obtained from ESI-MS (Table 4) and by Edman N-terminal peptide sequencing (Midwest Analytical, Inc., St. Louis MO). The oligomeric state of the protein (hexamer) was confirmed by size-exclusion chromatography and by ESI-MS under nondenaturing conditions (Supporting Information, vide infra).
Table 4.
XANES Analysis of Dithionite-Reduced WT-NiSOD and Mutant Enzymes
| Sample | Edge Energy (eV) | 1s→3d peak area (× 10−2 eV) | Geometry | Reference |
|---|---|---|---|---|
| WT | 8340.5(2) | 4.7(5) | planar, CN=4 | (47) |
| Y9F | 8339.5(2) | 5(2) | planar, CN=4 | This work |
| Y62F | 8339.3(2) | 3.2(4) | planar, CN=4 | This work |
| Y9FY62F | 8340.0(2) | 5.8(5) | SPY, CN = 5 | This work |
| D3A | 8342.0(2) | 4.5(5) | planar, CN=4 | This work |
Reconstitution of the recombinant, N-terminal processed apo-NiSOD was achieved by addition of a 3-fold excess of dithiothreitol (DTT) for 2 hours in an anaerobic chamber (Coy Laboratory Products Inc., Grass Lakes, MI) followed by addition of a 5-fold excess of NiCl2. The properties of the reconstituted enzyme were virtually identical to those previously reported for native enzymes (see results).
D3A-NiSOD
The mutation of Asp3 to Ala was made by PCR reaction. The PCR reaction used the WT-NiSOD gene along with the 3′SODNU and the mutagenesis primer 5′-GGTATTGAGGGTCGCCACTGCGCGCTGCCCTGCGGCG-3′. The PCR reaction product was gel purified (2% agarose) and the gene was ligated into the pET30 Xa/LIC vector and then transformed, overexpressed, purified and processed in the same manner as WT-NiSOD (vide supra).
To probe the nature of a protein based radical signal observed in the product of the reaction of D3A-NiSOD with H2O2, a sample of this mutant was prepared where all four tyrosine residues present were substituted by d4-Tyr. Thus, all four aromatic protons are substituted by deuterons in each Tyr residue. To incorporate d4Tyr into D3A-NiSOD, E. coli cells BL21 (DE3) pLysS were grown in LB broth until they reached an OD at 600 nm of 0.6. The cells were then spun down and resuspended in M9 media containing glyphosate (1 g/L), Trp, His, Phe and d4-Tyr (0.5 g/L), and grown for an additional 30 minutes before induction with 1 mL of IPTG (200 mg/mL). Cells were grown for an additional 3 hours and then harvested. d4Tyr-D3A-NiSOD was purified and isolated as described above for WT-NiSOD.
Y9F-NiSOD
The Y9F point mutation was made in two steps: Two fragments were created in two different PCR reactions (reactions A and B) using the WT gene. Reaction A utilized the 5′SODN15 primer and 5′-GCGGCGTGTTCGACCCTGCCCAG-3′, while reaction B used the 3′SODNU primer along with 5′-GGCCGTCCAGGAGAAGCTGGCCGGCAACGACGACG-3′. The PCR fragments were gel purified (2% agarose) and then combined with the 5′SODN15 and 3′SODNU primers in a third PCR reaction to generate the NiSOD gene with Tyr9 mutated to a Phe. The mutated gene was ligated into the pET30 Xa/LIC vector as described for WT-NiSOD (vide supra). The transformation, overexpression, isolation, purification and N-terminal processing of Y9F-NiSOD were performed in the same manner described for WT-NiSOD (vide supra).
Y62F- and Y9FY62F-NiSOD
Both Y62F- and Y9FY62F-NiSOD were made using the QuikChange (Stratagene™) mutagenesis kit. Table 2 shows the primers and the parental DNA that were used in the two PCR reactions. DNA isolated using the Qiagen® miniprep kit was sequenced at the Keck DNA facility (Yale University) to ensure that the correct mutation was present. Transformation, induction, protein purification and N-terminal processing were preformed as described above for WT NiSOD. The protein product was confirmed by the molecular weight of the fusion protein determined using ESI-MS (Table 4, vide infra).
Table 2.
NiSOD mutant Data Collection and Refinement Statistics
| Data Collection | ||||
|---|---|---|---|---|
| In House | Synchrotron | |||
| NiSOD mutant | Y9F (Cl) | Y9F (Cl) | Y9F (Br) | D3A |
| Wavelength (Å) | 1.5418 | 0.9184 | ||
| Resolution (Å) (last shell) | 50–2.01 (50–2.02) | 20.9–1.87 (1.94–1.87) | (1.94–1.87) | 21.57–1.90 (1.97–1.90) |
| Cell parameters (C2221) a, b, c (Å) | 60.09, 112.57, 111.87 | 59.87, 111.58, 111.55 | 59.99, 112.46, 110.96 | 59.99, 112.27, 111.70 |
| No. of observations | 118668 | 197247 | 127478 | 238695 |
| No. of unique observations (last shell) | 23158 (2175) | 28883 (2637) | 29651 (2757) | 28277 (1978) |
| Multiplicity (last shell) | 3.8 (2.7) | 6.9 (5.4) | 4.3 (3.2) | 8.0 (7.2) |
| Completeness (%)(last shell) | 99.2 (97.8) | 92.6 (84.8) | 94.2 (89.1) | 99.4 (95.5) |
| Rsym (last shell) | 0.050 (0.440) | 0.029 (0.361) | 0.038 (0.732) | 0.046 (0.929) |
| I/σI (last shell) | 21.9 (2.4) | 42.6 (4.5) | 28.5 (1.6) | 41.6 (2.3) |
| Refinement | ||||
| Rwork/Rfree (%) | 17.03/24.14 | 17.90/24.78 | 18.16/23.60 | |
| Ave B-factor (Å2) | 30.5 | 29.6 | 39.5 | |
| # of atoms | 3213 | 3249 | 3058 | |
| # of water | 396 | 453 | 318 | |
Quaternary Structure and Stability
The oligomeric states as well as the stabilities of WT and mutant NiSODs were determined by a combination of three techniques; size-exclusion chromatography, ESI-MS under non-denaturing conditions, and differential scanning calorimetry (DSC). Size-exclusion chromatography employed solutions of WT- or mutant-NiSODs that were applied to a HiLoad 16/60 Superdex 200 (GE lifesciences) column. The column was standardized with albumin (67 kDa), ovalbumin (43 kDa), chymotrypsinogen (25 kDa) and ribonuclease A (13.7 kDa). All reconstituted NiSOD samples eluted with molecular weights appropriate for homohexamers. In order to determine the MW of the expression products, ESI-MS was performed using a Bruker Esquire mass spectrometer on solutions of denatured proteins in aqueous ammonium acetate solution (final conc. 5 mM) with 3% acetic acid and 50% MeOH. To confirm the oligomeric state of the enzyme, ESI-MS was also performed on a JMS-700 MStation magnetic sector (double focusing) mass spectrometer under ‘nondenaturing conditions’. To prevent oligomeric breakdown, the orifice voltage was turned to 0 V and the skimmer voltage was set to 120 V. Protein solutions used were 60 μM in NiSOD monomer dissolved in 10 mM ammonium acetate solution. Under these conditions, WT NiSOD exhibits peaks characteristic of the hexameric holoprotein and little else (see supporting information).
Differential scanning calorimetry thermograms were measured with a Microcal VP-DSC with 0.5 mL sample and reference cells. NiSOD samples were concentrated to 2–5 μM hexamer (Y62F- and Y9FY62F-NiSOD) or 100–140 μM in hexamer (WT-, D3A-, Y9F-NiSOD) in buffer (50 mM Tris·HCl, pH 8.0), degassed under vacuum and syringed into the sample and reference cells. Spectra were collected at 30 psi between 6–100 °C at a scan rate of 30 °C/hour. Baseline correction and normalization was performed with the Microcal interface to the Origin graphing program. Peak maxima were taken as melting temperatures, Tm. The values of Tm obtained are shown in Table 3 and thermograms are included in supporting information.
Table 3.
Analytical data for WT-NiSOD and mutants.
| NiSOD sample | ESI-MS MWe (calc’d value) | Quaternary Structurec | Ni/protein | EPR as isolated (% Ni(III)) | Kinetics d | E1/2 (mV vs. NHE) | Tm (°C) | |
|---|---|---|---|---|---|---|---|---|
| kcalc x 108 M−1 s−1 @ pH 7.5 [range of O2− concen. (μM)] | Reductive Titration | Oxidative Titration | ||||||
| WT | 18,171.2a (18,169.6) | Hexamer | 0.88 | g = 2.30, 2.23, 2.01 Azz = 24.9G (51(2)) |
7.07 [1.93–5.97] | 290(4) | 279(6) | 84.8 |
| D3A | 13,160.9b (13,157.0) | Hexamer with decreased stability | 1.11 | g = 2.30, 2.24, 2.01 Azz = 24.9G (48(2)) |
2.09 [1.68–5.99] | 308(3) | 290(7) | 73.9 |
| Y9F | 13,183.4b (13,185.0) | Hexamer | 0.72 | g = 2.30, 2.23, 2.01 Azz = 24.9G (53(2)) |
3.71 [1.54–2.38] | 297(5) | 299(8) | 86.3 |
| Y62F | 18,154.1a (18,153.3) | Hexamer | 0.90 | g = 2.30,2.23, 2.01 Azz = 24.9G (48(2)) |
6.42 [2.09–9.8] | ND | ND | 82.4 |
|
Y9F Y62F |
18,137.5a (18,137.2) | Hexamer | 0.74 | g = 2.30,2.23, 2.01 Azz =24.4G (46(2)) |
2.41 [2.18–4.12] | ND | ND | 74.7 |
Fusion peptide,
Processed NiSOD,
from size-exclusion chromatography and/or ESI-MS under non-denaturing conditions (see experimental),
Rates determined under first-order conditions; kcalc = kobs/[Ni], where [Ni] = 2 μM. The value of kcalc was determined by averaging the values over the range of [O2−] where there is no apparent decrease in kobs.
ESI-MS measurements were conducted under denaturing conditions resulting in monomer MW of processed NiSOD or fusion peptide.
Electron Paramagnetic Resonance (EPR)
X-band EPR spectra were obtained on a Bruker ELEXSYS E-500 X-band spectrometer. Samples in polycarbonate XAS holders were inserted into a liquid N2 finger dewar. The data were collected over a 1000 Gauss field centered at 3000 Gauss. The modulation receiver and signal channel parameters were a modulation frequency of 100 kHz, a modulation amplitude of 10 Gauss, and a time constant of 327 ms. The microwave power was set to 20 mW with a frequency of 9.46 GHz. To determine the number of Ni(III) centers present in each mutant NiSOD, spin integration was performed on each EPR spectrum. The concentration of Ni in each sample was determined using ICP-OES (λ=231.604 nm) on a Perkin-Elmer Optima 4300 DV instrument. The resulting EPR spectra were then integrated twice and compared to a spin standard of known concentration, Bis-(pyridine-2, 6-bis(thiocarboxylate))nickel(III) (NiPDTC)(34), to determine the percentage of Ni centers that were in the oxidized state (Table 3).
Redox Titrations
A modification of the procedure of Reiter et al. was used to determine the E1/2 of WT-NiSOD and the various mutants(35). Solutions of NiSOD (3 mL, 100 – 200 μM) were prepared in 100 mM potassium phosphate buffer at pH 7.5 with 100 mM potassium chloride as a supporting electrolyte. Samples were degassed and kept under argon throughout the experiment. Electrochemical potentials were monitored with an Extech Instruments True RMS digital multimeter connected to a platinum wire and a Ag/AgCl reference electrode. Optical spectra were monitored on a Hewlett-Packard 8452A diode array spectrophotometer equipped with a thermostatted cell holder that held all samples at 25 °C.
Prior to the experiment, the samples were treated with KMnO4 (3 μl aliquots of 6 mM aqueous solution) until the absorbance at 380 nm was at a maximum, ensuring the enzyme was in its most oxidized form. Reductive titrations were performed by adding aliquots (1–5 μl) of reduced methylviologen (6 mM, electrochemically prepared with a Bioanalytical Systems (BAS) CV50W using a BAS bulk electrolysis cell under argon; concentration was confirmed by measuring the absorption of the MV+ solution at 606 nm, ε = 13700 cm−1M−1(36)) via a Hamilton gas-tight syringe. After complete reduction, the oxidative titrations were performed in a similar manner by the addition of small aliquots (1–5 μl) from a 6 mM KMnO4 stock solution (degassed).
The data was plotted as Absorbance (at 378 nm) vs. potential (converted to NHE) and was fit to equation 4, which was derived from the Nernst equation by Bernhardt et al.(37), where Aox is the absorbance of the solution when all the species are in the oxidized form, Ared is the absorbance of the solution when all the species are present in the reduced form, n is the number of electrons being transferred during the reaction, and E0 is the midpoint potential of the redox reaction. To obtain the reported E0 values for NiSOD samples, n was fixed at 1.0 and the other three parameters were allowed to vary.
| (4) |
X-Ray Absorption Spectroscopy
X-ray absorption spectroscopy (XAS) data collection and analysis was performed as previously described(38). Ni K-edge XAS data were collected on beamline X9B at the National Synchrotron Light Source (Brookhaven National Laboratory). Samples of frozen protein solutions (1–3 mM, based on Ni content, in 20 mM Tris•HCl, pH 8.0) were placed in polycarbonate holders inserted into aluminum blocks and held near 50 K using a liquid He cryostat. The ring conditions for data collection were 2.8 GeV and 120–300 mA. A sagittally focusing Si(111) double crystal monochromator and a 13-channel Ge fluorescence detector (Canberra) were used for data collection. X-ray absorption near-edge spectroscopy (XANES) data was collected from ± 200 eV relative to the Ni edge. The edge energy reported was taken to be the maximum of the first-derivative of the XANES spectrum. Extended X-ray absorption fine structure (EXAFS) was collected to 9307 eV (k = 16 Å−1). The X-ray energy for the K-edge of Ni was internally calibrated to 8331.6 eV using transmission data from a Ni foil. The data shown is the average of 4 – 8 scans and was analyzed using the EXAFS123 software package for XANES data(39) and the SixPack software package was used for EXAFS analysis(40). Scattering parameters for EXAFS fitting were generated using the FEFF 8 software package(41). The SixPack fitting software builds on the ifeffit engine(41, 42) and uses iterative FEFF calculations to fit EXAFS data during model refinement, and is thus an improvement over previous methods that employ a static set of calculated scattering parameters. To compare different models to the same data set, ifeffit uses three goodness of fit parameters, χ2, reduced χ2, and the R-factor. χ2 is given by Equation 4, where Nidp = number of independent data points, Nε2 = number of uncertainties to minimize; Re(fi) = real part of the EXAFS fitting function; Im(fi) = imaginary part of the EXAFS fitting function. Reduced χ2 = χ2/(Nind-Nvarys) (where Nvarys = the number of refining parameters) and represents the degrees of freedom in the fit. Ifeffit also calculates the R-factor for the fit, which is given by Equation 5 and is scaled to the magnitude of the data, making it proportional to χ2.
| (4) |
| (5) |
To compare different models (fits) the R-factor and reduced χ2 parameters can be assessed to determine which model is the best fit, in which case both parameters should be minimized. Although the R-factor will always improve with an increasing number of adjustable parameters, reduced χ2 will go through a minimum and then increase, indicating that the model is over fitting the data.
Crystallography
Crystallization experiments were performed using the hanging-drop vapor-diffusion method. All crystals grew after 2–3 weeks at 20°C. Crystals of Y9F-NiSOD (Cl) were obtained by mixing 1μL of the reservoir solution (0.05 M CaCl2, 0.1 M Bis-Tris (pH 6.5 with HCl) and 25% (v/v) PEG MME (Fluka) 550) with 1 μL of a protein solution at 8.0 mg/mL in a buffer of 50 mM Tris-HCl, pH 8.0. Prior to freezing and data collection, the crystals were transferred stepwise into a cryoprotectant solution containing 40% (v/v) PEG MME (Fluka) 550, 0.05 M CaCl2 and 0.1 M Bis-Tris (pH 6.5 with HCl).
To produce crystals of Y9F-NiSOD (Br) with bromide in place of chloride, the protein was exchanged into a buffer solution containing 50 mM Tris-HBr, pH 8.0. Crystals of Y9F-NiSOD (Br) were obtained by mixing 1μL of the reservoir solution (0.1 M Bis-Tris (pH 6.5 with HBr) and 30% (v/v) PEG MME (Fluka) 550) with 1 μL of a protein solution at 8.0 mg/mL. Prior to freezing and data collection, the crystals were transferred stepwise into a cryoprotectant solution containing 40% (v/v) PEG MME (Fluka) 550 and 0.1 M Bis-Tris (pH 6.5 with HBr).
Crystals of D3A-NiSOD were obtained by mixing 1μL of the reservoir solution (0.05 M MgCl2, 0.1 M HEPES (pH 7.5) and 30% (v/v) PEG MME (Fluka) 550) with 1 μL of a protein solution at 8.2 mg/mL in 50 mM Tris-HCl, pH 8.0. Prior to freezing and data collection, the crystals were transferred stepwise into a final cryoprotectant solution containing 40% (v/v) PEG MME (Fluka) 550, 0.5 M MgCl2 and 0.1 M Bis-Tris (pH 6.5).
Crystals were frozen in liquid nitrogen and then transferred into a gaseous nitrogen stream at 100K for X-ray data collection. Data collection was performed at the National Synchrotron Light Source (NSLS) beamline X6A at the Brookhaven National Laboratory. All data were collected at 13500 eV (0.9184Å), which is above the Br absorption edge energy. Each frame consisted of 0.75° rotation taken for exposures of 5 sec [for Y9F(Cl−) and Y9F(Br−)] or 15 sec (for D3A). X-ray images were integrated, indexed and scaled using HKL2000 software package(43) The diffraction data were scaled in space group C2221, leading to a solvent content of 47% and three subunits in the asymmetric unit. Data collection and refinement statistics are given in Table 2.
Molecular replacement calculations were performed in the CCP4 program MolRep(44) using a trimeric search model derived from the native NiSOD structure (PDB accession code 1T6Q)1. The starting model for manual building was the native structure with the first 10 amino acids and the solvent molecules omitted. Rigid body refinement and positional refinement with 3-fold non-crystallographic symmetry (NCS) imposed were performed using the CCP4 program Refmac5(44) and 2Fo – Fc and Fo – Fc electron density maps showed substantial density representing the omitted parts of the model. The entire model, including the N-terminal 10 amino acids, was built into the electron density using the O graphics program(45). Weighted 2Fo – Fc and Fo – Fc maps were examined at each stage of the refinement. In later rounds of refinement, heteroatoms, such as the Ni2+/3+, Cl−, and Br− ions, were assigned to electron density peaks. The experimental electron density was of very high quality except for the N-terminal seven residues, where three-fold averaging was imposed in order to aid in map interpretation. The final statistics for the model are presented in Table 2. To determine if the electron density near the Ni in the Y9F-NiSOD structure was Cl− or water, crystals were grown using Br− instead of Cl−. After the determination that the Cl− was replaced with Br−, the Y9F(Br) structure was not fully refined because the Y9F(Cl) data was of better quality. The quality of the model was assessed with PROCHECK(46).
Kinetics
The kinetics of superoxide dismutation was examined using pulse-radiolytic generation of superoxide and was carried out at Brookhaven National Laboratory as previously described(47). Samples of NiSOD (final concentration 2 μM, based on Ni concentration) were placed into a 2 cm cuvette that contained phosphate buffer (10 mM), formate (30 mM) and ethylenediaminetetraacetic acid (EDTA, 5 μM). The sample was then pulsed from a 2.0 MeV van de Graaff accelerator, varying the pulse width (100–2000 ns), resulting in a range of initial superoxide concentrations (2 – 40 μM). The disappearance of the absorbance at 260 nm that corresponds to O2− was fit to a first-order process (Supplemental Figure 7). Catalytic rate constants were determined by dividing the first order rate by the nickel concentration, assuming all of the nickel is bound and active in the enzyme. The rate of O2− disappearance in the absence of NiSOD was measured for the O2− concentrations and pHs used and was, in all cases, at least an order of magnitude slower than the enzymatically driven rate. The rate constants are reported on a per Ni basis as determined by the Ni content of each sample. Nickel content was determined by ICP-OES using the Ni wavelength of 231.604 nm.
Reactions with H2O2
Both WT- and D3A-NiSOD in 20mM TrisCl, pH = 8, were treated anaerobically with a 10 fold excess of H2O2 at RT. Peroxide concentration was determined by titration with a standard solution of potassium permanganate (48). After 10 minutes, the peroxide was buffer exchanged using spin concentrators (Vivaspin, 5k MW cutoff) and five 1:10 dilutions with buffer. The resulting enzyme solutions were examined by pulse radiolysis to determine if any activity was lost. A portion of each solution was set aside for MW determination by ESI-MS to determine if oxygen incorporation had occurred.
Results
Protein Characterization
Data regarding the molecular weight of expressed protein, the quaternary structure, the Tm, and Ni content following reconstitution of recombinant proteins are summarized in Table 3. All of the recombinant proteins had the MW expected for the amino acid substitutions involved, and all were present largely as homohexamers following reconstitution with NiCl2. In the case of D3A-NiSOD, a significant amount of monomeric protein was also observed in the non-denaturing mass spectrum (see supporting information), however, this was not observed by size-exclusion chromatography, indicating that the protein is not completely stable to the ESI-MS conditions. These results are consistent with the lower Tm value obtained from DSC analysis (Table 3). WT and Y9F had high thermal stability with Tm = 84.8 °C and 86.3 °C, respectively, whereas D3A had lower thermal stability, Tm = 73.9 °C, which may be attributed to weaker interactions between subunits in the hexamer (vide infra).
Electron Paramagnetic Resonance
EPR spectroscopy was used to examine the redox state of the nickel center in the resting enzymes and probe the electronic structure of the Ni(III) site in mutant NiSODs. The EPR data show that the Ni(III) center present in all the mutants is similar to that of WT-NiSOD. As isolated, native NiSOD has a distinctive rhombic EPR spectrum that arises from a five-coordinate, low-spin, S = 1/2 Ni(III) center, with the unpaired electron in dz2. X-band EPR spectra of the WT recombinant NiSOD, tyrosine mutants (Y9F, Y62F, Y9FY62F) and D3A (Table 4) also exhibit typical rhombic Ni(III) signals with strong N-hyperfine splittings on the gz feature (Az = 24.9 G) arising from the apical imidazole bound to the Ni(III) center(47, 49). The electronic structure of the Ni(III) center was not expected to be greatly perturbed by mutations in the second coordination sphere, and this was confirmed by the spectra obtained (Table 4). When Tyr9 is mutated to Phe, subtle hyperfine structure on the gy feature that is not resolved in spectrum of WT NiSOD is observed (see supporting information).
Careful integration of the Ni(III) signal in WT-NiSOD indicates that the EPR signal accounts for only half of the nickel present in the samples (Table 3). All samples were approximately 50:50, Ni(III):Ni(II). Excess potassium permanganate or hexachloroiridate were used in attempts to oxidize the remaining Ni(II) centers in as isolated WT-NiSOD. In both cases no additional oxidation was seen. These results do not coincide with a previous report in which, excess ferricyanide was used to reach a maximum oxidation of 72% Ni(III) in WT-NiSO (17). However, it should be noted that both permanganate and hexachloroiridate are stronger oxidants than ferricyanide, which has a redox potential (360 mV) that is barely above the midpoint potential of the enzyme.
In summary, the EPR results from the nickel center are consistent with a resting oxidized state that contains 50% Ni(III) and 50% Ni(II), that is not significantly perturbed by mutation of Tyr9 or Asp3.
X-Ray Absorption Spectroscopy
XAS studies were used to confirm that the nickel center was bound to the correct protein site in the reconstituted WT protein and to characterize the nickel site in the mutant NiSODs, as well as to supplement the results of the crystallographic investigations of the nickel sites in Y9F-NiSOD and in D3A-NiSOD and allow comparisons with other mutants. Since the oxidized enzymes always contain a mixture of oxidized and reduced nickel centers, the samples studied were all reduced with sodium dithionite prior to XAS data collection and were EPR silent. The results for the XAS experiments on the tyrosine mutants of NiSOD are summarized in Tables 4 and 5, and Figure 2.
Table 5.
EXAFS Analysis for Dithionite-Reduced NiSOD samples
| Sample | N | R (Å) | σ2 (×103 Å−2) | ΔE0 (eV) | Reduced χ2 | R factor |
|---|---|---|---|---|---|---|
| WTa | 2N | 1.91(1) | 1(2) | −3.4 | N/A | 0.64c |
| 2S | 2.160(4) | 0(4) | −3.2 | |||
| WTb | 2N | 1.83(4) | 10(4) | −17(3) | 22.8 | 0.11 |
| 2S | 2.16(2) | 7(1) | ||||
| 2C | 2.73(4) | |||||
| 2C | 2.90(5) | 3(5) | ||||
| Y9F | 2N | 1.95(2) | 5(2) | −7(2) | 22.2 | 0.06 |
| 1S | 2.23(2) | 3(1) | ||||
| 1S | 2.52(3) | 6(4) | ||||
| 2C | 3.00(4) | 4(4) | ||||
| Y62F | 1N | 1.90(3) | 5(4) | −10(2) | 11.2 | 0.04 |
| 1N | 2.2(2) | |||||
| 2S | 2.17(3) | 5(2) | ||||
| 3C | 2.85(3) | |||||
| 2C | 3.03(4) | 2(2) | ||||
| D3A | 3N | 2.08(2) | 2.3(9) | −7(1) | 9.04 | 0.04 |
| 2S | 2.13(1) | 8(3) | ||||
| 2C | 2.95(6) | 12(8) | ||||
| Y9FY62F | 2N | 2.02(6) | 12(6) | −5(3) | 14.7 | 0.09 |
| 1S | 2.20(2) | 2(1) | ||||
| 1S | 2.46(4) | 6(3) | ||||
| 2C | 3.02(5) | 6(6) |
Figure 2.

Comparison of XAS data for Tyrosine mutant NiSODs; WT(Yellow), Y9F(Blue), Y62F(Purple), Y9FY62F(Red), D3A(Green). Left: Normalized XANES spectra. Right: Unfiltered EXAFS data in solid colored line and fits (black) from Table 6. The range of the k-space fits is 2–12.5 Å−1 for all data.
XANES
XANES (X-ray absorption near edge structure) yields information about the coordination number and geometry of the nickel site. Nickel has vacancies in the 3d manifold, which give rise to transitions in the XANES region of the XAS spectrum that are dependent on the geometry/coordination number of the metal site. These transitions include the 1s → 3d and 1s → 4pz (plus shakedown contributions), which occur near 8331 and 8336 eV, respectively, in Ni(II) complexes(50). In WT-NiSOD and in the mutant NiSODs, both transitions are observed, which indicates either a square planar or five-coordinate pyramidal nickel center geometry(50). The peak areas of the 1s → 3d transition (Table 5) lie on the line between four-coordinate planar species (0–0.04 eV) and five-coordinate pyramidal species (0.04 – 0.09 eV); and indicate the sites that are most likely distorted four-coordinate planar in nature, consistent with what was previously reported for native Streptomyces seoulensis reduced NiSOD(47). The edge energies observed for the reduced species compare well with native S. seoulensis enzyme.
EXAFS
The EXAFS (Extended X-ray Absorption Fine Structure) region of the XAS spectrum provides information regarding the type of scattering atoms present and their distance from the central metal ion. Table 6 and Figure 2 compare the best fits obtained for WT- and mutant NiSODs. In order to evaluate the change in fitting method, data was collected for reduced WT-NiSOD and compared to previously published data(47). The fit is quite similar except for the Ni-N bond length is ~0.08 Å shorter than previously found. However, prior model studies indicate that it can be difficult to obtain a distance with low error for N-scattering atoms in a complex where the EXAFS is dominated by scattering from S atoms(50). The new fits include atoms in the second coordination sphere, which are main-chain carbon atoms from His1 and Cys2 as well as the β-carbon from Cys2 as seen in the two crystal structures of NiSOD(17, 21). These atoms are rigidly positioned by chelate rings, and their inclusion improves the fits. The single mutation, Y9F-NiSOD, alters the structure around the Ni center. The best fit for Y9F-NiSOD contains two shells of sulfur donors at different distances. The first sulfur donor lies at 2.23 Å while the other sulfur ligand is longer and lies at 2.52 Å. This splitting of the two sulfur ligands into two distances is also seen in the double mutation Y9FY62F-NiSOD, which has a similar fit to Y9F-NiSOD (Table 6). D3A- and Y62F-NiSOD have fits that are more similar to WT-NiSOD with both S-donors at the same Ni-S distance. For D3A-NiSOD, the Ni-S bond distance found was similar to WT-NiSOD (0.03 Å shorter) where Y62F-NiSOD had a slightly longer Ni-S bond (0.07 Å) than WT-NiSOD (Table 6). The WT- and D3A-NiSOD fits differ by the addition of one Ni-N vector in the D3A-NiSOD fit, and the Ni-N bonds are longer in D3A-NiSOD (2.08(2) Å versus 1.83(4) Å in WT-NiSOD). The fit obtained for Y62F-NiSOD is slightly different in that the best fit splits the two nitrogen scatterers into different shells with one ligand at 1.90 Å and the other at 2.2 Å.
Table 6.
Redox Potentials
| Sample | E0 mV (vs. NHE) | Aox | Ared | N* |
|---|---|---|---|---|
| WT – Reduction | 290(4) | 0.76(1) | 0.19(1) | 1 |
| WT – Oxidation | 279(6) | 0.91(2) | 0.16(5) | 1 |
| D3A – Reduction | 308(3) | 0.815(9) | 0.24(1) | 1 |
| D3A – Oxidation | 290(7) | 0.91(3) | 0.16(6) | 1 |
| Y9F – Reduction | 297(5) | 0.54(2) | 0.15(2) | 1 |
| Y9F – Oxidation | 299(8) | 1.12(9) | 0.06(6) | 1 |
This value was held constant in fitting the absorption data to obtain E0.
N is the number of electrons per Ni(III) center.
The data indicates that when Tyr9 is altered by loss of the OH group, that different Ni-S distances are obtained. This structural perturbation is confirmed by single crystal diffraction studies (vide infra) and may result from the alteration of an H-bonding network in the Ni-hook domain. Alternatively, the results may indicate that the nickel site is a mixture of low- and high-spin Ni centers, given the similarity of the two distances to low-spin(51) and high spin(50, 51) nickel thiolate model complexes.
Protein Structure
NiSOD crystallographic results
The crystal structures of two NiSOD mutants, Y9F and D3A, were determined to 1.9 Å. Both the Y9F and D3A NiSOD mutant structures are present as hexamers with a trimer in the crystallographic asymmetric unit. In an effort to understand the structure and function of NiSOD, the structures of Y9F- and D3A-NiSOD were compared to WT-NiSOD, including both the holo and apo forms (PDB codes 1T6U, 1T6I, and 1T6Q).
Y9F crystal structure
The Y9F crystal structure revealed the presence of an anion-binding site near the active site in NiSOD. The WT-NiSOD structure showed the presence of two water molecules making hydrogen bonds to the hydroxyl of the Tyr9 side chain. In our NiSOD Y9F mutant, electron density appears at similar locations to those two water molecules in the WT-NiSOD structure. During the initial interpretation of the electron density, water molecules were placed at these locations in the structure. However, when a water molecule was refined in one of these locations, Fo-Fc electron density maps showed residual electron density and positive peaks. When that water molecule was replaced with a chloride ion, the refinement then led to lower Rfree values and the positive peak in the Fo-Fc map disappeared. The location of the chloride ion is ideally suited to anion binding, with the main chain amide nitrogen atoms of residues 3, 6, and 7 pointing toward the chloride and less than 4 Å away.
To confirm the identity of the chloride, and to probe the specificity of the putative anion-binding pocket, chloride was substituted by bromide in the crystallization of Y9F-NiSOD. Diffraction data was collected above the bromine absorption edge energy from crystals of both bromide and chloride containing Y9F-NiSOD [NiSOD(Br) and NiSOD(Cl) respectively]. The anomalous difference electron density maps from NiSOD(Br) diffraction data revealed strong density (greater than 8 standard deviations) in the anion-binding site, unambiguously identifying the bromide ion. In the NiSOD(Cl) crystals, no peak appeared at this location, as chloride has a negligible anomalous signal at this energy. As an internal control, the nickel ion appears in both anomalous maps, because nickel has a slight anomalous contribution (~ 2 e− vs. ~ 4e− for bromine) at this energy. To test if the anion-binding site is a feature of the Y9F structure alone, the same location in the WT- NiSOD structure was examined. Intriguingly, the coordinates of the WT- NiSOD contain a water molecule at this position, but Fo-Fc maps from that diffraction data show a positive peak, suggesting that the wild type crystals may also contain some amount of chloride ion in the anion-binding pocket.
D3A crystal structure
The mutation D3A leads to the loss of two ion pairs across the interface between neighboring molecules in the hexamer. As a result, 12 ionic interactions are lost when comparing the WT structure to the D3A mutant, providing a plausible explanation for the lower thermal stability of the D3A mutant protein. The carboxyl group of Asp3 lost upon mutating it to Ala leads to the loss of a salt bridge between Asp3 of one chain and Lys89 of a neighboring chain. The loss of the carboxyl group from the side chain of residue 3 produces a gap in the interface between monomers, allowing the Nζ of Lys89 to move towards Ala3 about 2.0Å to fill the space. Lys89 is critical to the network of salt bridges across the interface between monomers in the hexamer. The movement of the Lys89 side chain causes the neighboring Glu49 side chain to move by 2.0Å toward Lys89. This movement of Glu49 weakens the ionic interaction between Glu49 and His53 across the interface between monomers. Therefore mutating Asp3 to Ala results in the loss of two ionic interactions across each interchain interface (Asp3:Lys89 and Glu49:His53). With six of these interfaces in the hexamer, the loss of twelve ion pairs per hexamer could account for the decrease of the melting temperature of the mutant from 84.8 to 73.9 °C.
As a result of the disruption in the intersubunit salt bridges in the D3A structure, the “nickel-hook” region is subtly perturbed. Most notably, Tyr9 has moved ~ 1 Å closer to the nickel (Tyr9-O-Ni; WT-NiSOD = 5.47 Å; D3A-NiSOD = 4.26 Å), and a water molecule (W1) has been displaced from the anion-binding pocket.
Redox chemistry
EPR and MCD spectroscopic methods have been used to demonstrate that the nickel centers in the resting oxidized and reduced proteins are low-spin Ni(III) and diamagnetic Ni(II) centers, respectively(25). We have used potentiometric titrations monitoring the absorbance at 380 nm assigned to a CysS → Ni LMCT transition in the Ni(III) complex(25) to measure the potential of this redox process (Figure 5). Using samples that were pre-oxidized with KMnO4, the data from the initial reductive titration were fit to Equation 4 to yield values of E0 that are shown in Table 6. Reoxidation of the reduced protein with KMnO4 shows that the process is reversible. The potentials obtained from fitting the oxiditative titrations are within experimental error of those obtained from the reductive titrations (Table 6). The slight increase in absorbance in the re-oxidized enzyme likely reflects a small concentration of the protein due to solvent loss during the long incubation under Ar that was required to stabilize the potential at each point (ca. 30 min. per point). The E0 value obtained for WT NiSOD (290(4) mV vs. N.H.E) is approximately midway between the potentials for the oxidation of O2− to O2.
Figure 5.
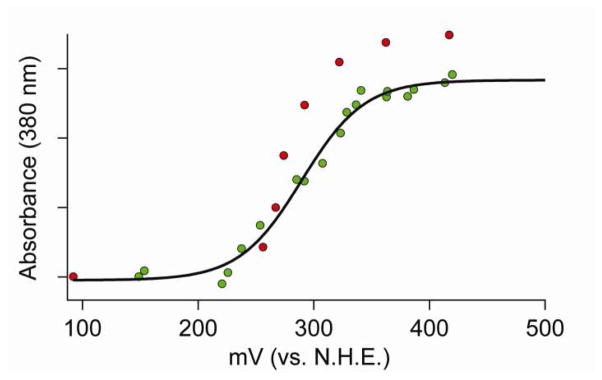
Redox titrations of WT-NiSOD. The green points are the initial reduction of oxidized to reduced NiSOD and the red points were obtained by re-oxidation of the same sample. The black line depicts the best fit of the reductive titration (green dots) using equation 4.
These values are quite similar to the potentials obtained from other SODs (see discussion section). The potentials obtained for the Y9F- and D3A-NiSOD mutants are very similar to the potential of the WT-NiSOD, consistent with the high catalytic rates observed for these mutants (vide infra). This result is similar to that found for MnSOD, where mutation of Tyr34 does not affect the redox potential of the Mn center(52).
Kinetics
The rate of dismutation of superoxide by NiSOD was determined by monitoring the decrease in absorbance of the superoxide anion at 260 nm (ε = 2000 M−1 cm−1)(53) generated by pulse radiolysis. Table 3 shows the calculated catalytic rate constant for the different tyrosine mutants of NiSOD as compared to WT. Y9F-NiSOD exhibits a slightly perturbed value of kcalc that is ~63% of the activity of WT-NiSOD. This value is slightly less than the value obtained from S. seoulensis Y9F-NiSOD(21) (78%) using a construct in which the SOD from S. lividans was knocked out and replaced with a mutated copy of S. seoulensis(21). The SOD activity in that study was measured by the standard cytochrome C assay in cell extract(21). However, the studies presented here reveal that at high substrate concentrations (above a 5x excess), Y9F-NiSOD begins to exhibit saturation behavior. The disappearance of the absorbance at 260 nm at high superoxide concentration relative to the enzyme concentration no longer fits a first order process and begins to exhibit a linear component suggesting saturation behavior. The half-life for superoxide disappearance at high substrate concentrations in the presence of Y9F-NiSOD is lowered to ~ 41% of that in the presence of lower superoxide concentrations. Saturation kinetics is not a feature of WT-NiSOD, which cannot be saturated under these conditions. Saturation behavior is seen in MnSODs, although in that case for both the WT- and mutants (54, 55).
The other Tyr mutant, Y62F-NiSOD, has a catalytic rate similar to WT-NiSOD, and does not saturate, demonstrating that this residue is not intimately involved in the redox mechanism of NiSOD. This is not surprising as Tyr62 sits approximately 13 Å from the Ni center(17, 21). The double mutant, Y9FY62F-NiSOD, is generally similar to the Y9F mutant (kcalc ~ 35% of WT) and displays evidence of saturation at high substrate concentrations, consistent with the mutation at Tyr9.
The crystal structure of Y9F-NiSOD (vide supra) shows that one of the water molecules found in the active site pocket near the metal site of WT-NiSOD has been replaced by a small anion (Cl− or Br−) from the buffer solution. Mutation of Tyr9 may lead to an opening of the binding pocket, which would allow small anions to compete for the binding site, placing a negative charge in the path of the negatively charged superoxide and leading to diminished catalytic rates or a modification of the electrostatic component of the rate constant. To test this hypothesis, ionic strength measurements were carried out using Cl− and ClO4-. Figure 6 shows a plot of the log of the calculated rate versus the square root of the ionic strength for WT, Y9F and Y62F. The results are similar to those found for native NiSOD from S. seoulensis(47) and do not show a change from WT-NiSOD behavior as the ionic strength increases. These results suggest that small anions are not competing with superoxide during catalysis for the binding site.
Figure 6.
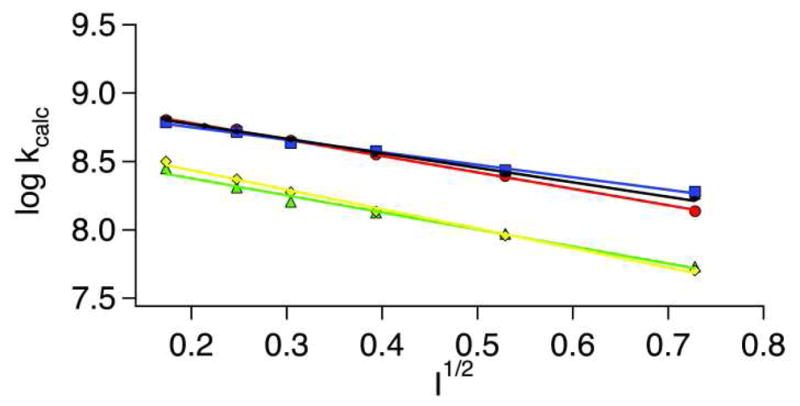
Ionic strength dependence of NiSOD catalysis. Y9F- and WT-NiSOD were reacted with both NaCl (red-WT and yellow-Y9F) and NaClO4 (blue-WT and green-Y9F), while Y62F was only reacted with NaCl (black). Pulse radiolytic conditions were the same as those mentioned in Materials and Methods, except 0.5 M – 0.0156 M of either NaCl or NaClO4 was added to the solution being monitored to probe the effect of ionic strength.
Mutation of Asp3 to Ala results in a 50% decrease in activity at pH 7.5 when compared to WT-NiSOD. Upon further investigation it was also determined that D3A-NiSOD was much more sensitive to peroxide than WT-NiSOD. To examine the nature of this sensitivity, both D3A- and WT-NiSOD were incubated with H2O2 for 5 minutes, and then the reductant/inhibitor was dialyzed away. These samples were then tested for SOD activity using pulse-radiolytic generation of O2−. WT-NiSOD retained full activity, whereas the activity of D3A-NiSOD was below the detectable limit (data not shown). ESI-MS of D3A-NiSOD before and after H2O2 incubation were identical (see supporting information) ruling out the possibility that the peroxide was causing an oxidative modification of the protein, such as conversion of the cysteine thiolate ligands to sulfinates(24), which would likely result in a loss of activity. However, incubation of D3A-NiSOD with peroxide did result in the production of a tyrosyl radical EPR signal (vide infra), an oxidative modification that does not affect the MW of the enzyme.
As isolated, D3A-NiSOD has the same EPR parameters as WT-NiSOD (Table 4). However, when treated with H2O2 (a reductant and inhibitor of NiSOD(56)) under anaerobic conditions, a signal attributable to a protein-based radical at g = 2.004 emerges with a 1H hyperfine splitting pattern that resembles a tyrosine radical (Figure 7)(57). Integration of this signal versus Ni(PDTC) showed that ~8% of the enzyme possessed the tyrosine radical. This assignment was confirmed by replacing all four tyrosine residues with d4-tyrosine, where the protons on the aromatic ring are substituted by deuterons. The resulting hyperfine is clearly altered, consistent with the assignment of the hyperfine to a tyrosyl radical. (The deuterated sample is ~50% less concentrated than the non-deuterated sample.) This signal is not observed to any significant extent in WT-NiSOD or any of the other mutants, and indicates that the interaction of the enzyme with peroxide is fundamentally altered by the D3A mutation.
Figure 7.

X-band EPR data from D3A-NiSOD. (A) As-isolated D3A. (B) H2O2 treated D3A. (C) Enlargement of the region near g = 2.00 expanded (solid line) and a simulation with parameters gx=gy=gz=2.004, Axx=Ayy=5 G, Azz=20 G (dashed line-red). (D) H2O2 treated D3A-NiSOD (d4-Tyr), showing alteration of hyperfine due to tyrosine ring protons. All spectra are shown with the same intensity scale with the exception of D, which has a scale half as large.
Discussion
Redox chemistry of the nickel site
The protein environments of the catalytic metal sites in SODs are responsible for achieving at least three features of these enzymes: adjusting the redox potential of the metal to a value that is optimum for catalysis (ca. 300 mV)(8), controlling anion access to the active site(27), and providing a source of protons for formation of H2O2(17, 21). The O2/O2− reduction potential has been determined to be −160 mV (vs. N.H.E.) and the O2−/H2O2 potential lies at 870 mV (vs. N.H.E.)(58). The midpoint of these two potentials (365 mV vs. N.H.E) is the potential where an optimal SOD catalyst would lie. Nickel is an unusual choice of metal for an SOD because the reduction potential of Niaq(III)/(II) is ≥ 2 V (1.94 V - low spin Ni(III) and 2.24 -high spin Ni(III))(6), and lies well outside the biologically relevant range. Nonetheless, the redox titrations of NiSOD presented above establish that the redox potential of the nickel site is ~290 mV, appropriate for SOD catalysis. The other classes of SODs (Cu/Zn, Mn and Fe) all have reduction potentials near 300 mV (vs. N.H.E.) with deviations up to ~100 mV(59–63). This demonstrates that the protein environment is critical for tuning the reduction potential of each metal to the optimal potential for SOD catalysis. The reduction potential for hexa-aqua complexes of Mn (1.5 V) and Fe (0.77 V)(8, 64) requires a smaller adjustment by the protein than Ni does. The suppression of the Em in Mn- and Fe-SOD is achieved by a hydrogen bonding network that couples the protonation state of an aqua ligand to the redox process of the metal(8). In the case of NiSOD, no aqua ligand exists in any crystal structure, and thus the protonation state of such a ligand is not available to adjust the redox potential of Ni(III)/(II). Furthermore, the shift in potential for the Fe(III)/(II) redox couple in FeSOD has been attributed to the protonation state of the aqua/hydroxo ligand(65), such a shift would not be adequate to optimize the potential of a nickel center in a similar ligand environment. Instead, NiSOD relies on a different ligand environment, one that includes cysteine ligands, a feature of every redox active nickel enzyme that utilizes the Ni(III)/(II) redox couple(66). Thus, NiSOD uses a unique strategy to accomplish the same goal, a ~300 mV redox potential, that is found in every other SOD.
EPR integration of the Ni(III) signals in the resting, oxidized enzyme account for only 50% of the Ni present in WT-NiSOD and in the mutant enzymes, indicating that NiSOD contains a 50:50 mixture of Ni(II) and Ni(III). The amount of Ni present in NiSOD was determined by ICP-OES (see experimental) after extensive buffer exchanging and treatment with chelex to remove any non-specifically bound metal. EXAFS analysis confirms that all of the Ni is bound in the active site, since each Ni center has two S-donor ligands, and there are only two cysteine residues in the protein. Oxidation of resting, oxidized WT-NiSOD with KMnO4 failed to increase the intensity of the EPR signal arising from the five-coordinate Ni(III) center (see supporting information), indicating that the 50:50 mixture is, in fact, the fully oxidized enzyme. Although the present data do not show why one nickel center is redox active and the other one is not, a likely possibility is that the apical His1 imidazole ligand plays a role. The two nickel centers differ by coordination of the imidazole ligand (His-on vs. His-off) and it seems likely that the Ni(II/III) redox potential of the four-coordinate planar center is much higher than for the five-coordinate complex. In this regard, it is noteworthy that mutation of the apical His ligand to Gln(49) or Ala(67) yields mutants that have a resting oxidation state that contains only EPR silent Ni(II). Other than a possible role in maintaining charge neutrality in the hexamer during catalysis, the biochemical rationale for the equal mix of the two oxidation states remains to be elucidated but would seem to require some cooperativity between active site centers in order to maintain the equal balance of oxidized and reduced nickel ions. Until now, the mechanism of communication between active sites within the hexamer that are spaced ~25 Å apart has not been clear, but the structure of D3A-NiSOD suggests a mode for maintaining the redox balance. As described above, a network of ion pairs connect Asp3 to Lys89 to Glu49 to His53 across the interface between neighboring monomers (Figure 4) derived from different trimer subunits. His53 also lies near a two-fold symmetry axis of the hexamer (which contains 32 point symmetry). The Nδ of His53 hydrogen bonds to a water molecule that lies directly on the molecular 2-fold axis. Thus, the His53 side chains from two monomers related by a molecular dyad make a water mediated hydrogen bond, raising the possibility that information about one active site nickel may be communicated to a neighboring active site nickel.
Figure 4.
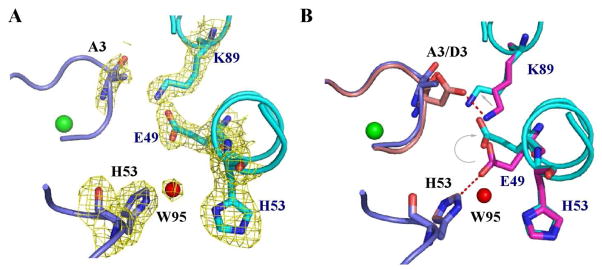
Salt bridge changes in the D3A-NiSOD mutant. A. The D3A-NiSOD mutant is shown at the interface between neighboring monomers (in blue and cyan) in the hexamer. A weighted 2Fo-Fc map shows the electron density in the interface. B. Superposition of this interface from D3A-NiSOD (in blue and cyan) onto wild type (pink and magenta) shows the movement of Glu49 and Lys89. Images were generated in PyMOL (75).
Human MnSOD (hMnSOD) is a homotetramer and forms a dimer of dimers. A recent study examined the H-bonding network between Glu162 and His163 (a metal ligand) by mutating Glu162 to both an Ala and Asp. This H-bonding network connects neighboring monomers across a dimer interface and links two Mn centers(54). Both mutations retained tetrameric structure and specificity for Mn over Fe (in contrast, studies of the analogous mutation made in E. coli MnSOD (EcMnSOD), a homodimer, show the mutant enzyme preferred Fe over Mn(55)). Both the human Glu162 mutations have diminished activity compared to WT-hMnSOD(54), but each helps to elucidate fine points of how hMnSOD carries out O2− dismutation. By performing pH titrations on the enzyme and monitoring the electronic absorption spectrum, the pKa of the system can be determined. WT-hMnSOD has a pKa of ~9.5, which has been attributed to ionization of Tyr34(32, 68, 69). In the E162D-hMnSOD mutant the pKa decreases slightly to 8.7(2), while the E162A-hMnSOD pKa is closer to the pKa of Tyr in water (10.1(1))(54). This can be explained by the interaction of the Glu162 with Tyr34, which is located 6.2 Å away. The H-bonding network extends from the carboxylate of Glu162 to His143 through a solvent molecule to Tyr34. This network is maintained in the E162D-hMnSOD mutant but is disrupted in the E162A-hMnSOD mutant. This is analogous to the backbone amide of Asp3 in NiSOD, which is connected to the phenol proton on Tyr9 through a H-bonding network involving ordered water in the pocket. This network is perturbed in the D3A-NiSOD mutant and perturbed kinetics and peroxide sensitivity is observed. (It is noteworthy that in hMnSOD, there is no functional difference between E162A and E162D mutants despite the structural difference. In NiSOD, there is a functional difference.)
The roles of Tyr9 in NiSOD catalysis
Mutations of conserved second coordination sphere residues, Tyr9, Asp3, as well as the more distant Tyr62, do not lead to major perturbations of the protein structure, or of either the electronic structure or redox chemistry of the nickel site. However, both the Tyr9 and Asp3 mutations have significant effects on catalysis. In the case of Y9F, there is a small decrease in kcalc and the enzyme shows the onset of saturation behavior that is not a feature of WT-NiSOD under the conditions studied. In the case of D3A-NiSOD, the loss of activity is more pronounced, the enzyme is somewhat less thermally stable, and exhibits distinct reactivity with H2O2.
Much of the altered functions of Y9F- and D3A-NiSOD can be accounted for in terms of the roles played by the Tyr9 residue in catalysis. The structure of WT-NiSOD reveals no water molecules coordinated to either the five-coordinate pyramidal “His-on” Ni(III) center or the reduced four-coordinate planar “His-off” Ni(II) center. Tyr9 lies near the vacant sixth coordination position with a Ni-O-Tyr9 distance of 5.47 Å. The position of Tyr9 is such that the phenol proton is engaged in hydrogen bonds with two ordered water molecules, W1 at 2.56 Å and W2 at 2.84 Å, that also accept hydrogen bonds from amide protons from Cys6 (W1) and Asp3 (W2)(17, 21). In the Y9F mutant, loss of the phenol group leads to a small perturbation in the position of phenyl ring, opening up the existing anion binding pocket (Figure 3) and W1 is replaced by Cl−. This is the first crystallographic characterization of an anion complex of NiSOD and clearly identifies a likely binding site for the substrate, O2−. That Cl− is not a nickel ligand is evidenced by the 3.5 Å distance, the absence of a Ni-Cl vector in the EXAFS spectrum and the lack of chlorine hyperfine (even at 200 mM Cl−) in the EPR spectrum, particularly on gz which exhibits large N-hyperfine (Az = 25 G) from the apical H1 imidazole ligand in the dz21 ground state. Hyperfine splitting from the enzyme (likely due to the amide and or N-terminal amine N-donor ligands) is partially resolved on gy in Y9F-NiSOD. Similar hyperfine on the gy feature has also been observed in azide-inhibited NiSOD(17). When 15N-labeled azide was used, the same EPR spectrum was observed, indicating that the splitting was due to an electronic and/or structural perturbation rather than to azide binding(17). Further, the location of the Cl− in the Y9F-NiSOD structure is in the same location that was previously suggested for azide (anion) binding(17). The Cl− anion has hydrogen bonding interactions with both of the amides from Cys6 (N-Cl = 3.4 Å) and Asp3 (N-Cl = 3.4 Å), which provide a mechanism for a small perturbation of the nickel site. The Cl− is positioned such that it could interact with the phenol group of Tyr9 in WT-NiSOD, which suggests possible roles for Tyr9 in the release of H2O2, and in controlling access of anions to the active site. The size of the pocket in WT-NiSOD is appropriate for only small anions and is positioned near to the nickel site but does not allow anions to bind to the metal. Since there is no evidence of anion binding to Ni in either His-on (Ni(III)) sites or in His-off (Ni(II)) sites, and assuming that O2- binds to the same site, the structural data support an outer-sphere redox reaction for both half-reactions as illustrated in (Scheme 1).
Figure 3.

NiSOD hook domain. A. WT-NiSOD N-terminal region showing the active site Ni ion (green sphere) and two water molecules (red spheres). B. The same region of the Y9F-NiSOD mutant. A chloride ion (orange sphere) at the anion-binding site replaces a water molecule. C. The same region of the Y9F-NiSOD(Br) structure. An anomalous difference map (red cage), contoured at 4σ, from data measured above the bromide absorption edge energy. Only the bromide and nickel show an anomalous signal. D. The same region of the Y9F-NiSOD(Cl) structure. An anomalous difference map (black cage), contoured at 4σ, from data measured above the bromide absorption edge. Only the nickel shows an anomalous signal. The Ni centers are modeled as the “His-off” state, the actual state is most likely a mixture of “His-on” and “His-off”, but higher B-factors led to poorly resolved electron density at the N-terminus. Images were generated in PyMOL (75).
Scheme 1.

Proposed outer-sphere mechanism for NiSOD. Ni(III) is indicated by the His-on complex, while Ni(II) is denoted as a His-off complex, although removal of the His1 during turnover may not occur. Superoxide is denoted by the orange molecule and water is denoted by red spheres. Images were generated in PyMOL (75).
In an outer-sphere mechanism the superoxide would displace the two water molecules (Scheme 1 – I) and sit between the backbone amides of Asp3 and Cys6 (Scheme 1 – II). Electron transfer would occur yielding molecular oxygen and Ni(II) (Scheme 1 – II → III). The second half of the reaction starts the same as the first half, with superoxide binding in the substrate binding pocket (Scheme 1 – IV). Electron transfer occurs concomitant with proton transfers, leading to formation of peroxide and Ni(III). Loss of the phenol group and opening up the anion-binding pocket leads to a small perturbation in the position of phenyl ring and increased occupancy of the site by Cl− and Br− in the absence of O2− (Figure 3). When the bound peroxide is protonated, the phenol group could play a role in destabilizing the binding of the product, H2O2, in the anion binding site and inhibiting the direct interaction with the nickel site and its oxidation-sensitive cysteine thiolate ligands. Slower peroxide release in the absence of the phenol group could account for the decrease in activity and the saturation behavior observed in Y9F-NiSOD kinetics.
It has been shown that binding of azide to Ec MnSOD is temperature dependent and at low temperature, a six-coordinate complex derived from complexation of azide is formed, whereas at room temperature the active site is five-coordinate and lacks an azide ligand, resembling the resting enzyme(70, 71),(31). One hypothesis speculates that a “dead-end” complex of Ec MnSOD forms when superoxide binds to Mn2+-SOD causing oxidation of the Mn center and reduction of superoxide to peroxide with concomitant transfer of a proton from the bound water to yield a Mn center with bound hydroperoxide and OH−(31). The Y34F mutation changes the way the enzyme interacts with anions. When azide is added to Y34F-Mn3+-SOD the enzyme resembles the low temperature six-coordinate N3-Mn3+-SOD complex, and shows no temperature dependence(71). Thus, the “gateway” tyrosine seems to play a critical role in controlling anion access to the metal site in both Fe- and Mn-SODs(15, 16, 71) by inhibiting anion binding at the metal center. The same function for Y9 appears to be a feature of NiSOD catalysis. Second sphere residues such as Tyr34 also tune the lability of the peroxide product in MnSOD. This is seen in that the activity of Y34F-MnSOD is more product inhibited than WT-MnSOD(68).
Another possible explanation might be that mutation of Tyr9 and loss of W1 from the active site perturbs a hydrogen bonding network that is important for supplying protons for the formation of H2O2(17, 21). This has also been proposed for MnSOD, where the H-bonding network in the second coordination sphere has been implicated as the source of protons for the release of H2O2(68, 72). Studies on Tyr34 from human MnSOD (hMnSOD) shows that replacing the Tyr with a Phe has little effect on the protein fold or the geometry at the active site, in fact the thermal stability of the mutant is increased when compared to WT-hMnSOD(68). Tyr34 is not essential for MnSOD catalysis, as at low substrate concentrations (compared to Km) Y34F-MnSOD has similar catalytic rates to WT-MnSOD. This is shown by the fact that the steady state constant, kcat/Km, is the nearly identical for both Y34F- and WT-MnSOD and is near the diffusion limit of 109 M−1s−1. The only major perturbation is the decrease in maximal rate, kcat, which is approximately 10 fold lower for the mutant enzyme(68). This implies that Tyr34 may be involved in peroxide release or proton donation either directly to the metal site or indirectly through a H-bonding network that ties the Tyr34 to the Mn active site(68). Mutation of Tyr34 has also been examined in FeSOD from E. coli (EcFeSOD) and Y34F-FeSOD results in a rate decrease of ~ 60% when compared to WT-FeSOD(33). The “gateway” Tyr residue in FeSOD also seems to play a key role in controlling anion binding to the metal site, which differs between the reduced and oxidized state in FeSOD(15).
The importance of Tyr9 in controlling the access of H2O2 to the active site is supported by studies of D3A-NiSOD. Although this mutant exhibits reduced activity relative to WT-NiSOD, redox titration shows that this is not due to alteration of the redox potential of the nickel site. Further, the similar EPR spectrum obtained from Ni(III) centers in the oxidized enzyme show that the electronic structure of the nickel site is not perturbed by the D3A mutation. Nonetheless, this mutant exhibits a drastically altered interaction with H2O2. Unlike WT-NiSOD, which shows inhibition (activity can be restored to normal levels upon removal of peroxide) by H2O2, D3A-NiSOD is inactivated (loss of activity is irreversible) by exposure to H2O2. This inactivation does not result from oxygenation of the cysteine residues, but appears to involve the oxidation of Tyr9 to a tyrosyl radical as detected by EPR. The crystal structure of D3A-NiSOD reveals that in addition to the loss of two intersubunit interactions per monomer resulting in a hexamer with lower thermal stability, the position of Tyr9 is altered. In D3A-NiSOD, the tyrosine is positioned such that the phenol group is ~1Å closer to the nickel site (Figure 8). In this configuration, it is in a position that would be less effective in inhibiting access of H2O2 to the nickel site, and could even stabilize formation of a Ni-OOH peroxo- or superoxo- adduct that results in oxidation of Tyr9.
Figure 8.

Superposition of the Ni-hook domains of WT- (blue), Y9F- (purple) and D3A- (Orange) NiSODs showing the position of Y9 in each. Image was generated in PyMOL (75).
Mutation of Tyr34 in MnSOD leads to a more product inhibited form of the enzyme, which is pronounced in hMnSOD and not as evident in E. coli MnSOD (Ec MnSOD)(73). To probe this inhibition further, Phe66 from hMnSOD was studied because it contributes to the local environment of Tyr34(74). Phe66 also lies at a dimer interface of hMnSOD and when this was mutated to an Ala, there were no major perturbations as the enzyme acted very similar to WT-hMnSOD. F66L-hMnSOD showed a decrease in product inhibition and had characteristics similar to that of Ec MnSOD. Crystallographic characterization of F66L-hMnSOD has provided insight into why this particular mutant had decreased product inhibition. The substitution of Phe 66 by Leu had an effect on two residues: First, Gln 119 moved approximately 1 Å further from the Mn site, but maintained the same distance between residues 66 and 119 as seen in WT-hMnSOD. Tyr34 also had a subtle movement of 0.3 Å away from the Mn center. This movement is small and has a high uncertainty due to the resolution of the structure (2.2 Å). This is consistent with the data presented here in that inward movement of the “gateway” tyrosine (Tyr9-NiSOD Tyr34-MnSOD), leads to a more product sensitive enzyme. When the corresponding residue swings away from the metal center, this inhibition is decreased in hMnSOD and is not seen in NiSOD.
The angle of the aromatic ring plane of residue 9 (Figure 8) may also act as a sensor for the presence of bound metal. When comparing the location of Y9 in the holo- and apo-NiSOD structures, the angle of the residue 9 aromatic ring plane changes by 15° between the two structures. This difference is consistent in all 12 copies of the holoenzyme monomer in PDB 1T6U and all 6 copies of the apoenzyme in 1T6I and 1T6Q. Thus, the aromatic ring may also be a sensor for the presence of a metal in the active site of the enzyme. In the case of the Y9F-NiSOD mutant structure, the angle of the aromatic ring of residue 9 is more consistent with the apo structures, producing a structure intermediate between the active holoenzyme and the inactive apoenzyme. The Y9F mutant is the first structure where the angle of residue #9 side chain is decoupled from the presence of metal in the active site of the enzyme. This observation is consistent with the result that the Y9F mutation does not bind nickel as efficiently as the wild type enzyme (the crystals have only 60% occupancy). Additionally, the eight N-terminal residues that comprise the Ni-hook domain have higher B factors and lower quality density than the rest of the structure, suggesting some heterogeneity in the region around the Ni center. It appears that the Ni is required to properly form a well ordered static “Ni-hook” region at the N-terminus.
Structural homology between MnSOD and NiSOD
The analogous functional roles ascribed to Tyr9 in NiSOD and Tyr34 in MnSOD are also reflected in their structural relationship with their respective metal centers. Both tyrosine phenol oxygen atoms lie at a M-O distance of ca. 5.5 Å, and are engaged in well-defined hydrogen bonding interactions. In addition, both are close to a second aromatic residue. In the case of MnSOD, Tyr34 lies between Phe66 and the manganese center. The F66L-MnSOD mutation in hMnSOD results in perturbation of the positions of water molecules and hydrogen bonding interactions that leads to a mutant enzyme that is less sensitive to peroxide inhibition(74). In WT-NiSOD, Tyr62 lies ~ 13 Å from the nickel center and Tyr9 lies between it and the metal. Neither the structure of the nickel site (XAS, EPR) nor the kinetic properties of Y62F-NiSOD differ from significantly from WT-NiSOD. Although no similar function has been found for NiSOD-Tyr62 and human MnSOD-Phe66, they share the same spatial relationship to the respective Tyr residues near the metal sites. This is clearly shown in Figure 9, which shows a superposition of the active site of human MnSOD structure (PDB 1LUV) and WT-NiSOD (PDB 1T6U), where Phe66 from MnSOD and Tyr62 from NiSOD were constrained to overlap. Thus, it can be seen that despite the lack of any primary, secondary, tertiary, or quaternary structure, the two enzymes have arrived at the same mechanism for controlling access of anions and H2O2 to the active site.
Figure 9.
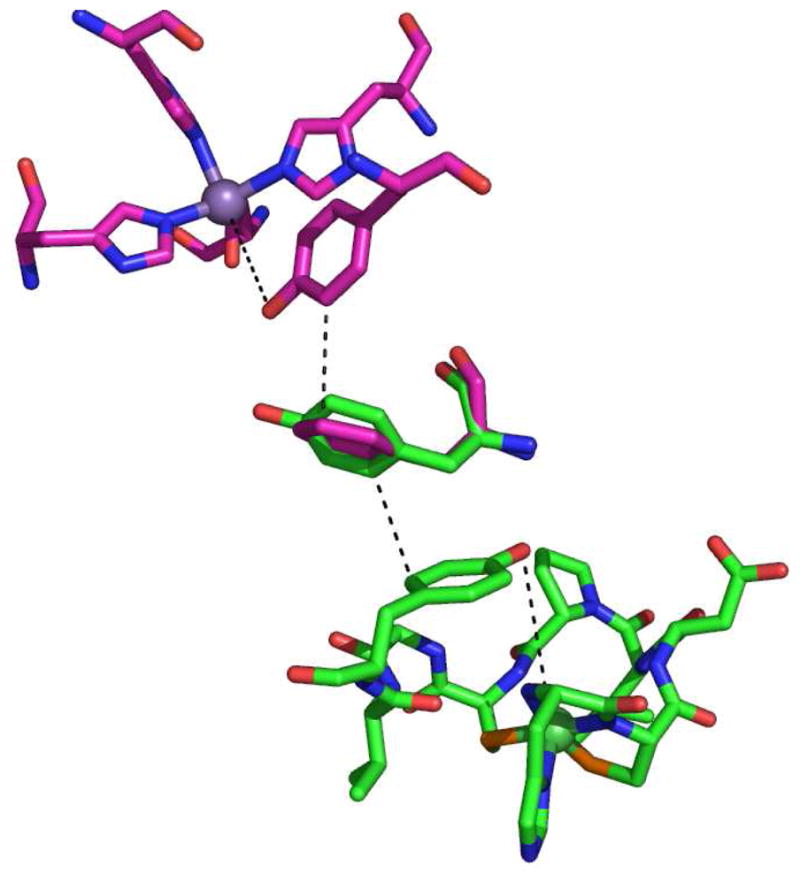
Superposition of the active site structures of hMnSOD (pink) and S. coelicolor NiSOD (green) showing the position of aromatic residues. Image was generated in PyMOL (75).
Supplementary Material
Table 1.
Mutagenic Primers for Y62F- and Y9FY62F-NiSOD
| Mutant | Forward Primer | Reverse Primer | Parental DNA |
|---|---|---|---|
| Y62F-NiSOD | 5′-GCTGTGGAGCGACTTCTTCAAGCCCCCGC-3′ | 5′-GCGGGGGCTTGAAGAAGTCGCTCCACAGC-3′ | WT-NiSOD |
| Y9FY62F-NiSOD | 5′-GCTGTGGAGCGACTTCTTCAAGCCCCCGC-3′ | 5′-GCGGGGGCTTGAAGAAGTCGCTCCACAGC-3′ | Y9F-NiSOD |
Acknowledgments
This work was supported by NSF grants MCB-0321482 and CHE-0809188 to MJM. SCG acknowledges the University of Massachusetts and the Charles H. Hood Foundation for support. The U.S. Department of Energy, Division of Materials Sciences and Division of Chemical Sciences supported XAS and x-ray diffraction data collection at the National Synchrotron Light Source at Brookhaven National Laboratory. The NIH supports Beamlines X3B and X6A at NSLS. Pulse radiolysis studies were carried out at the Center for Radiation Chemistry Research at BNL, which is funded under contract DE-AC02-98CH10886 with the U.S. Department of Energy and supported by its Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences.
Abbreviations
- SOD
Superoxide Dismutase
- XAS
X-Ray Absorption Spectroscopy
- EPR
Electron Paramagnetic Resonance
- NHE
Normal Hydrogen Electrode
- ROS
Reactive Oxygen Species
- PDTC
Bis-(pyridine-2, 6-bis(thiocarboxylate))nickel(III)
- EXAFS
Extended X-Ray Absorption Spectroscopy
- XANES
X-Ray Absorption Near Edge Structure
- ICP-OES
Inductively Coupled Plasma – Optical Emission Spectroscopy
- ESI-MS
Electrospray Ionization- Mass Spectrometry
- DSC
Differential Scanning Calorimetry
- Amp
Ampicillin
- Cam
Chloramphenicol
- OD
Optical Density
- FPLC
Fast Protein Liquid Chromatography
- IPTG
Isopropyl β-D-1-thiogalactopyranoside
Footnotes
Supporting information available (http://pubs.acs.org): Amino acid sequence alignments for selected NiSODs, DSC thermograms, ESI-MS under non-denaturing conditions, EPR spectra of WT- and mutant NiSODs and as-isolated and KMnO4-oxidized WT-NiSOD, Tables of EXAFS fits.
References
- 1.Cabelli DE, Riley D, Rodriguez JA, Valentine JS, Zhu H. Models of Superoxide Dismutases. In: Meunier B, editor. Biomimetic Oxidations Catalyzed by Transition Metal Complexes. Ch 10 1998. [Google Scholar]
- 2.Fridovich I. Superoxide radical and superoxide dismutases. Annu Rev Biochem. 1995;64:97–112. doi: 10.1146/annurev.bi.64.070195.000525. [DOI] [PubMed] [Google Scholar]
- 3.Miller AF, Sorkin D. Superoxide Dismutases: A Molecular Perspective. Comments Mol Cell Biophys. 1997;9:1–48. [Google Scholar]
- 4.Touati D. Superoxide Dismutases in Bacteria and Pathogen Protists. In: Scandalios JG, editor. Oxidative Stress and the Molecular Biology of Antioxidant Defenses. Cold Spring Harbor Laboratory Press; NY: 1997. pp. 447–493. [Google Scholar]
- 5.Lee JW, Roe JH, Kang SO. Nickel-containing superoxide dismutase. Superoxide Dismutase. 2002:90–101. doi: 10.1016/s0076-6879(02)49324-x. [DOI] [PubMed] [Google Scholar]
- 6.Uudsemaa M, Tamm T. Density-functional theory calculations of aqueous redox potentials of fourth-period transition metals. J Phys Chem A. 2003;107:9997–10003. [Google Scholar]
- 7.Jackson TA, Brunold TC. Combined spectroscopic/computational studies on Fe- and Mn-dependent superoxide dismutases: Insights into second-sphere tuning of active site properties. Acc Chem Res. 2004;37:461–470. doi: 10.1021/ar030272h. [DOI] [PubMed] [Google Scholar]
- 8.Miller AF. Redox tuning over almost 1 V in a structurally conserved active site: lessons from Fe-containing superoxide dismutase. Acc Chem Res. 2008;41:501–510. doi: 10.1021/ar700237u. [DOI] [PubMed] [Google Scholar]
- 9.Vance CK, Miller AF. Simple proposal that can explain the inactivity of metal-substituted superoxide dismutases. J Am Chem Soc. 1998;120:461–467. [Google Scholar]
- 10.Rulisek L, Jensen KP, Lundgren K, Ryde U. The reaction mechanism of iron and manganese superoxide dismutases studied by theoretical calculations. J Comp Chem. 2006;27:1398–1414. doi: 10.1002/jcc.20450. [DOI] [PubMed] [Google Scholar]
- 11.Carrasco R, Morgenstern-Badarau I, Cano J. Two proton-one electron coupled transfer in iron and manganese superoxide dismutases: A density functional study. Inorganica Chimica Acta. 2007;360:91–101. [Google Scholar]
- 12.Miller AF, Padmakumar K, Sorkin DL, Karapetian A, Vance CK. Proton-coupled electron transfer in Fe-superoxide dismutase and Mn-superoxide dismutase. J Inorg Biochem. 2003;93:71–83. doi: 10.1016/s0162-0134(02)00621-9. [DOI] [PubMed] [Google Scholar]
- 13.Fisher CL, Cabelli DE, Hallewell RA, Beroza P, Lo TP, Getzoff ED, Tainer JA. Computational, pulse-radiolytic, and structural investigations of lysine-136 and its role in the electrostatic triad of human Cu,Zn superoxide dismutase. Proteins-Structure Function and Genetics. 1997;29:103–112. doi: 10.1002/(sici)1097-0134(199709)29:1<103::aid-prot8>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 14.Vance CK, Miller AF. Spectroscopic comparisons of the pH dependencies of Fe-substituted (Mn)superoxide dismutase and Fe-superoxide dismutase. Biochemistry. 1998;37:5518–5527. doi: 10.1021/bi972580r. [DOI] [PubMed] [Google Scholar]
- 15.Miller AF, Sorkin DL, Padmakumar K. Anion binding properties of reduced and oxidized iron-containing superoxide dismutase reveal no requirement for tyrosine 34. Biochemistry. 2005;44:5969–5981. doi: 10.1021/bi0476331. [DOI] [PubMed] [Google Scholar]
- 16.Tabares LC, Cortez N, Un S. Role of tyrosine-34 in the anion binding equilibria in manganese(II) superoxide dismutases. Biochemistry. 2007;46:9320–9327. doi: 10.1021/bi700438j. [DOI] [PubMed] [Google Scholar]
- 17.Barondeau DP, Kassmann CJ, Bruns CK, Tainer JA, Getzoff ED. Nickel Superoxide Dismutase Structure and Mechanism. Biochemistry. 2004;43:8038–8047. doi: 10.1021/bi0496081. [DOI] [PubMed] [Google Scholar]
- 18.Borgstahl GEO, Pokross M, Chehab R, Sekher A, Snell EH. Cryo-trapping the six-coordinate, distorted-octahedral active site of manganese superoxide dismutase. J Mol Bio. 2000;296:951–959. doi: 10.1006/jmbi.1999.3506. [DOI] [PubMed] [Google Scholar]
- 19.Kerfeld CA, Yoshida S, Tran KT, Yeates TO, Cascio D, Bottin H, Berthomieu C, Sugiura M, Boussac A. The 1.6 angstrom resolution structure of Fe-superoxide dismutase from the thermophilic cyanobacterium Thermosynechococcus elongatus. J Biol Inorg Chem. 2003;8:707–714. doi: 10.1007/s00775-003-0469-0. [DOI] [PubMed] [Google Scholar]
- 20.Strange RW, Antonyuk S, Hough MA, Doucette PA, Rodriguez JA, Hart PJ, Hayward LJ, Valentine JS, Hasnain SS. The structure of holo and metal-deficient wild-type human Cu, Zn superoxide dismutase and its relevance to familial amyotrophic lateral sclerosis. J Mol Bio. 2003;328:877–891. doi: 10.1016/s0022-2836(03)00355-3. [DOI] [PubMed] [Google Scholar]
- 21.Wuerges J, Lee JW, Yim YI, Yim HS, Kang SO, Carugo KD. Crystal structure of nickel-containing superoxide dismutase reveals another type of active site. Proc Natl Acad Sci USA. 2004;101:8569–8574. doi: 10.1073/pnas.0308514101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chohan BS, Maroney MJ. Selective oxidations of a dithiolate complex produce a mixed sulfonate/thiolate complex. Inorg Chem. 2006;45:1906–1908. doi: 10.1021/ic051982h. [DOI] [PubMed] [Google Scholar]
- 23.Grapperhaus CA, Darensbourg MY. Oxygen Capture by Sulfur in Nickel Thiolates. Acc Chem Res. 1998;31:451–459. [Google Scholar]
- 24.Kumar M, Colpas GJ, Day RO, Maroney MJ. Ligand Oxidation In A Nickel Thiolate Complex - A Model For The Deactivation Of Hydrogenase By O2. J Am Chem Soc. 1989;111:8323–8325. [Google Scholar]
- 25.Fiedler AT, Bryngelson PA, Maroney MJ, Brunold TC. Spectroscopic and Computational Studies of Ni Superoxide Dismutase: Electronic Structure Contributions to Enzymatic Function. J Am Chem Soc. 2005;127:5449–5462. doi: 10.1021/ja042521i. [DOI] [PubMed] [Google Scholar]
- 26.Pelmenschikov V, Siegbahn PEM. Nickel superoxide dismutase reaction mechanism studied by hybrid density functional methods. J Am Chem Soc. 2006;128:7466–7475. doi: 10.1021/ja053665f. [DOI] [PubMed] [Google Scholar]
- 27.Bertini I, Banci L, Piccioli M, Luchinat C. Spectroscopic Studies on Cu2zn2sod - a Continuous Advancement of Investigation Tools. Coord Chem Revs. 1990;100:67–103. [Google Scholar]
- 28.Banci L, Bertini I, Cabelli DE, Hallewell RA, Luchinat C, Viezzoli MS. Advances in the understanding of the structure-function relationship in Cu,Zn superoxide dismutase. Free Radic Res Commun. 1991;12–13(Pt 1):239–251. doi: 10.3109/10715769109145792. [DOI] [PubMed] [Google Scholar]
- 29.Getzoff ED, Cabelli DE, Fisher CL, Parge HE, Viezzoli MS, Banci L, Hallewell RA. Faster superoxide dismutase mutants designed by enhancing electrostatic guidance. Nature. 1992;358:347–351. doi: 10.1038/358347a0. [DOI] [PubMed] [Google Scholar]
- 30.Getzoff ED, Tainer JA, Weiner PK, Kollman PA, Richardson JS, Richardson DC. Electrostatic Recognition Between Superoxide And Copper, Zinc Superoxide-Dismutase. Nature. 1983;306:287–290. doi: 10.1038/306287a0. [DOI] [PubMed] [Google Scholar]
- 31.Jackson TA, Karapetian A, Miller AF, Brunold TC. Spectroscopic and computational studies of the azide-adduct of manganese superoxide dismutase: definitive assignment of the ligand responsible for the low-temperature thermochromism. J Am Chem Soc. 2004;126:12477–12491. doi: 10.1021/ja0482583. [DOI] [PubMed] [Google Scholar]
- 32.Maliekal J, Karapetian A, Vance C, Yikilmaz E, Wu Q, Jackson T, Brunold TC, Spiro TG, Miller AF. Comparison and contrasts between the active site PKs of Mn-superoxide dismutase and those of Fe-superoxide dismutase. J Am Chem Soc. 2002;124:15064–15075. doi: 10.1021/ja027319z. [DOI] [PubMed] [Google Scholar]
- 33.Hunter T, Ikebukuro K, Bannister WH, Bannister JV, Hunter GJ. The conserved residue tyrosine 34 is essential for maximal activity of iron-superoxide dismutase from Escherichia coli. Biochemistry. 1997;36:4925–4933. doi: 10.1021/bi9629541. [DOI] [PubMed] [Google Scholar]
- 34.Kruger HJ, Peng G, Holm RH. Low-Potential Nickel(Iii,Ii) Complexes - New Systems Based on Tetradentate Amidate Thiolate Ligands and the Influence of Ligand Structure on Potentials in Relation to the Nickel Site in Nife-Hydrogenases. Inorg Chem. 1991;30:734–742. [Google Scholar]
- 35.Reiter TA, Rusnak F. Electrochemical studies of the mono-Fe, Fe-Zn, and Fe-Fe metalloisoforms of bacteriophage lambda protein phosphatase. Biochemistry. 2004;43:782–790. doi: 10.1021/bi0356956. [DOI] [PubMed] [Google Scholar]
- 36.Watanabe T, Honda K. Measurement of the Extinction Coefficient of the Methyl Viologen Cation Radical and the Efficiency of Its Formation by Semiconductor Photocatalysis. J Phys Chem. 1982;86:2617–2619. [Google Scholar]
- 37.Bernhardt PV, Chen KI, Sharpe PC. Transition metal complexes as mediator-titrants in protein redox potentiometry. J Biol Inorg Chem. 2006;11:930–936. doi: 10.1007/s00775-006-0148-z. [DOI] [PubMed] [Google Scholar]
- 38.Leitch S, Bradley MJ, Rowe JL, Chivers PT, Maroney MJ. Nickel-specific response in the transcriptional regulator, Escherichia coli NikR. J Am Chem Soc. 2007;129:5085–5095. doi: 10.1021/ja068505y. [DOI] [PubMed] [Google Scholar]
- 39.Padden KM, Krebs JF, MacBeth CE, Scarrow RC, Borovik AS. Immobilized metal complexes in porous organic hosts: development of a material for the selective and reversible binding of nitric oxide. J Am Chem Soc. 2001;123:1072–1079. doi: 10.1021/ja003282b. [DOI] [PubMed] [Google Scholar]
- 40.Webb SM. SIXPack: A graphical user interface for XAS analysis using IFEFFIT. Physica Scripta. 2005;T115:1011–1014. [Google Scholar]
- 41.Ankudinov AL, Ravel B, Rehr JJ, Conradson SD. Real-space multiple-scattering calculation and interpretation of x-ray-absorption near-edge structure. Physical Review B. 1998;58:7565–7576. [Google Scholar]
- 42.Zabinsky SI, Rehr JJ, Ankudinov A, Albers RC, Eller MJ. Multiple-Scattering Calculations of X-Ray-Absorption Spectra. Physical Review B. 1995;52:2995–3009. doi: 10.1103/physrevb.52.2995. [DOI] [PubMed] [Google Scholar]
- 43.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CW, JRMS, editors. Methods in Enzymology : Macromolecular Crystallography, part A. Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 44.Collaborative Computational Project, N. The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 45.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 46.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallog. 1993;26:283–291. [Google Scholar]
- 47.Choudhury SB, Lee JW, Davidson G, Yim YI, Bose K, Sharma ML, Kang SO, Cabelli DE, Maroney MJ. Examination of the Nickel Site Structure and Reaction Mechanism in Streptomyces seoulensis Superoxide Dismutase. Biochemistry. 1999;38:3744–3752. doi: 10.1021/bi982537j. [DOI] [PubMed] [Google Scholar]
- 48.Solvay Chemicals, Inc. Technical Data Sheet HH-121. 2004. Revised April 2004. [Google Scholar]
- 49.Bryngelson PA, Arobo SE, Pinkham JL, Cabelli DE, Maroney MJ. Expression, reconstitution, and mutation of recombinant Streptomyces coelicolor NiSOD. J Am Chem Soc. 2004;126:460–461. doi: 10.1021/ja0387507. [DOI] [PubMed] [Google Scholar]
- 50.Colpas GJ, Maroney MJ, Bagyinka C, Kumar M, Willis WS, Suib SL, Baidya N, Mascharak PK. X-Ray Spectroscopic Studies of Nickel-Complexes, with Application to the Structure of Nickel Sites in Hydrogenases. Inorg Chem. 1991;30:920–928. [Google Scholar]
- 51.Colpas GJ, Kumar M, Day RO, Maroney MJ. Structural Investigations of Nickel-Complexes with Nitrogen and Sulfur Donor Ligands. Inorg Chem. 1990;29:4779–4788. [Google Scholar]
- 52.Leveque VJ, Vance CK, Nick HS, Silverman DN. Redox properties of human manganese superoxide dismutase and active-site mutants. Biochemistry. 2001;40:10586–10591. doi: 10.1021/bi010792p. [DOI] [PubMed] [Google Scholar]
- 53.Rabani J, Nielsen SO. Absorption Spectrum and Decay Kinetics of O2- and Ho2 in Aqueous Solutions by Pulse Radiolysis. J Phys Chem. 1969;73:3736. [Google Scholar]
- 54.Quint PS, Domsic JF, Cabelli DE, McKenna R, Silverman DN. Role of a glutamate bridge spanning the dimeric interface of human manganese superoxide dismutase. Biochemistry. 2008;47:4621–4628. doi: 10.1021/bi7024518. [DOI] [PubMed] [Google Scholar]
- 55.Whittaker MM, Whittaker JW. A glutamate bridge is essential for dimer stability and metal selectivity in manganese superoxide dismutase. J Biol Chem. 1998;273:22188–22193. doi: 10.1074/jbc.273.35.22188. [DOI] [PubMed] [Google Scholar]
- 56.Szilagyi RK, Bryngelson PA, Maroney MJ, Hedman B, Hodgson KO, Solomon EI. S K-edge X-ray absorption spectroscopic investigation of the Ni-containing superoxide dismutase active site: New structural insight into the mechanism. J Am Chem Soc. 2004;126:3018–3019. doi: 10.1021/ja039106v. [DOI] [PubMed] [Google Scholar]
- 57.Barry BA, Einarsdottir O. Insights into the structure and function of redox- active tyrosines from model compounds. J Phys Chem B. 2005;109:6972–6981. doi: 10.1021/jp044749y. [DOI] [PubMed] [Google Scholar]
- 58.Fee JA, Valentine JS. Chemistry of O2−. In: Michelson AM, McCord JM, Fridovich I, editors. Superoxide and superoxide dismutases. Academic Press; London and New York: 1977. pp. 25–28. [Google Scholar]
- 59.Ge B, Scheller FW, Lisdat F. Electrochemistry of immobilized CuZnSOD and FeSOD and their interaction with superoxide radicals. Biosensors & Bioelectronics. 2003;18:295–302. doi: 10.1016/s0956-5663(02)00174-4. [DOI] [PubMed] [Google Scholar]
- 60.Leveque VJP, Vance CK, Nick HS, Silverman DN. Redox properties of human manganese superoxide dismutase and active-site mutants. Biochemistry. 2001;40:10586–10591. doi: 10.1021/bi010792p. [DOI] [PubMed] [Google Scholar]
- 61.St Clair CS, Gray HB, Valentine JS. Spectroelectrochemistry of Copper-Zinc Superoxide-Dismutase. Inorg Chem. 1992;31:925–927. [Google Scholar]
- 62.Vance CK, Miller AF. Novel insights into the basis for Escherichia coli superoxide dismutase’s metal ion specificity from Mn-substituted FeSOD and its very high E-m. Biochemistry. 2001;40:13079–13087. doi: 10.1021/bi0113317. [DOI] [PubMed] [Google Scholar]
- 63.Barrette WC, Jr, Sawyer DT, Fee JA, Asada K. Potentiometric titrations and oxidation-reduction potentials of several iron superoxide dismutases. Biochemistry. 1983;22:624–627. doi: 10.1021/bi00272a015. [DOI] [PubMed] [Google Scholar]
- 64.Lange NA, Dean JA. Lange’s Handbook of chemistry. 13. McGraw-Hill; New York: 1985. [Google Scholar]
- 65.Yikilmaz E, Porta J, Grove LE, Vahedi-Faridi A, Bronshteyn Y, Brunold TC, Borgstahl GE, Miller AF. How can a single second sphere amino acid substitution cause reduction midpoint potential changes of hundreds of millivolts? J Am Chem Soc. 2007;129:9927–9940. doi: 10.1021/ja069224t. [DOI] [PubMed] [Google Scholar]
- 66.Maroney MJ. Structure/function relationships in nickel metallobiochemistry. Curr Opin Chem Biol. 1999;3:188–199. doi: 10.1016/S1367-5931(99)80032-5. [DOI] [PubMed] [Google Scholar]
- 67.Ryan KC, Maroney MJ. Unpublished Results. [Google Scholar]
- 68.Guan Y, Hickey MJ, Borgstahl GEO, Hallewell RA, Lepock JR, O’Connor D, Hsieh YS, Nick HS, Silverman DN, Tainer JA. Crystal structure of Y34F mutant human mitochondrial manganese superoxide dismutase and the functional role of tyrosine 34. Biochemistry. 1998;37:4722–4730. doi: 10.1021/bi972394l. [DOI] [PubMed] [Google Scholar]
- 69.Hsu JL, Hsieh YS, Tu CK, Oconnor D, Nick HS, Silverman DN. Catalytic properties of human manganese superoxide dismutase. J Biol Chem. 1996;271:17687–17691. doi: 10.1074/jbc.271.30.17687. [DOI] [PubMed] [Google Scholar]
- 70.Whittaker MM, Whittaker JW. Low-temperature thermochromism marks a change in coordination for the metal ion in manganese superoxide dismutase. Biochemistry. 1996;35:6762–6770. doi: 10.1021/bi960088m. [DOI] [PubMed] [Google Scholar]
- 71.Whittaker MM, Whittaker JW. Mutagenesis of a proton linkage pathway in Escherichia coli manganese superoxide dismutase. Biochemistry. 1997;36:8923–8931. doi: 10.1021/bi9704212. [DOI] [PubMed] [Google Scholar]
- 72.Edwards RA, Whittaker MM, Whittaker JW, Baker EN, Jameson GB. Outer sphere mutations perturb metal reactivity in manganese superoxide dismutase. Biochemistry. 2001;40:15–27. doi: 10.1021/bi0018943. [DOI] [PubMed] [Google Scholar]
- 73.Abreu IA, Hearn A, An H, Nick HS, Silverman DN, Cabelli DE. The kinetic mechanism of manganese-containing superoxide dismutase from Deinococcus radiodurans: a specialized enzyme for the elimination of high superoxide concentrations. Biochemistry. 2008;47:2350–2356. doi: 10.1021/bi7016206. [DOI] [PubMed] [Google Scholar]
- 74.Zheng J, Domsic JF, Cabelli D, McKenna R, Silverman DN. Structural and Kinetic Study of Differences between Human and Escherichia coli Manganese Superoxide Dismutases. Biochemistry. 2007;46:14830–14837. doi: 10.1021/bi7014103. [DOI] [PubMed] [Google Scholar]
- 75.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; Palo Alto, CA: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.